
New Antifungal Agents with Azole Moieties



 and
and
Abstract
1. Introduction
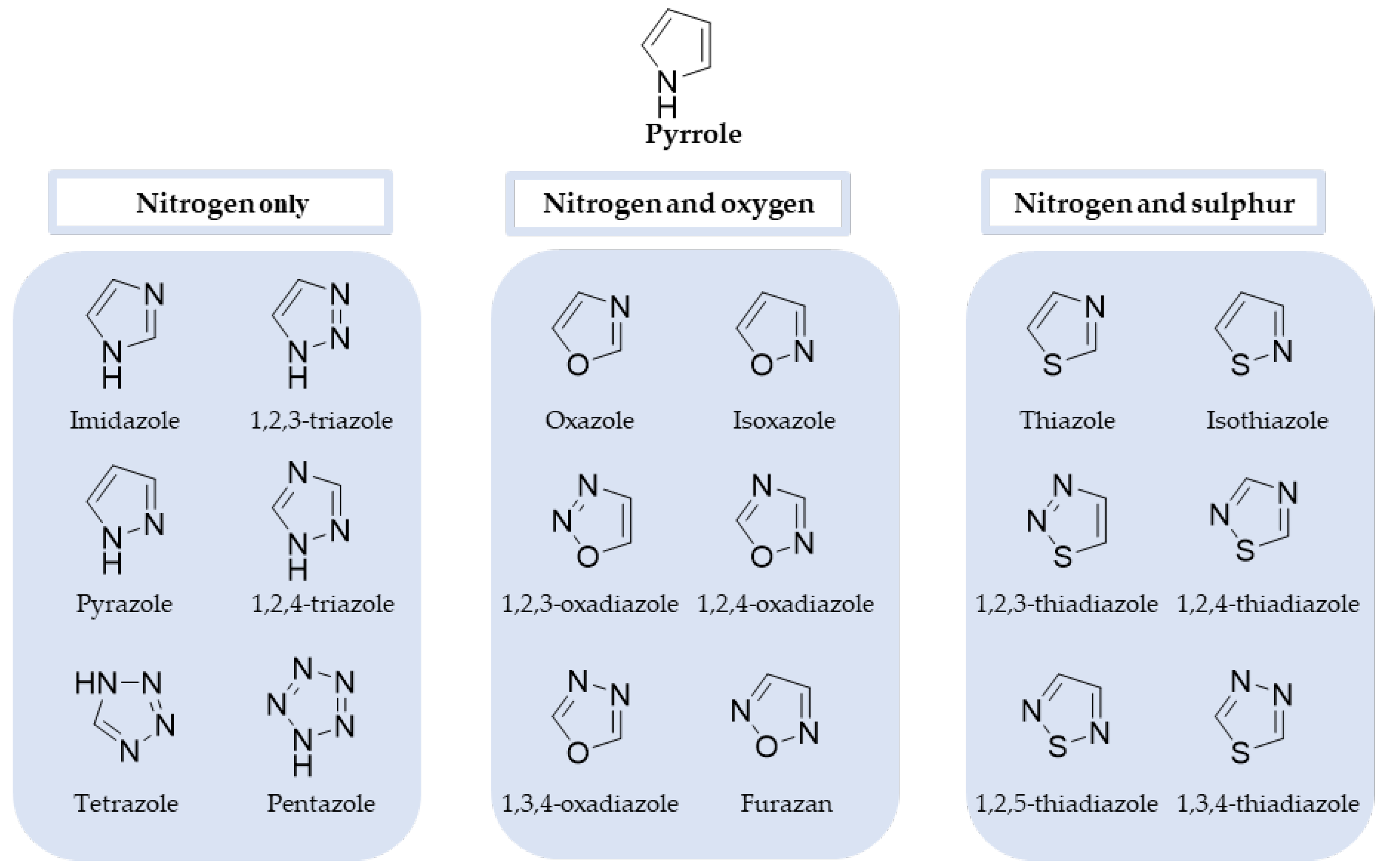
