
Assessing Clinical Potential of Old Antibiotics against Severe Infections by Multi-Drug-Resistant Gram-Negative Bacteria Using In Silico Modelling




Abstract
1. Introduction
2. Methods
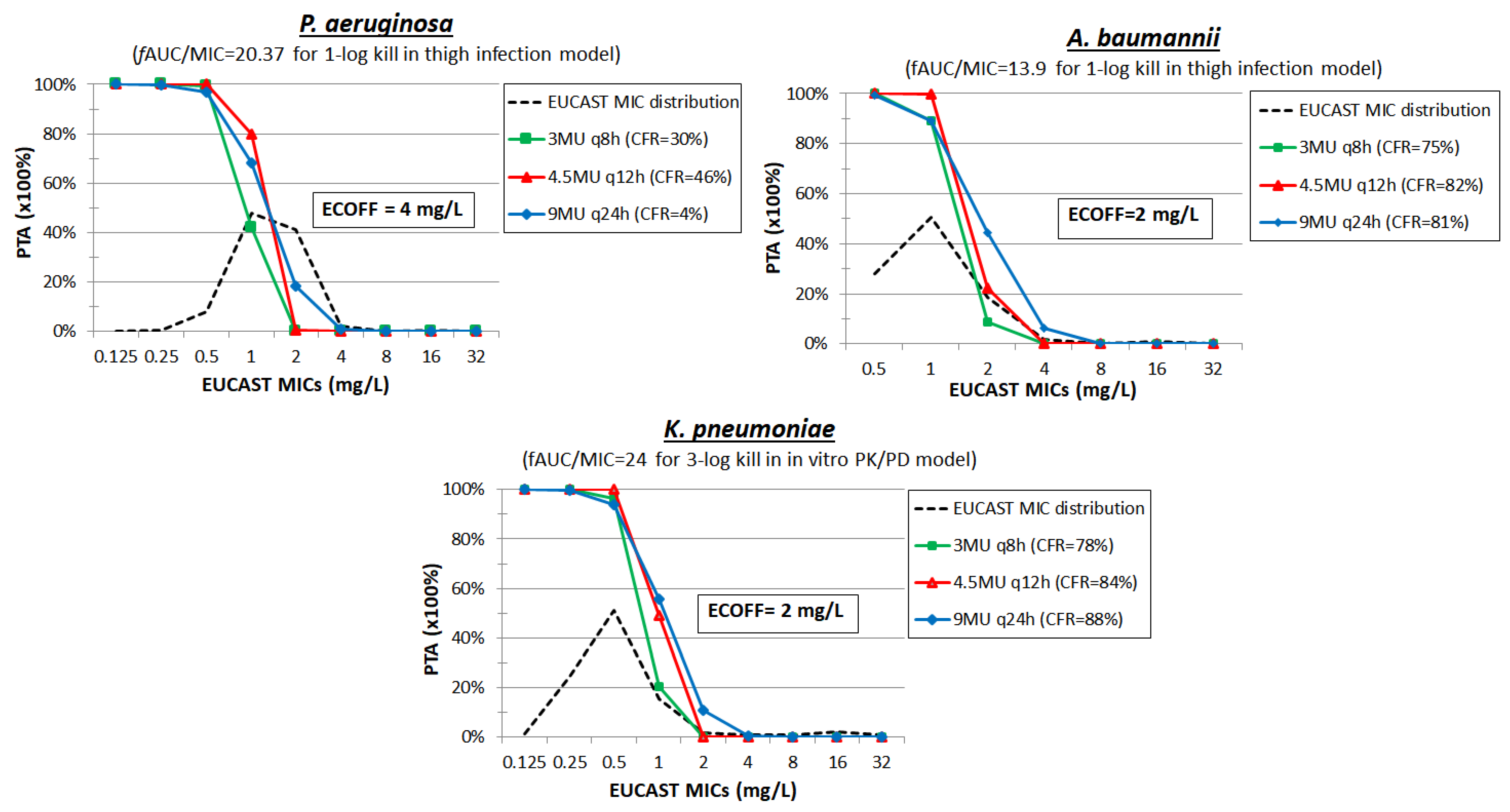
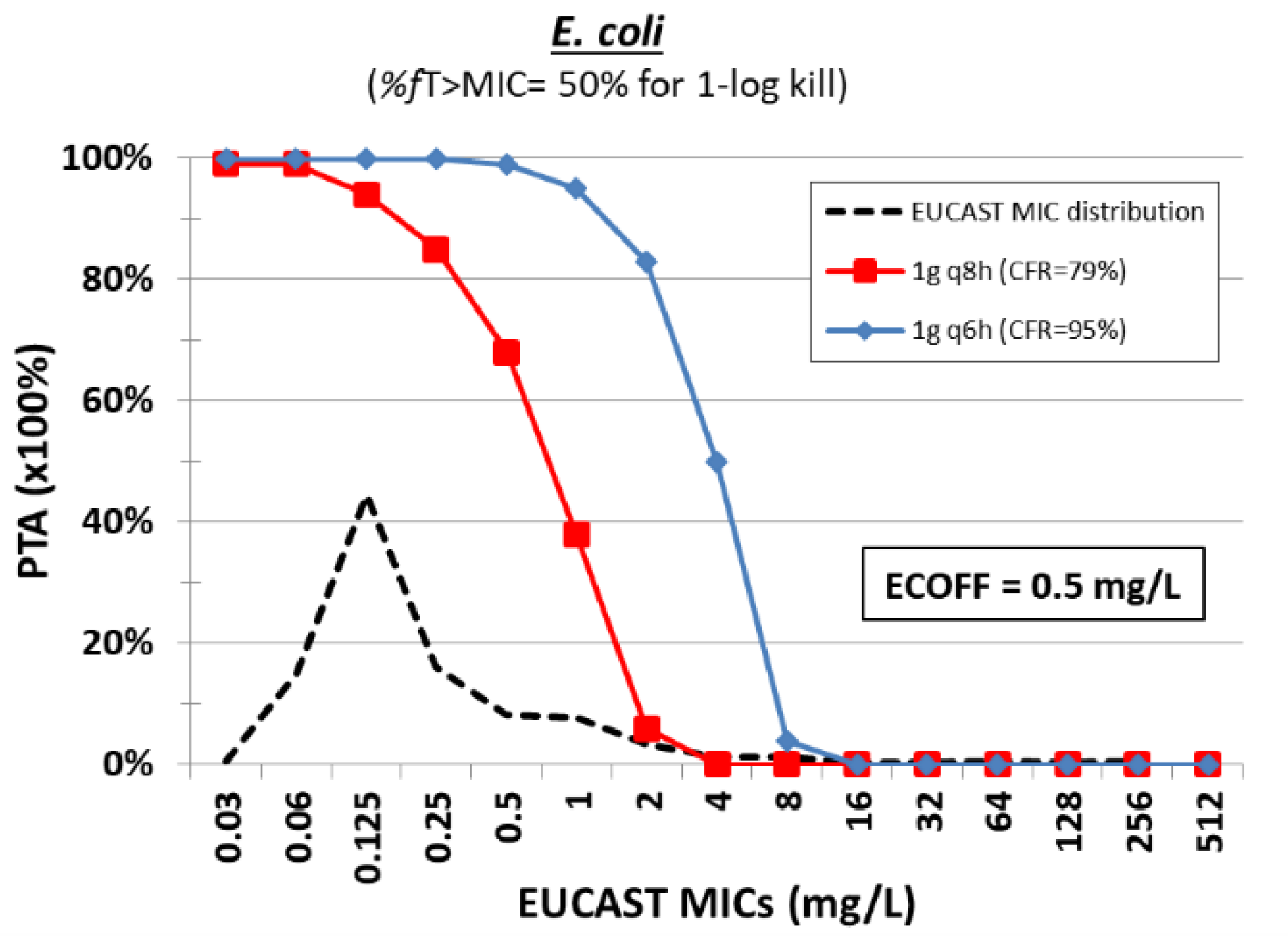
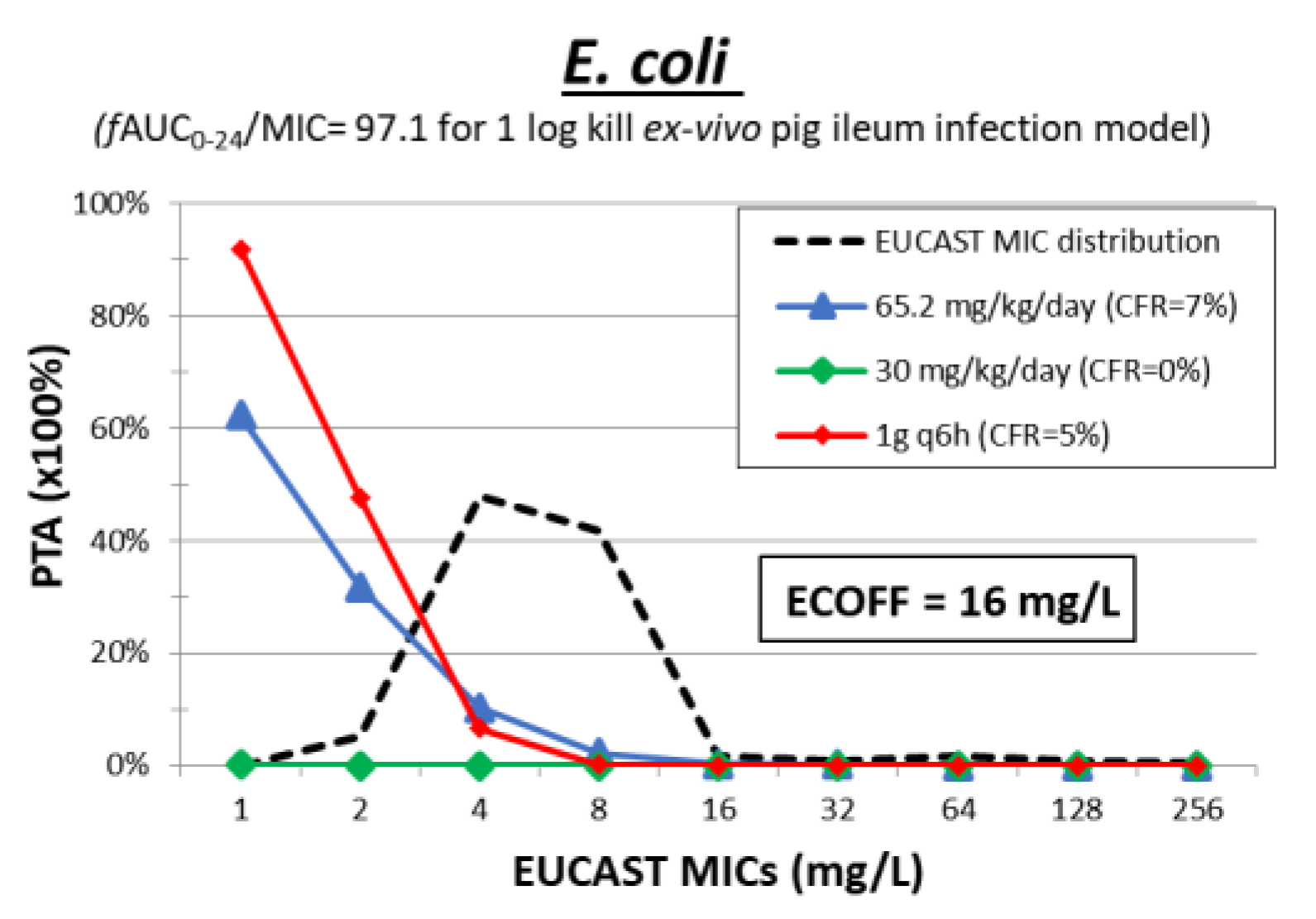
3. Colistin
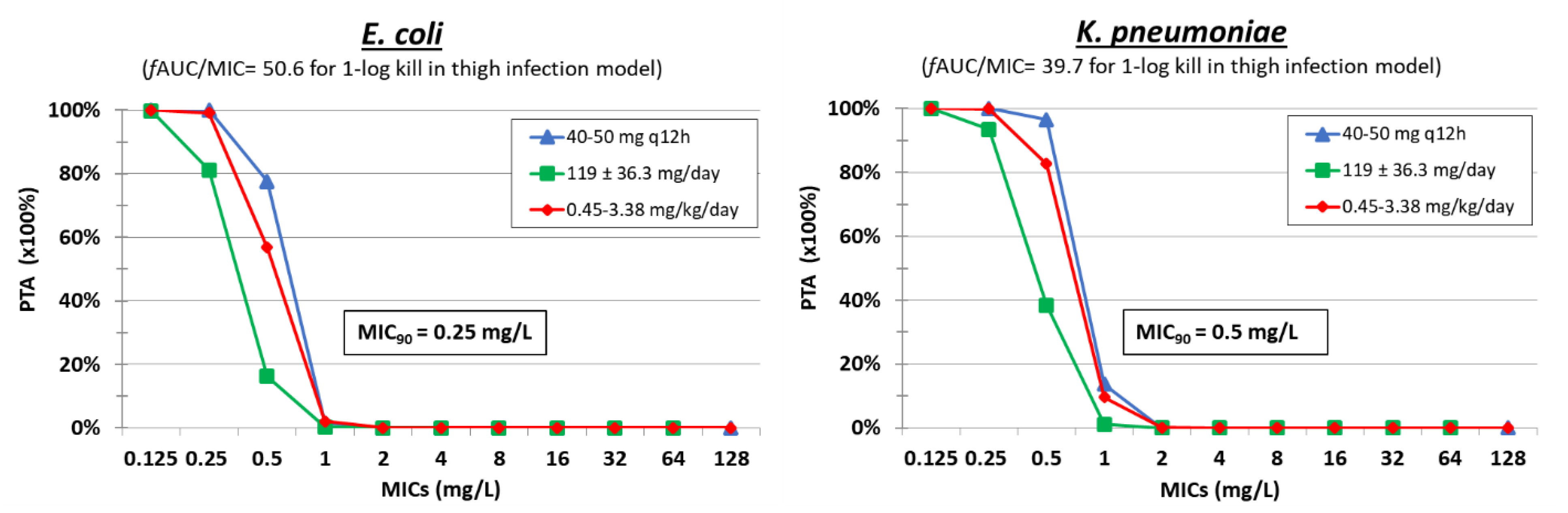
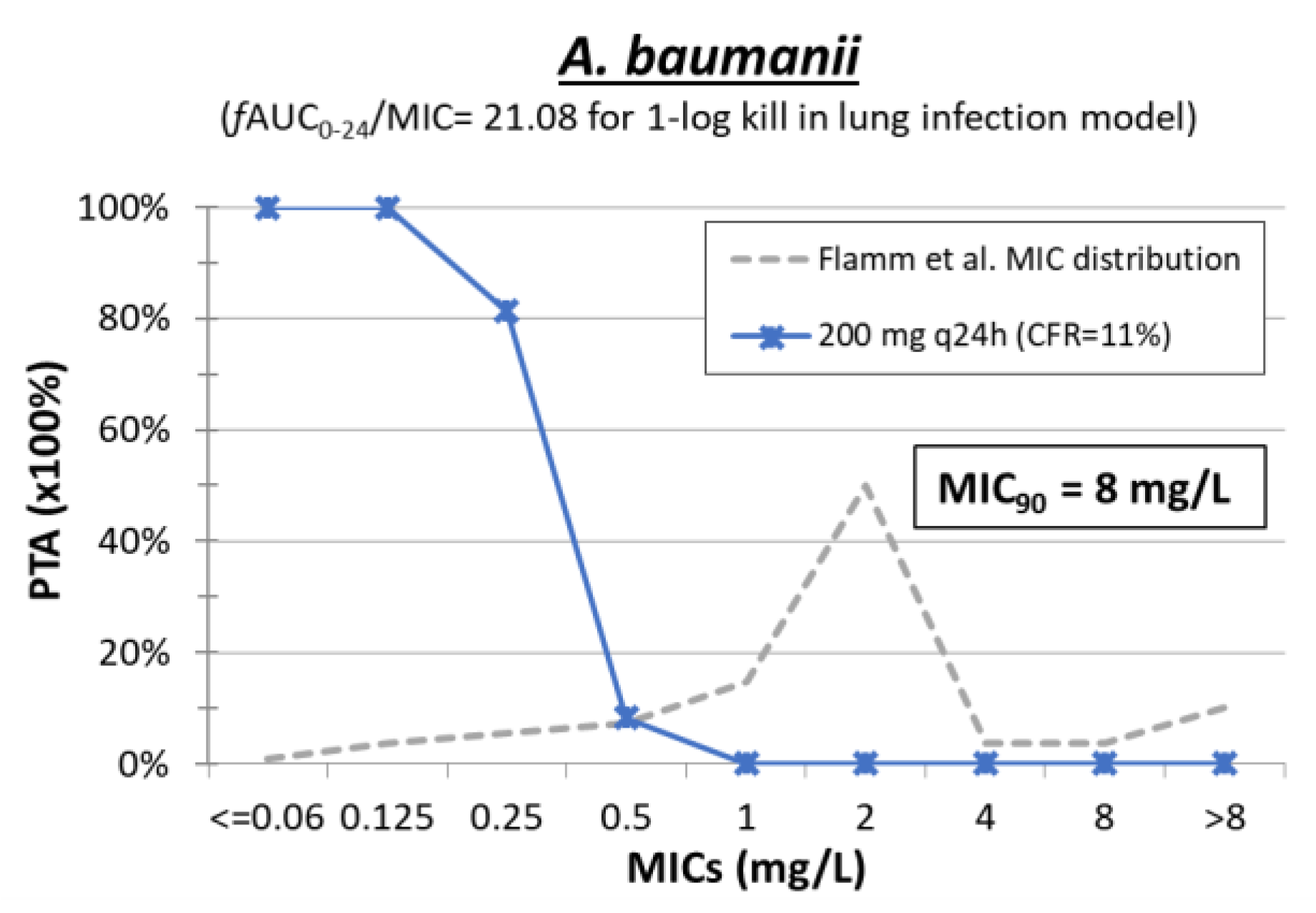
4. Polymyxin B
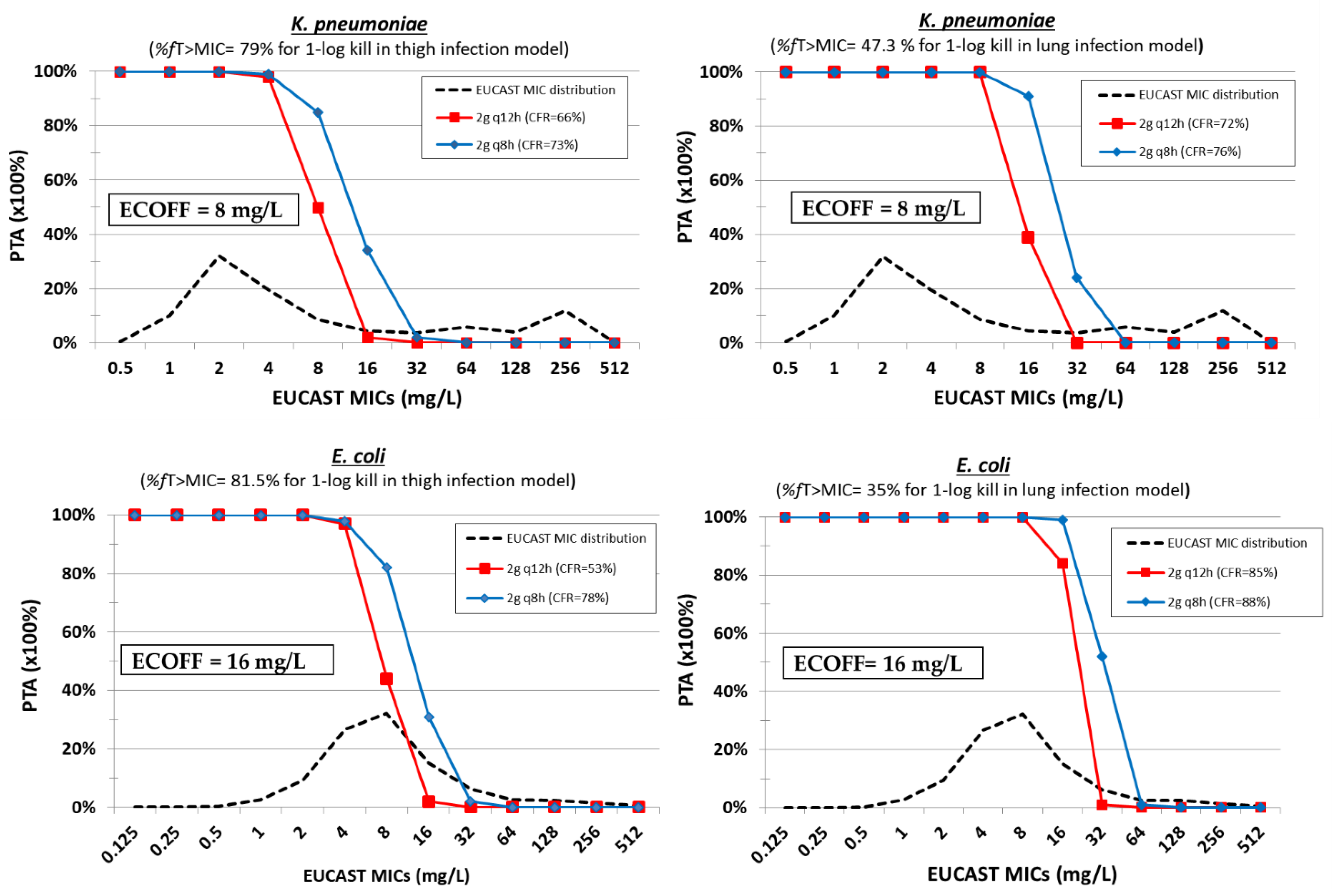
5. Temocillin
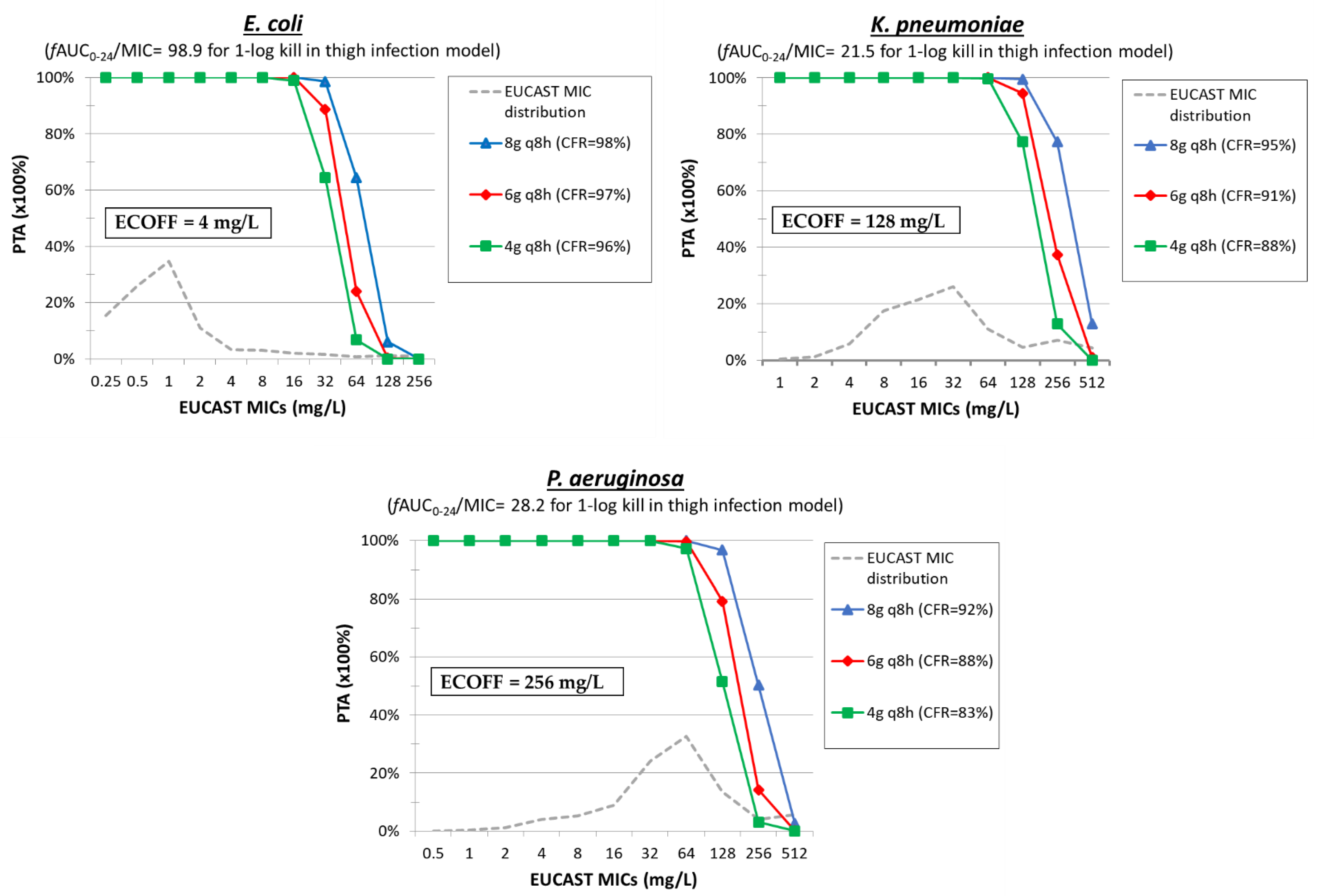
6. Fosfomycin
7. Mecillinam
8. Nitrofurantoin
9. Minocycline
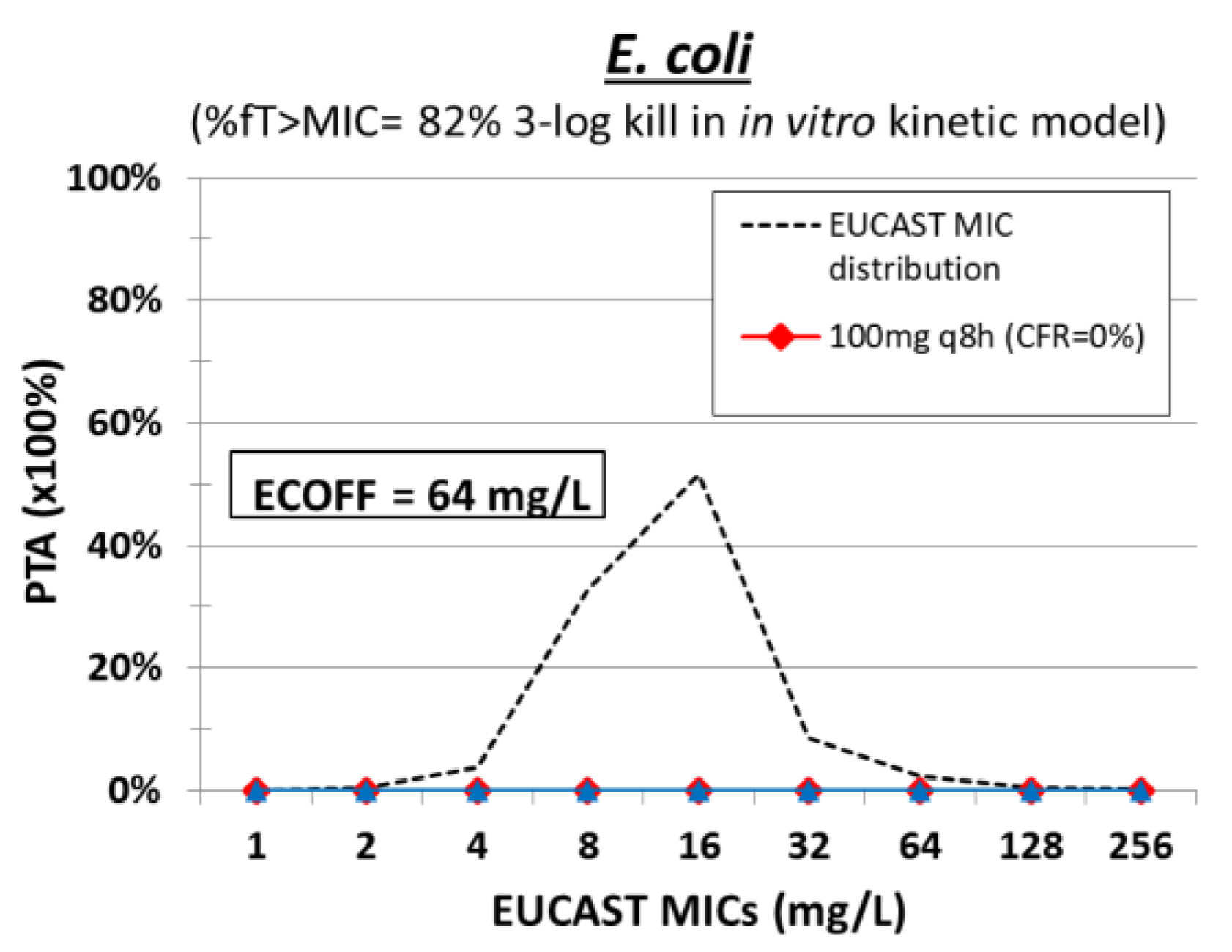
10. Chloramphenicol
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ECDC; WHO. Antimicrobial Resistance Surveillance in Europe; ECDC: Stockholm, Sweden, 2022; ISBN 9789294985521. [Google Scholar]
- Theuretzbacher, U.; Paul, M. Revival of old antibiotics: Structuring the re-development process to optimize usage. Clin. Microbiol. Infect. 2015, 21, 878–880. [Google Scholar] [CrossRef]
- Giske, C.G. Contemporary resistance trends and mechanisms for the old antibiotics colistin, temocillin, fosfomycin, mecillinam and nitrofurantoin. Clin. Microbiol. Infect. 2015, 21, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.E.; Theuretzbacher, U.; Mouton, J.W. Use of old antibiotics now and in the future from a pharmacokinetic/pharmacodynamic perspective. Clin. Microbiol. Infect. 2015, 21, 881–885. [Google Scholar] [CrossRef]
- Cassir, N.; Rolain, J.-M.; Brouqui, P. A new strategy to fight antimicrobial resistance: The revival of old antibiotics. Front. Microbiol. 2014, 5, 551. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Van Bambeke, F.; Cantó, R.; Giske, C.G.; Mouton, J.W.; Nation, R.L.; Paul, M.; Turnidge, J.D.; Kahlmeter, G. Reviving old antibiotics. J. Antimicrob. Chemother. 2015, 70, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Grammatikos, A.P.; Michalopoulos, A. Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev. Anti. Infect. Ther. 2008, 6, 593–600. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Use of Pharmacokinetics and Pharmacodynamics in the Development of Antibacterial Medicinal Products; European Medicines Agency: London, UK, 2016; pp. 1–17. [Google Scholar]
- Bulitta, J.B.; Hope, W.W.; Eakin, A.E.; Guina, T.; Tam, V.H.; Louie, A.; Drusano, G.L.; Hoover, J.L. Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data To Support Translation to Humans. Antimicrob. Agents Chemother. 2019, 63, e02307-18. [Google Scholar] [CrossRef]
- Tsala, M.; Vourli, S.; Kotsakis, S.; Daikos, G.; Tzouvelekis, L.; Zerva, L.; Miriagou, V.; Meletiadis, J. Pharmacokinetic-pharmacodynamic modeling of meropenem against VIM producing Klebsiella pneumoniae isolates: Clinical implications. J. Med. Microbiol. 2015, 65, 211–218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mouton, J.W.; Dudley, M.N.; Cars, O.; Derendorf, H.; Drusano, G.L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: An update. J. Antimicrob. Chemother. 2005, 55, 601–607. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Colistin: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2010. [Google Scholar]
- Sader, H.S.; Farrell, D.J.; Castanheira, M.; Flamm, R.K.; Jones, R.N. Antimicrobial activity of ceftolozane/tazobactam tested against Pseudomonas aeruginosa and Enterobacteriaceae with various resistance patterns isolated in European hospitals (2011–2012). J. Antimicrob. Chemother. 2014, 69, 2713–2722. [Google Scholar] [CrossRef]
- Mikhail, S.; Singh, N.B.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Castanheira, M.; Rybak, M.J. Evaluation of the Synergy of Ceftazidime-Avibactam in Combination with Meropenem, Amikacin, Aztreonam, Colistin, or Fosfomycin against Well-Characterized Multidrug-Resistant Klebsiella pneumoniae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e02233-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Xie, X.; Wu, X.H.; Li, X.; Zhao, Z.; Luo, S.; Wan, Z.; Liu, J.; Fu, L.; et al. In vitro and in vivo assessment of the antibacterial activity of colistin alone and in combination with other antibiotics against Acinetobacter baumannii and Escherichia coli. J. Glob. Antimicrob. Resist. 2020, 20, 351–359. [Google Scholar] [CrossRef]
- Thet, K.T.; Lunha, K.; Srisrattakarn, A.; Lulitanond, A.; Tavichakorntrakool, R.; Kuwatjanakul, W.; Charoensri, N.; Chanawong, A. Colistin heteroresistance in carbapenem-resistant Acinetobacter baumannii clinical isolates from a Thai university hospital. World J. Microbiol. Biotechnol. 2020, 36, 102. [Google Scholar] [CrossRef]
- Gales, A.C.; Jones, R.N.; Sader, H.S. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: Results from the SENTRY Antimicrobial Surveillance Program (2006–2009). J. Antimicrob. Chemother. 2011, 66, 2070–2074. [Google Scholar] [CrossRef]
- Kuti, J.L.; Wang, Q.; Chen, H.; Li, H.; Wang, H.; Nicolau, D.P. Defining the potency of amikacin against Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii derived from Chinese hospitals using CLSI and inhalation-based breakpoints. Infect. Drug Resist. 2018, 11, 783. [Google Scholar] [CrossRef]
- Bratu, S.; Tolaney, P.; Karumudi, U.; Quale, J.; Mooty, M.; Nichani, S.; Landman, D. Carbapenemase-producing Klebsiella pneumoniae in Brooklyn, NY: Molecular epidemiology and in vitro activity of polymyxin B and other agents. J. Antimicrob. Chemother. 2005, 56, 128–132. [Google Scholar] [CrossRef]
- Cielo, N.C.; Belmonte, T.; Raro, O.H.F.; da Silva, R.M.C.; Wink, P.L.; Barth, A.L.; da Cunha, G.R.; Mott, M.P.; Riche, C.V.W.; Dias, C.; et al. Polymyxin B broth disk elution: A feasible and accurate methodology to determine polymyxin B susceptibility in Enterobacterales. Diagn. Microbiol. Infect. Dis. 2020, 98, 115099. [Google Scholar] [CrossRef]
- Gales, A.C.; Jones, R.N.; Sader, H.S. Global assessment of the antimicrobial activity of polymyxin B against 54,731 clinical isolates of Gram-negative bacilli: Report from the SENTRY antimicrobial surveillance programme (2001–2004). Clin. Microbiol. Infect. 2006, 12, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Hermes, D.M.; Pormann Pitt, C.; Lutz, L.; Teixeira, A.B.; Ribeiro, V.B.; Netto, B.; Martins, A.F.; Zavascki, A.P.; Barth, A.L. Evaluation of heteroresistance to polymyxin B among carbapenem-susceptible and -resistant Pseudomonas aeruginosa. J. Med. Microbiol. 2013, 62, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, C.; Wang, Q.; Wang, X.; Chen, H.; Li, H.; Zhang, F.; Wang, H. Evaluation of the in vitro activity of new polymyxin B analogue SPR206 against clinical MDR, colistin-resistant and tigecycline-resistant Gram-negative bacilli. J. Antimicrob. Chemother. 2020, 75, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Zykov, I.N.; Sundsfjord, A.; Småbrekke, L.; Samuelsen, Ø. The antimicrobial activity of mecillinam, nitrofurantoin, temocillin and fosfomycin and comparative analysis of resistance patterns in a nationwide collection of ESBL-producing Escherichia coli in Norway 2010–2011. Infect. Dis. 2016, 48, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Villalobos, H.; Malaviolle, V.; Frankard, J.; de Mendonça, R.; Nonhoff, C.; Struelens, M.J. In vitro activity of temocillin against extended spectrum beta-lactamase-producing Escherichia coli. J. Antimicrob. Chemother. 2006, 57, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Tärnberg, M.; Östholm-Balkhed, Å.; Monstein, H.J.; Hällgren, A.; Hanberger, H.; Nilsson, L.E. In vitro activity of beta-lactam antibiotics against CTX-M-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Hope, R.; Fagan, E.J.; Warner, M.; Woodford, N.; Potz, N. Activity of temocillin against prevalent ESBL- and AmpC-producing Enterobacteriaceae from south-east England. J. Antimicrob. Chemother. 2006, 57, 1012–1014. [Google Scholar] [CrossRef]
- Glupczynski, Y.; Huang, T.D.; Berhin, C.; Claeys, G.; Delmée, M.; Ide, L.; Ieven, G.; Pierard, D.; Rodriguez-Villalobos, H.; Struelens, M.; et al. In vitro activity of temocillin against prevalent extended-spectrum beta-lactamases producing Enterobacteriaceae from Belgian intensive care units. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.; Fantin, B. Pharmacokinetics and Pharmacodynamics of Temocillin. Clin. Pharmacokinet. 2018, 57, 287–296. [Google Scholar] [CrossRef]
- Aprile, A.; Scalia, G.; Stefani, S.; Mezzatesta, M.L. In vitro fosfomycin study on concordance of susceptibility testing methods against ESBL and carbapenem-resistant Enterobacteriaceae. J. Glob. Antimicrob. Resist. 2020, 23, 286–289. [Google Scholar] [CrossRef]
- Kaase, M.; Szabados, F.; Anders, A.; Gatermann, S.G. Fosfomycin susceptibility in carbapenem-resistant Enterobacteriaceae from Germany. J. Clin. Microbiol. 2014, 52, 1893–1897. [Google Scholar] [CrossRef]
- Lai, B.; Zheng, B.; Li, Y.; Zhu, S.; Tong, Z. In vitro susceptibility of Escherichia coli strains isolated from urine samples obtained in mainland China to fosfomycin trometamol and other antibiotics: A 9-year surveillance study (2004–2012). BMC Infect. Dis. 2014, 14, 66. [Google Scholar] [CrossRef]
- Wootton, M.; Walsh, T.R.; Macfarlane, L.; Howe, R.A. Activity of mecillinam against Escherichia coli resistant to third-generation cephalosporins. J. Antimicrob. Chemother. 2010, 65, 79–81. [Google Scholar] [CrossRef]
- Marrs, E.C.L.; Day, K.M.; Perry, J.D. In vitro activity of mecillinam against Enterobacteriaceae with NDM-1 carbapenemase. J. Antimicrob. Chemother. 2014, 69, 2873–2875. [Google Scholar] [CrossRef] [PubMed]
- Wentao, N.; Guobao, L.; Jin, Z.; Junchang, C.; Rui, W.; Zhancheng, G.; Youning, L. In vitro activity of minocycline combined with aminoglycosides against Klebsiella pneumoniae carbapenemase-producing K. pneumoniae. J. Antibiot. 2018, 71, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Li, G.; Zhao, J.; Cui, J.; Wang, R.; Gao, Z.; Liu, Y. Use of Monte Carlo simulation to evaluate the efficacy of tigecycline and minocycline for the treatment of pneumonia due to carbapenemase-producing Klebsiella pneumoniae. Infect. Dis. 2018, 50, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Shortridge, D.; Castanheira, M.; Sader, H.S.; Pfaller, M.A. In Vitro Activity of Minocycline against U.S. Isolates of Acinetobacter baumannii-Acinetobacter calcoaceticus Species Complex, Stenotrophomonas maltophilia, and Burkholderia cepacia Complex: Results from the SENTRY Antimicrobial Surveillance Program, 2014. Antimicrob. Agents Chemother. 2019, 63, e01154-19. [Google Scholar] [CrossRef]
- Huang, L.Y.; Chen, T.L.; Lu, P.L.; Tsai, C.A.; Cho, W.L.; Chang, F.Y.; Fung, C.P.; Siu, L.K. Dissemination of multidrug-resistant, class 1 integron-carrying Acinetobacter baumannii isolates in Taiwan. Clin. Microbiol. Infect. 2008, 14, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Tsala, M.; Vourli, S.; Georgiou, P.-C.; Pournaras, S.; Tsakris, A.; Daikos, G.L.; Mouton, J.W.; Meletiadis, J. Exploring colistin pharmacodynamics against Klebsiella pneumoniae: A need to revise current susceptibility breakpoints. J. Antimicrob. Chemother. 2018, 73, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Cheah, S.-E.; Wang, J.; Nguyen, V.T.T.; Turnidge, J.D.; Li, J.; Nation, R.L. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: Smaller response in lung infection. J. Antimicrob. Chemother. 2015, 70, 3291–3297. [Google Scholar] [CrossRef]
- Ram, K.; Sheikh, S.; Bhati, R.K.; Tripathi, C.D.; Suri, J.C.; Meshram, G.G. Steady-state pharmacokinetic and pharmacodynamic profiling of colistin in critically ill patients with multi-drug–resistant gram-negative bacterial infections, along with differences in clinical, microbiological and safety outcome. Basic Clin. Pharmacol. Toxicol. 2021, 128, 128–140. [Google Scholar] [CrossRef]
- Plachouras, D.; Karvanen, M.; Friberg, L.E.; Papadomichelakis, E.; Antoniadou, A.; Tsangaris, I.; Karaiskos, I.; Poulakou, G.; Kontopidou, F.; Armaganidis, A.; et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by gram-negative bacteria. Antimicrob Agents Chemother 2009, 53, 3430–3436. [Google Scholar] [CrossRef]
- Daikos, G.L.; Skiada, A.; Pavleas, J.; Vafiadi, C.; Salatas, K.; Tofas, P.; Tzanetou, K.; Markogiannakis, A.; Thomopoulos, G.; Vafiadi, I.; et al. Serum bactericidal activity of three different dosing regimens of colistin with implications for optimum clinical use. J. Chemother. 2010, 22, 175–178. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Zhu, Z.Y.; Liu, Y.P.; Xu, P.; Li, X.; Xie, Y.L.; Yao, H.C.; Yang, L.; Zhang, B.K.; et al. Population Pharmacokinetics of Polymyxin B and Dosage Optimization in Renal Transplant Patients. Front. Pharmacol. 2021, 12, 2184. [Google Scholar] [CrossRef] [PubMed]
- Manchandani, P.; Thamlikitkul, V.; Dubrovskaya, Y.; Babic, J.T.; Lye, D.C.; Lee, L.S.; Tam, V.H. Population Pharmacokinetics of Polymyxin B. Clin. Pharmacol. Ther. 2018, 104, 534–538. [Google Scholar] [CrossRef]
- Sandri, A.M.; Landersdorfer, C.B.; Jacob, J.; Boniatti, M.M.; Dalarosa, M.G.; Falci, D.R.; Behle, T.F.; Bordinhão, R.C.; Wang, J.; Forrest, A.; et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: Implications for selection of dosage regimens. Clin. Infect. Dis. 2013, 57, 524–531. [Google Scholar] [CrossRef]
- De Joung, R.; Hens, R.; Basma, V.; Mouton, J.W.; Tulkens, P.M.; Carryn, S. Continuous versus intermittent infusion of temocillin, a directed spectrum penicillin for intensive care patients with nosocomial pneumonia: Stability, compatibility, population pharmacokinetic studies and breakpoint selection. J. Antimicrob. Chemother. 2008, 61, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Laterre, P.F.; Wittebole, X.; Van De Velde, S.; Muller, A.E.; Mouton, J.W.; Carryn, S.; Tulkens, P.M.; Dugernier, T. Temocillin (6 g daily) in critically ill patients: Continuous infusion versus three times daily administration. J. Antimicrob. Chemother. 2015, 70, 891–898. [Google Scholar] [CrossRef]
- Hampel, B.; Feike, M.; Koeppe, P.; Lode, H. Pharmacokinetics of temocillin in volunteers. Drugs 1985, 29 (Suppl. 5), 99–102. [Google Scholar] [CrossRef]
- Merino-Bohórquez, V.; Docobo-Pérez, F.; Sojo, J.; Morales, I.; Lupión, C.; Martín, D.; Cameán, M.; Hope, W.; Pascual; Rodríguez-Baño, J. Population pharmacokinetics and pharmacodynamics of fosfomycin in non–critically ill patients with bacteremic urinary infection caused by multidrug-resistant Escherichia coli. Clin. Microbiol. Infect. 2018, 24, 1177–1183. [Google Scholar] [CrossRef]
- Roholt, K. Pharmacokinetic studies with mecillinam and pivmecillinam. J. Antimicrob. Chemother. 1977, 3, 71–81. [Google Scholar] [CrossRef]
- Gambertoglio, J.G.; Barriere, S.L.; Lin, E.T.; Conte, J.E. Pharmacokinetics of mecillinam in health subjects. Antimicrob. Agents Chemother. 1980, 18, 952–956. [Google Scholar] [CrossRef]
- Huttner, A.; Wijma, R.A.; Stewardson, A.J.; Olearo, F.; Von Dach, E.; Harbarth, S.; Brüggemann, R.J.M.; Mouton, J.W.; Muller, A.E. The pharmacokinetics of nitrofurantoin in healthy female volunteers: A randomized crossover study. J. Antimicrob. Chemother. 2019, 74, 1656–1661. [Google Scholar] [CrossRef]
- Lodise, T.P.; Van Wart, S.; Sund, Z.M.; Bressler, A.M.; Khan, A.; Makley, A.T.; Hamad, Y.; Salata, R.A.; Silveira, F.P.; Sims, M.D.; et al. Pharmacokinetic and Pharmacodynamic Profiling of Minocycline for Injection following a Single Infusion in Critically Ill Adults in a Phase IV Open-Label Multicenter Study (ACUMIN). Antimicrob. Agents Chemother. 2021, 65, e01809-20. [Google Scholar] [CrossRef]
- Slaughter, R.L.; Pieper, J.A.; Cerra, F.B.; Brodsky, B.; Koup, J.R. Chloramphenicol sodium succinate kinetics in critically ill patients. Clin. Pharmacol. Ther. 1980, 28, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Acharya, G.P.; Davis, T.M.E.; Ho, M.; Harris, S.; Chataut, C.; Acharya, S.; Tuhladar, N.; Kafle, K.E.; Pokhrel, B.; Nosten, F.; et al. Factors affecting the pharmacokinetics of parenteral chloramphenicol in enteric fever. J. Antimicrob. Chemother. 1997, 40, 91–98. [Google Scholar] [CrossRef] [PubMed]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Temocillin: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2010. [Google Scholar]
- Dudhani, R.V.; Turnidge, J.D.; Coulthard, K.; Milne, R.W.; Rayner, C.R.; Li, J.; Nation, R.L. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob. Agents Chemother. 2010, 54, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, V.A.; Aranzana-Climent, H.; van der Spek, B.C.M.; de Winter, W.; Couet, J.; Meletiadis, A.E.; van den Muller, S.B. Pharmacokinetics and pharmodynamic properties of polymyxin B in murine infection models. 2022; Unpublished. [Google Scholar]
- Landersdorfer, C.B.; Wang, J.; Wirth, V.; Chen, K.; Kaye, K.S.; Tsuji, B.T.; Li, J.; Nation, R.L. Pharmacokinetics/pharmacodynamics of systemically administered polymyxin B against Klebsiella pneumoniae in mouse thigh and lung infection models. J. Antimicrob. Chemother. 2018, 73, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.E.; Raaphorst, M.; van der Meijden, A.; de Winter, B.C.M.; Meletiadis, J.; van den Muller, S.B. Pharmacodynamics of temocillin in neutropenic murine infection models. 2022, ahead of printing. 2022; ahead of printing. [Google Scholar]
- Lepak, A.J.; Zhao, M.; Vanscoy, B.; Taylor, D.S.; Ellis-Grosse, E.; Ambrose, P.G.; Andes, D.R. In Vivo Pharmacokinetics and Pharmacodynamics of ZTI-01 (Fosfomycin for Injection) in the Neutropenic Murine Thigh Infection Model against Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e00476-17. [Google Scholar] [CrossRef]
- Komp Lindgren, P.; Klockars, O.; Malmberg, C.; Cars, O. Pharmacodynamic studies of nitrofurantoin against common uropathogens. J. Antimicrob. Chemother. 2015, 70, 1076–1082. [Google Scholar] [CrossRef]
- Tarazi, Z.; Sabet, M.; Dudley, M.N.; Griffith, D.C. Pharmacodynamics of Minocycline against Acinetobacter baumannii in a Rat Pneumonia Model. Antimicrob. Agents Chemother. 2019, 63, e01671-18. [Google Scholar] [CrossRef]
- Lei, Z.; Liu, Q.; Khaliq, H.; Cao, J.; He, Q. Resistant cutoff values and optimal scheme establishments for florfenicol against Escherichia coli with PK-PD modeling analysis in pigs. J. Vet. Pharmacol. Ther. 2019, 42, 324–335. [Google Scholar] [CrossRef]
- Paul, M.; Daikos, G.L.; Durante-Mangoni, E.; Yahav, D.; Carmeli, Y.; Benattar, Y.D.; Skiada, A.; Andini, R.; Eliakim-Raz, N.; Nutman, A.; et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: An open-label, randomised controlled trial. Lancet. Infect. Dis. 2018, 18, 391–400. [Google Scholar] [CrossRef]
- Nation, R.L.; Li, J.; Cars, O.; Couet, W.; Dudley, M.N.; Kaye, K.S.; Mouton, J.W.; Paterson, D.L.; Tam, V.H.; Theuretzbacher, U.; et al. Framework for optimisation of the clinical use of colistin and polymyxin B: The Prato polymyxin consensus. Lancet. Infect. Dis. 2015, 15, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Trimble, M.J.; Mlynárčik, P.; Kolář, M.; Hancock, R.E.W. Polymyxin: Alternative Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025288. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nation, R.L.; Milne, R.W.; Turnidge, J.D.; Coulthard, K. Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int. J. Antimicrob. Agents 2005, 25, 11–25. [Google Scholar] [CrossRef]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Bishburg, E.; Bishburg, K. Minocycline--an old drug for a new century: Emphasis on methicillin-resistant Staphylococcus aureus (MRSA) and Acinetobacter baumannii. Int. J. Antimicrob. Agents 2009, 34, 395–401. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Q.; Feng, M.; Sun, T.; Yang, J.; Zhang, X. Population Pharmacokinetics of Polymyxin B in Obese Patients for Resistant Gram-Negative Infections. Front. Pharmacol. 2021, 12, 754844. [Google Scholar] [CrossRef]
- Chen, N.; Guo, J.; Xie, J.; Xu, M.; Hao, X.; Ma, K.; Rao, Y. Population pharmacokinetics of polymyxin B: A systematic review. Ann. Transl. Med. 2022, 10, 231. [Google Scholar] [CrossRef]
- Kubin, C.J.; Nelson, B.C.; Miglis, C.; Scheetz, M.H.; Rhodes, N.J.; Avedissian, S.N.; Cremers, S.; Yin, M.T. Population Pharmacokinetics of Intravenous Polymyxin B from Clinical Samples. Antimicrob. Agents Chemother. 2018, 62, e01493-17. [Google Scholar] [CrossRef]
- Avedissian, S.N.; Liu, J.; Rhodes, N.J.; Lee, A.; Pais, G.M.; Hauser, A.R.; Scheetz, M.H. A Review of the Clinical Pharmacokinetics of Polymyxin B. Antibiotics 2019, 8, 31. [Google Scholar] [CrossRef]
- Kvitko, C.H.; Rigatto, M.H.; Moro, A.L.; Zavascki, A.P. Polymyxin B versus other antimicrobials for the treatment of pseudomonas aeruginosa bacteraemia. J. Antimicrob. Chemother. 2011, 66, 175–179. [Google Scholar] [CrossRef]
- Fang, J.; Li, H.; Zhang, M.; Shi, G.; Liu, M.; Wang, Y.; Bian, X. Efficacy of Ceftazidime-Avibactam Versus Polymyxin B and Risk Factors Affecting Clinical Outcomes in Patients With Carbapenem-Resistant Klebsiella pneumoniae Infections a Retrospective Study. Front. Pharmacol. 2021, 12, 780940. [Google Scholar] [CrossRef] [PubMed]
- Ouderkirk, J.P.; Nord, J.A.; Turett, G.S.; Kislak, J.W. Polymyxin B nephrotoxicity and efficacy against nosocomial infections caused by multiresistant gram-negative bacteria. Antimicrob. Agents Chemother. 2003, 47, 2659–2662. [Google Scholar] [CrossRef] [PubMed]
- Rigatto, M.H.; Falci, D.R.; Zavascki, A.P. Clinical Use of Polymyxin B. Adv. Exp. Med. Biol. 2019, 1145, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Tulkens, P.M. Temocillin revived. J. Antimicrob. Chemother. 2009, 63, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Villalobos, H.; Bogaerts, P.; Berhin, C.; Bauraing, C.; Deplano, A.; Montesinos, I.; de Mendonça, R.; Jans, B.; Glupczynski, Y. Trends in production of extended-spectrum beta-lactamases among Enterobacteriaceae of clinical interest: Results of a nationwide survey in Belgian hospitals. J. Antimicrob. Chemother. 2011, 66, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Adams-Haduch, J.M.; Potoski, B.A.; Sidjabat, H.E.; Paterson, D.L.; Doi, Y. Activity of temocillin against KPC-producing Klebsiella pneumoniae and Escherichia coli. Antimicrob. Agents Chemother. 2009, 53, 2700–2701. [Google Scholar] [CrossRef]
- Chalhoub, H.; Pletzer, D.; Weingart, H.; Braun, Y.; Tunney, M.M.; Elborn, J.S.; Rodriguez-Villalobos, H.; Plésiat, P.; Kahl, B.C.; Denis, O.; et al. Mechanisms of intrinsic resistance and acquired susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients to temocillin, a revived antibiotic. Sci. Rep. 2017, 7, 40208. [Google Scholar] [CrossRef]
- Titelman, E.; Iversen, A.; Kahlmeter, G.; Giske, C.G. Antimicrobial susceptibility to parenteral and oral agents in a largely polyclonal collection of CTX-M-14 and CTX-M-15-producing Escherichia coli and Klebsiella pneumoniae. APMIS 2011, 119, 853–863. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.; Seo, M.R.; Wie, S.H.; Cho, Y.K.; Lim, S.K.; Lee, J.S.; Kwon, K.T.; Lee, H.; Cheong, H.J.; et al. Clinical characteristics of community-acquired acute pyelonephritis caused by ESBL-producing pathogens in South Korea. Infection 2013, 41, 603–612. [Google Scholar] [CrossRef]
- Balakrishnan, I.; Awad-El-Kariem, F.M.; Aali, A.; Kumari, P.; Mulla, R.; Tan, B.; Brudney, D.; Ladenheim, D.; Ghazy, A.; Khan, I.; et al. Temocillin use in England: Clinical and microbiological efficacies in infections caused by extended-spectrum and/or derepressed AmpC β-lactamase-producing Enterobacteriaceae. J. Antimicrob. Chemother. 2011, 66, 2628–2631. [Google Scholar] [CrossRef]
- Martinez-Beltran, J.; Loza, E.; Gomez-Alferez, A.; Romero-Vivas, J.; Bouza, E. Temocillin. In vitro activity compared with other antibiotics. Drugs 1985, 29 (Suppl. 5), 91–97. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.; Leysour De Rohello, F.; Dahyot, S.; Etienne, M.; Tiret, I.; Gillibert, A.; Pestel-Caron, M.; Caron, F. Efficacy of temocillin against MDR Enterobacterales: A retrospective cohort study. J. Antimicrob. Chemother. 2021, 76, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Athanasaki, F.; Voulgaris, G.L.; Triarides, N.A.; Vardakas, K.Z. Resistance to fosfomycin: Mechanisms, Frequency and Clinical Consequences. Int. J. Antimicrob. Agents 2019, 53, 22–28. [Google Scholar] [CrossRef] [PubMed]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Fosfomycin: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2022. [Google Scholar]
- Parker, S.; Lipman, J.; Koulenti, D.; Dimopoulos, G.; Roberts, J.A. What is the relevance of fosfomycin pharmacokinetics in the treatment of serious infections in critically ill patients? A systematic review. Int. J. Antimicrob. Agents 2013, 42, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Cao, M.; Hu, Y.; Zhang, R.; Xiao, Y.; Chen, G. Prevalence and mechanisms of fosfomycin resistance among KPC-producing Klebsiella pneumoniae clinical isolates in China. Int. J. Antimicrob. Agents 2021, 57, 106226. [Google Scholar] [CrossRef]
- Rodríguez-Gascón, A.; Canut-Blasco, A. Deciphering pharmacokinetics and pharmacodynamics of fosfomycin. Rev. Esp. Quimioter. 2019, 32, 19–24. [Google Scholar]
- Parker, S.L.; Frantzeskaki, F.; Wallis, S.C.; Diakaki, C.; Giamarellou, H.; Koulenti, D.; Karaiskos, I.; Lipman, J.; Dimopoulos, G.; Roberts, J.A. Population Pharmacokinetics of Fosfomycin in Critically Ill Patients. Antimicrob. Agents Chemother. 2015, 59, 6471–6476. [Google Scholar] [CrossRef]
- Kaye, K.S.; Rice, L.B.; Dane, A.L.; Stus, V.; Sagan, O.; Fedosiuk, E.; Das, A.F.; Skarinsky, D.; Eckburg, P.B.; Ellis-Grosse, E.J. Fosfomycin for Injection (ZTI-01) Versus Piperacillin-tazobactam for the Treatment of Complicated Urinary Tract Infection Including Acute Pyelonephritis: ZEUS, A Phase 2/3 Randomized Trial. Clin. Infect. Dis. 2019, 69, 2045–2056. [Google Scholar] [CrossRef]
- Nicolle, L.E. Pivmecillinam in the treatment of urinary tract infections. J. Antimicrob. Chemother. 2000, 46, 35–39. [Google Scholar] [CrossRef]
- Kerrn, M.B.; Frimodt-Møller, N.; Espersen, F. Urinary concentrations and urine ex-vivo effect of mecillinam and sulphamethizole. Clin. Microbiol. Infect. 2004, 10, 54–61. [Google Scholar] [CrossRef]
- Sullivan, Å.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B.G. The mechanism of action of mecillinam. J. Antimicrob. Chemother. 1977, 3 (Suppl. B), 13–19. [Google Scholar] [CrossRef] [PubMed]
- Thulin, E.; Sundqvist, M.; Andersson, D.I. Amdinocillin (Mecillinam) resistance mutations in clinical isolates and laboratory-selected mutants of Escherichia coli. Antimicrob. Agents Chemother. 2015, 59, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Perea, E.J.; Palomares, J.C.; García-Iglesias, M.C. In vitro evaluation of new penicillins and cephalosporins upon P. aeruginosa and their interaction with mecillinam. Chemotherapy 1980, 26, 282–288. [Google Scholar] [CrossRef]
- Thabaut, A.; Durosoir, J.L.; Saliou, P. Comparative in vitro antibacterial activity of seven semi-synthetic penicillins against aerobic gram-negative bacteria and enterococci. Infection 1982, 10 (Suppl. 3), S249–S256. [Google Scholar] [CrossRef]
- Jansåker, F.; Frimodt-Møller, N.; Benfield, T.L.; Knudsen, J.D. Mecillinam for the treatment of acute pyelonephritis and bacteremia caused by Enterobacteriaceae: A literature review. Infect. Drug Resist. 2018, 11, 761–771. [Google Scholar] [CrossRef]
- Dewar, S.; Reed, L.C.; Koerner, R.J. Emerging clinical role of pivmecillinam in the treatment of urinary tract infection in the context of multidrug-resistant bacteria. J. Antimicrob. Chemother. 2014, 69, 303–308. [Google Scholar] [CrossRef][Green Version]
- Gupta, K.; Hooton, T.M.; Naber, K.G.; Wullt, B.; Colgan, R.; Miller, L.G.; Moran, G.J.; Nicolle, L.E.; Raz, R.; Schaeffer, A.J.; et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: A 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin. Infect. Dis. 2011, 52, e103–e120. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Mecillinam: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2022. [Google Scholar]
- Neu, H.C. Amdinocillin: A novel penicillin. Antibacterial activity, pharmacology and clinical use. Pharmacotherapy 1985, 5, 1–10. [Google Scholar] [CrossRef]
- Shah, R.R.; Wade, G. Reappraisal of the risk/benefit of nitrofurantoin: Review of toxicity and efficacy. Advers. Drug React. Acute Poisoning Rev. 1989, 8, 183–201. [Google Scholar]
- Wijma, R.A.; Fransen, F.; Muller, A.E.; Mouton, J.W. Optimizing dosing of nitrofurantoin from a PK/PD point of view: What do we need to know? Drug Resist. Updat. 2019, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mc Osker, C.C.; Fitzpatrick, P.M. Nitrofurantoin: Mechanism of action and implications for resistance development in common uropathogens. J. Antimicrob. Chemother. 1994, 33 (Suppl. A), 23–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Wang, F.; Wang, C.; Chen, L.; Liu, H.; Lu, H.; Wen, H.; Zhou, T. Unravelling mechanisms of nitrofurantoin resistance and epidemiological characteristics among Escherichia coli clinical isolates. Int. J. Antimicrob. Agents 2018, 52, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Fransen, F.; Melchers, M.J.B.; Meletiadis, J.; Mouton, J.W. Pharmacodynamics and differential activity of nitrofurantoin against ESBL-positive pathogens involved in urinary tract infections. J. Antimicrob. Chemother. 2016, 71, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Stewardson, A.J.; Vervoort, J.; Adriaenssens, N.; Coenen, S.; Godycki-Cwirko, M.; Kowalczyk, A.; Huttner, B.D.; Lammens, C.; Malhotra-Kumar, S.; Goossens, H.; et al. Effect of outpatient antibiotics for urinary tract infections on antimicrobial resistance among commensal Enterobacteriaceae: A multinational prospective cohort study. Clin. Microbiol. Infect. 2018, 24, 972–979. [Google Scholar] [CrossRef]
- Stewardson, A.J.; Gaïa, N.; François, P.; Malhotra-Kumar, S.; Delémont, C.; Martinez de Tejada, B.; Schrenzel, J.; Harbarth, S.; Lazarevic, V.; Vervoort, J.; et al. Collateral damage from oral ciprofloxacin versus nitrofurantoin in outpatients with urinary tract infections: A culture-free analysis of gut microbiota. Clin. Microbiol. Infect. 2015, 21, 344-e1. [Google Scholar] [CrossRef]
- De Greeff, S.C.; Mouton, J.W. NethMap 2018: Consumption of Antimicrobial Agents and Antimicrobial Resistance among Medically Important Bacteria in the Netherlands/MARAN 2018: Monitoring of Antimicrobial Resistance and Antibiotic Usage in Animals in the Netherlands in 2017; National Institute for Public Health and the Environment: Bilthoven, The Netherlands, 2018. [CrossRef]
- Mezzatesta, M.L.; La Rosa, G.; Maugeri, G.; Zingali, T.; Caio, C.; Novelli, A.; Stefani, S. In vitro activity of fosfomycin trometamol and other oral antibiotics against multidrug-resistant uropathogens. Int. J. Antimicrob. Agents 2017, 49, 763–766. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Laing, N.M.; Nichol, K.A.; Palatnick, L.P.; Noreddin, A.; Hisanaga, T.; Johnson, J.L.; Hoban, D.J. Antibiotic activity against urinary tract infection (UTI) isolates of vancomycin-resistant enterococci (VRE): Results from the 2002 North American Vancomycin Resistant Enterococci Susceptibility Study (NAVRESS). J. Antimicrob. Chemother. 2003, 52, 382–388. [Google Scholar] [CrossRef][Green Version]
- Alamri, A.; Hassan, B.; Hamid, M. Susceptibility of hospital-acquired uropathogens to first-line antimicrobial agents at a tertiary health-care hospital, Saudi Arabia. Urol. Ann. 2021, 13, 166. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Nitrofurantoin: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2022. [Google Scholar]
- Wijma, R.A.; Huttner, A.; Koch, B.C.P.; Mouton, J.W.; Muller, A.E. Review of the pharmacokinetic properties of nitrofurantoin and nitroxoline. J. Antimicrob. Chemother. 2018, 73, 2916–2926. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Minocycline: Rationale for the EUCAST Clinical Breakpoints, version 1.0; EUCAST: Växjö, Sweden, 2022. [Google Scholar]
- Asadi, A.; Abdi, M.; Kouhsari, E.; Panahi, P.; Sholeh, M.; Sadeghifard, N.; Amiriani, T.; Ahmadi, A.; Maleki, A.; Gholami, M. Minocycline, focus on mechanisms of resistance, antibacterial activity, and clinical effectiveness: Back to the future. J. Glob. Antimicrob. Resist. 2020, 22, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Karageorgopoulos, D.E.; Falagas, M.E. Current control and treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet. Infect. Dis. 2008, 8, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.J.; Garavaglia-Wilson, A. A review of intravenous minocycline for treatment of multidrug-resistant Acinetobacter infections. Clin. Infect. Dis. 2014, 59 (Suppl. 6), S374–S380. [Google Scholar] [CrossRef] [PubMed]
- Scheetz, M.H.; Qi, C.; Warren, J.R.; Postelnick, M.J.; Zembower, T.; Obias, A.; Noskin, G.A. In vitro activities of various antimicrobials alone and in combination with tigecycline against carbapenem-intermediate or -resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2007, 51, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Fedorko, J.; Katz, S.; Allnoch, H. In vitro activity of minocycline, a new tetracycline. Am. J. Med. Sci. 1968, 255, 252–258. [Google Scholar] [CrossRef]
- Agwuh, K.N.; MacGowan, A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J. Antimicrob. Chemother. 2006, 58, 256–265. [Google Scholar] [CrossRef]
- Watanabe, A.; Anzai, Y.; Niitsuma, K.; Saito, M.; Yanase, K.; Nakamura, M. Penetration of minocycline hydrochloride into lung tissue and sputum. Chemotherapy 2001, 47, 1–9. [Google Scholar] [CrossRef]
- Summary Product Information MINOCIN® Minocycline For Injection 100 Mg/Vial Intravenous. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/050444s047lbl.pdf (accessed on 14 October 2022).
- Welling, P.G.; Shaw, W.R.; Uman, S.J.; Tse, F.L.; Craig, W.A. Pharmacokinetics of minocycline in renal failure. Antimicrob. Agents Chemother. 1975, 8, 532–537. [Google Scholar] [CrossRef]
- Zhou, J.; Ledesma, K.R.; Chang, K.T.; Abodakpi, H.; Gao, S.; Tama, V.H. Pharmacokinetics and Pharmacodynamics of Minocycline against Acinetobacter baumannii in a Neutropenic Murine Pneumonia Model. Antimicrob. Agents Chemother. 2017, 61, e02371-16. [Google Scholar] [CrossRef]
- Alfouzan, W.A.; Noel, A.R.; Bowker, K.E.; Attwood, M.L.G.; Tomaselli, S.G.; MacGowan, A.P. Pharmacodynamics of minocycline against Acinetobacter baumannii studied in a pharmacokinetic model of infection. Int. J. Antimicrob. Agents 2017, 50, 715–717. [Google Scholar] [CrossRef]
- Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Joslyn, D.A.; Burkholder, P.R. Chloromycetin, a New Antibiotic From a Soil Actinomycete. Science 1947, 106, 417. [Google Scholar] [CrossRef]
- Rajput, A.; Saxena, R.; Singh, K.P.; Kumar, V.; Singh, S.; Gupta, A.; Singh, R.K. Prevalence and antibiotic resistance pattern of metallo-beta-lactamase-producing Pseudomonas aeruginosa from burn patients—experience of an Indian tertiary care hospital. J. Burn Care Res. 2010, 31, 264–268. [Google Scholar] [CrossRef]
- Ahangarzadeh Rezaee, M.; Langarizadeh, N.; Aghazadeh, M. First report of class 1 and class 2 integrons in multidrug-resistant Klebsiella pneumoniae isolates from northwest Iran. Jpn. J. Infect. Dis. 2012, 65, 256–259. [Google Scholar] [CrossRef]
- Burke, J.T.; Wargin, W.A.; Sherertz, R.J.; Sanders, K.L.; Blum, M.R.; Sarubbi, F.A. Pharmacokinetics of intravenous chloramphenicol sodium succinate in adult patients with normal renal and hepatic function. J. Pharmacokinet. Biopharm. 1982, 10, 601–614. [Google Scholar] [CrossRef]
- Zayyad, H.; Eliakim-Raz, N.; Leibovici, L.; Paul, M. Revival of old antibiotics: Needs, the state of evidence and expectations. Int. J. Antimicrob. Agents 2017, 49, 536–541. [Google Scholar] [CrossRef]
- Salem, A.H.; Zhanel, G.G.; Ibrahim, S.A.; Noreddin, A.M. Monte Carlo simulation analysis of ceftobiprole, dalbavancin, daptomycin, tigecycline, linezolid and vancomycin pharmacodynamics against intensive care unit-isolated methicillin-resistant Staphylococcus aureus. Clin. Exp. Pharmacol. Physiol. 2014, 41, 437–443. [Google Scholar] [CrossRef]
- Ontong, J.C.; Ozioma, N.F.; Voravuthikunchai Id, S.P.; Chusri Id, S. Synergistic antibacterial effects of colistin in combination with aminoglycoside, carbapenems, cephalosporins, fluoroquinolones, tetracyclines, fosfomycin, and piperacillin on multidrug resistant Klebsiella pneumoniae isolates. PLoS ONE 2021, 16, e0244673. [Google Scholar] [CrossRef]
- Tsala, M.; Vourli, S.; Georgiou, P.-C.; Pournaras, S.; Daikos, G.L.; Mouton, J.W.; Meletiadis, J. Triple combination of meropenem, colistin and tigecycline was bactericidal in a dynamic model despite mere additive interactions in chequerboard assays against carbapenemase-producing Klebsiella pneumoniae isolates. J. Antimicrob. Chemother. 2019, 74, 387–394. [Google Scholar] [CrossRef]
- Verbist, L.; Verhaegen, J. Effect of temocillin in combination with other beta-lactam antibiotics. Antimicrob. Agents Chemother. 1984, 25, 142–144. [Google Scholar] [CrossRef][Green Version]
- Berleur, M.; Guérin, F.; Massias, L.; Chau, F.; Poujade, J.; Cattoir, V.; Fantin, B.; de Lastours, V. Activity of fosfomycin alone or combined with temocillin in vitro and in a murine model of peritonitis due to KPC-3- or OXA-48-producing Escherichia coli. J. Antimicrob. Chemother. 2018, 73, 3074–3080. [Google Scholar] [CrossRef]
- Singkham-in, U.; Chatsuwan, T. Synergism of imipenem with fosfomycin associated with the active cell wall recycling and heteroresistance in Acinetobacter calcoaceticus-baumannii complex. Sci. Rep. 2022, 12, 230. [Google Scholar] [CrossRef]
- Albiero, J.; Mazucheli, J.; Dos Reis Barros, J.P.; Dos Anjos Szczerepa, M.M.; Belini Nishiyama, S.A.; Carrara-Marroni, F.E.; Sy, S.; Fidler, M.; Sy, S.K.B.; Bronharo Tognim, M.C. Pharmacodynamic Attainment of the Synergism of Meropenem and Fosfomycin Combination against Pseudomonas aeruginosa Producing Metallo-β-Lactamase. Antimicrob. Agents Chemother. 2019, 63, e00126-19. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Compound | No. of Isolates (Ref.) | Mechanism b | MIC Range (mg/L) | MIC50 (mg/L) | MIC90 (mg/L) | ECOFF c |
|---|---|---|---|---|---|---|
| Colistin | ||||||
| E. coli | 6014 a | NA | 2–128 | 0.5 | 1 | 2 |
| 715 [13] | ESBL | NA | ≤0.25 | 0.5 | ||
| K. pneumoniae | 1841 a | NA | 0.125–512 | 0.5 | 1 | 2 |
| 21 [14] | CRKP | 0.25–2 | ||||
| 633 [13] | ESBL | NA | 0.5 | 2 | ||
| P. aeruginosa | 19,270 a | NA | 0.06–128 | 1 | 2 | 4 |
| 698 [13] | MDR | NA | 1 | 2 | ||
| A. baumannii | 2879 a | NA | 0.5–32 | 1 | 2 | [2] |
| 106 [15] | NA | 0.125–32 | 0.5 | 1 | ||
| 75 [16] | CRAB | 0.5–2 | 1 | 2 | ||
| Polymyxin B | ||||||
| E. coli | 17,035 [17] | NA | NA | ≤0.5 | ≤0.5 | |
| 86 [18] | All including ESBL | 0.064–8 | 0.125 | 0.25 | ||
| K. pneumoniae | 96 [19] | CRE | 0.5–16 | 2 | 2 | |
| 173 [20] | CRE | ≤0.125–>64 | 2 | 32 | ||
| 186 [18] | All including ESBL | 0.064–4 | 0.25 | 0.5 | ||
| P. aeruginosa | 8705 [21] | NA | ≤1–>8 | ≤1 | 2 | |
| 124 [22] | CRPsA included | NA | 1 | 2 | ||
| A. baumannii | 18 [23] | OXA-23 | 0.06–0.5 | 0.25 | 0.25 | |
| 131 [18] | All including CRAB | 0.125–>32 | 0.25 | 0.5 | ||
| Temocillin | ||||||
| E. coli | 5702 a | NA | 0.125–512 | 8 | 32 | 16 |
| 105 [24] | ESBL | 2–32 | 8 | 16 | ||
| 162 [25] | ESBL | 1–64 | 8 | 16 | ||
| 198 [26] | CTX-M | 0.125–>128 | 8 | 16 | ||
| 293 [27] | CTX-M | 2–64 | 8 | 16 | ||
| 40 [27] | AmpC | 2-64 | 8 | 16 | ||
| K. pneumoniae | 605 a | NA | 0.5–512 | 2 | 256 | 8 |
| 23 [28] | Non-CTX-M ESBL | 4–32 | 16 | 32 | ||
| 199 [27] | CTX-M | 2–64 | 8 | 32 | ||
| P. aeruginosa | 104 [29] | NA | 64–≥256 | ≥256 | ≥256 | |
| A. baumannii | 51 [29] | NA | 2–≥256 | ≥256 | ≥256 | |
| Fosfomycin | ||||||
| E. coli | 2351 a | NA | 0.125–512 | 1 | 4 | 4 |
| 35 [30] | ESBL | ≤0.5–1024 | 1 | 32 | ||
| 24 [31] | KPC, MBL, OXA-48 | ≤0.25–256 | 1 | 256 | ||
| 528 [32] | ESBL | NA | NA | 2 | ||
| K. pneumoniae | 1396 a | NA | 0.5–512 | 32 | 256 | 128 |
| 50 [30] | KPC | 8–>2048 | 32 | >2048 | ||
| 27 [30] | NDM, OXA-48 | 16–>2048 | 512 | >2048 | ||
| 50 [31] | KPC, MBL, OXA-48 | 0.5–>1024 | 16 | 256 | ||
| P. aeruginosa | 701 a | NA | 1–512 | 64 | 128 | 256 |
| Mecillinam | ||||||
| E. coli | 1502 a | NA | 0.03–512 | 0.125 | 2 | [0.5] |
| 198 [26] | CTX-M | 0.125–>128 | 1 | 8 | ||
| 30 [33] | Resistance to C3G | 0.5 | 4 | |||
| 29 [34] | NDM | 0.5–32 | 4 | 8 | ||
| K. pneumoniae | 175 a | NA | 0.03–512 | 0.25 | 128 | |
| 24 [34] | NDM | 2–>32 | 8 | >32 | ||
| Nitrofurantoin | ||||||
| E. coli | 4000 a | NA | 1–256 | 16 | 16 | 64 |
| 105 [24] | ESBL | 2–512 | 16 | 64 | ||
| 528 [32] | ESBL | NA | NA | 64 | ||
| Minocycline | ||||||
| E. coli | 1498 a | NA | 0.125–64 | 1 | 16 | 4 |
| K. pneumoniae | 938 a | NA | 0.125–64 | 2 | 16 | 8 |
| 70 [35] | KPC | 0.06–64 | 4 | 16 | ||
| 164 [36] | CP-Kp | NA | 8 | 32 | ||
| A. baumannii | 539 [37] | MDR | 0.06–8 | 2 | >8 | |
| 401 [37] | XDR | 0.06–8 | 2 | >8 | ||
| Chloramphenicol | ||||||
| E. coli | 45,852 a | NA | 1–256 | 4 | 8 | 16 |
| K. pneumoniae | 65 a | NA | 1–256 | 4 | 8 | |
| A. baumannii | 202 [38] | MDR | ≤2–≥32 | ≥32 | ≥32 |
| Drug | Dosage Regimen | Patient Population (Ref.) | CL (L/h) | VD in L (Mean ± SD) | % Unbound | AUC0–24 (mg·h/L) (Mean ± SD) | Cmax (mg/L) (Mean ± SD) | t1/2 (h) (Mean ± SD) |
|---|---|---|---|---|---|---|---|---|
| Colistin | 3 MU q8h | 13 ICU patients [43] | 40% | 50.18 ± 10.74 | 3.34 ± 0.35 | 7.8 ± 0.76 | ||
| 4.5 MU q12h | 13 ICU patients [43] | 40% | 60.71 ± 12.0 | 2.98 ± 0.27 | 8.8 ± 0.55 | |||
| 9 MU q24h | 13 ICU patients [43] | 40% | 72.93 ± 38.57 | 5.83 ± 0.87 | 9.6 ± 0.62 | |||
| Polymyxin B | 40–50 mg q12h | 50 renal transplant patients [44] | 1.18 ± 0.1 | 12.09 ± 1.58 | 74.6 ± 17.81 | 8.15 | ||
| 119 ± 36.3 mg q12h–q24h | 35 adult patients [45] | 2.5 ± 1.1 | 34.3 ± 16.4 | 52.3 ± 14.8/ 45.1 ± 17.3 | 10.1 | |||
| 0.45–3.38 mg/kg q12h–q24h | 24 critically ill patients [46] | 1.4 | V1 = 6.3, V2 = 23.1 | 42 | 66.9 ± 21.6 | 2.79 ± 0.90 d | 11.9 | |
| Temocillin | 2g q12h | 10 ICU patients [47] | 2.44 ± 0.39 | 14.3 ± 0.87 | 23.7 ± 6.15 | 1856 ± 282 | 147 ± 12 | 4.3 ± 0.3 |
| 2g q8h | 14 critically ill patients [48] | 3.69 ± 0.45 | V1 = 14 ± 2.51 V2 = 21.7 ± 4.52 | 41 | 1764 | 170 | ||
| 0.5g | 10 healthy volunteers [49] | 1.5 ± 0.09 | 10.5 ± 0.7 | 12 | 344.1 ± 18.7 | 77.9 ± 28.4 | 5.2 ± 0.3 | |
| 1 g | 10 healthy volunteers [49] | 1.78 ± 0.08 | 11.9 ± 0.7 | 14 | 573.3 ± 27.8 | 160.8 ± 58.2 | 5.0 ± 0.2 | |
| 2g | 10 healthy volunteers [49] | 2.62 ± 0.16 | 16.8 ± 0.7 | 37 | 784.5 ± 47.1 | 236.1 ± 93.3 | 5.0 ± 0.2 | |
| Fosfomycin | 4g q6h | 16 non-critically ill [50] | 2.43 ± 1.64 | 13.69 ± 2.81 | 100 c | 5215.08 ± 1972.2 | 422.6 ± 86.8 | |
| Mecillinam | 400 mg | 9 subjects [51] | 90–95 c | 22 ± 5 | 28 ± 5 | |||
| 200 mg | 9 subjects [51] | 90–95 c | 9.9 ± 1.5 | 12 ± 2 | ||||
| 10 mg/kg | 12 healthy volunteers [52] | 14.7 ± 1.4 | 16.1 ± 2.8 | 90–95 c | 61 | 0.85 ± 0.14 | ||
| Nitrofurantoin (oral) | 50 mg q6h | 12 healthy adult female [53] | 36.4 ± 11.4 | 100.0 ± 49.6 | 25–50 c | 4.43 ± 0.96 | 0.326 ± 0.081 | 2.3 ± 1.8 |
| 100 mg q8h | 12 healthy adult female [53] | 46.2 ± 18.6 | 103.8 ± 65.9 | 25–50 c | 6.49 ± 2.9 | 0.69 ± 0.35 | 1.7 ± 0.6 | |
| Minocycline | 200 mg | 55 critically ill patients [54] | 5.24 ± 2.63 | 146 ± 57 | 30 ± 12 | 24.3 ± 7.88 | 2.58 ± 1.33 | T1/2,α = 1.36 ± 0.456 T1/2,β = 23.4 ± 9.53 |
| Chloramphenicol Sodium succinate | 65.2 (32.3–114.4) mg/kg/day | 10 critically ill patients [55] | 21.24 ± 23.34 | 21 ± 8.4 | ~40 *a | 468 ± 498 | 1.20 ± 1.15 | |
| 30 mg/kg | 7 patients [56] | 22.08 ± 10.32 | 133 ± 56 | 34–63 *b | 72 ± 32 | 16.2 ± 9.1 | ||
| 1 g q6h | 8 patients [57] | 7.72 ± 1.87 | 23.1 ± 9.1 | ~40 *a | 518 | 8.4-26.0 | 0.57 ± 0.12 |
| Drug | Infection Model (Ref.) | Dose | Species | PK/PD INDEX | Stasis | 1-Log Kill | 2-Log Kill | 3-Log Kill |
|---|---|---|---|---|---|---|---|---|
| Colistin | Thigh [58] | 5–160 mg/kg/day q3h–q24h | P. aeruginosa (n = 3) MIC = 0.5–1 mg/L | fAUC/MIC | 13.35 ± 4.57 | 20.37 ± 4.13 | 31.63 ± 4.27 | 58.37 ± 7.27 |
| Lung [58] | 5–160 mg/kg/day q3h–q24h | P. aeruginosa (n = 3) MIC = 0.5–1 mg/L | fAUC/MIC | 5.31 ± 1.18 | 14.83 ± 2.35 | 40.13 ± 5.01 | 127 ± 19.29 | |
| In vitro [39] | fCmax 9, 3 and 1.5 mg/L q8h–q24 h | K. pneumoniae MDR(n = 2) MIC = 0.5–2 mg/L | fAUC/MIC | 10 ± 2.1 | 14 ± 2.4 | 18 ± 3.1 | 24 ± 4.7 | |
| Polymyxin B | Thigh [59] | 4–512 mg/kg q6h, q12h, q24h | E. coli MDR (n = 4) MIC = 1 mg/L | fAUC/MIC | 63.5 ± 34.8 | 50.6 ± 3.8 | ||
| Thigh [59] | 4–128 mg/kg q6h, 8–256 mg/kg q12h, 256– 512 mg/kg q24h | K. pneumoniae MDR (n = 5) MIC = 0.5–2 mg/L | fAUC/MIC | 11.6 ± 22.1 | 39.7 ± 14.4 | |||
| Thigh [60] | 0.5–120 mg/kg/day | K. pneumoniae (n = 3) MIC = 0.25–1 mg/L | fAUC/MIC | 6.73 ± 6.23 | 16.37 ± 12.17 | |||
| Temocillin | Thigh [61] | 8–512 mg/kg q2h, 16–512 mg/kg q4h | E. coli ESBL (n = 4) MIC = 8–16 mg/L | %fT > MIC | 66 ± 9.9 | 81.5 ± 14.4 | ||
| K. pneumoniae ESBL (n = 4) MIC = 8–64 mg/L | %fT > MIC | 63 ± 27.9 | 79 ± 6.4 | |||||
| Lung [61] | 16–1024 mg/kg q2h, 32–1024 mg/kg q4h | E. coli ESBL (n = 4) MIC = 8–16 mg/L | %fT > MIC | 27.8 ± 13.8 | 35 ± 18.3 | 42.8 ± 23 | ||
| K. pneumoniae ESBL (n = 4) MIC = 8–64 mg/L | %fT > MIC | 35.8 ± 23.6 | 47.3 ± 21.4 | |||||
| Fosfomycin | Thigh [62] | 12.5–6400 mg/kg/day q3h–q24h | E. coli ESBL (n = 5) MIC = 1–16 mg/L | fAUC0–24/MIC | 23.7 ± 15.3 | 98.9 ± 78.4 | ||
| Thigh [62] | 12.5–6400 mg/kg/day q3h–q24h | K. pneumoniae NDM (n = 3) MIC = 4–16 mg/L | fAUC0–24/MIC | 11.1 ± 19.5 | 21.5 (n = 1) | |||
| Thigh [62] | 12.5–6400 mg/kg/day q3h–q24h | P. aeruginosa (n = 2) MIC = 8–16 mg/L | fAUC0–24/MIC | 14.6 ± 4.7 | 28.2 ± 17.82 | |||
| Mecillinam | EUCAST RD | Enterobacterales | %fT > MIC | 30–35% | ~50% | |||
| Nitrofurantoin | In vitro kinetic model [63] | 16 mg/L | E. coli (n = 1) MIC = 2 mg/L | %fT > MIC | 72% | 82% | ||
| Minocycline | Lung [64] | 0.46–180 mg/kg/day q12h | A. baumannii (n = 6) MIC = 0.03–4 mg/L | fAUC0–24/MIC | 13.75 ± 3.76 | 21.08 ± 7.24 | ||
| Florfenicol * | Ex-vivo pig ileum [65] | 30 mg/kg Single dose | E. coli (n = 1) MIC = 8 mg/L | AUC0–24/MIC | 82.83 ± 3.52 | 97.1 ± 4.12 | 101.6 ± 7.74 |
| Old Antibiotics | E. coli | K. pneumoniae | P. aeruginosa | A. baumannii |
|---|---|---|---|---|
| Colistin | - | - | - | - |
| Polymyxin B | ✓ | ✓ | - | ✓ |
| Temocillin | ✓ | ✓ | - | - |
| Fosfomycin | ✓ | ✓ | - | - |
| Mecillinam | ✓ | - | - | - |
| Minocycline | - | - | - | - |
| Nitrofurantoin | - | - | - | - |
| Chloramphenicol | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paranos, P.; Vourli, S.; Pournaras, S.; Meletiadis, J. Assessing Clinical Potential of Old Antibiotics against Severe Infections by Multi-Drug-Resistant Gram-Negative Bacteria Using In Silico Modelling. Pharmaceuticals 2022, 15, 1501. https://doi.org/10.3390/ph15121501
Paranos P, Vourli S, Pournaras S, Meletiadis J. Assessing Clinical Potential of Old Antibiotics against Severe Infections by Multi-Drug-Resistant Gram-Negative Bacteria Using In Silico Modelling. Pharmaceuticals. 2022; 15(12):1501. https://doi.org/10.3390/ph15121501
Chicago/Turabian StyleParanos, Paschalis, Sophia Vourli, Spyros Pournaras, and Joseph Meletiadis. 2022. "Assessing Clinical Potential of Old Antibiotics against Severe Infections by Multi-Drug-Resistant Gram-Negative Bacteria Using In Silico Modelling" Pharmaceuticals 15, no. 12: 1501. https://doi.org/10.3390/ph15121501
APA StyleParanos, P., Vourli, S., Pournaras, S., & Meletiadis, J. (2022). Assessing Clinical Potential of Old Antibiotics against Severe Infections by Multi-Drug-Resistant Gram-Negative Bacteria Using In Silico Modelling. Pharmaceuticals, 15(12), 1501. https://doi.org/10.3390/ph15121501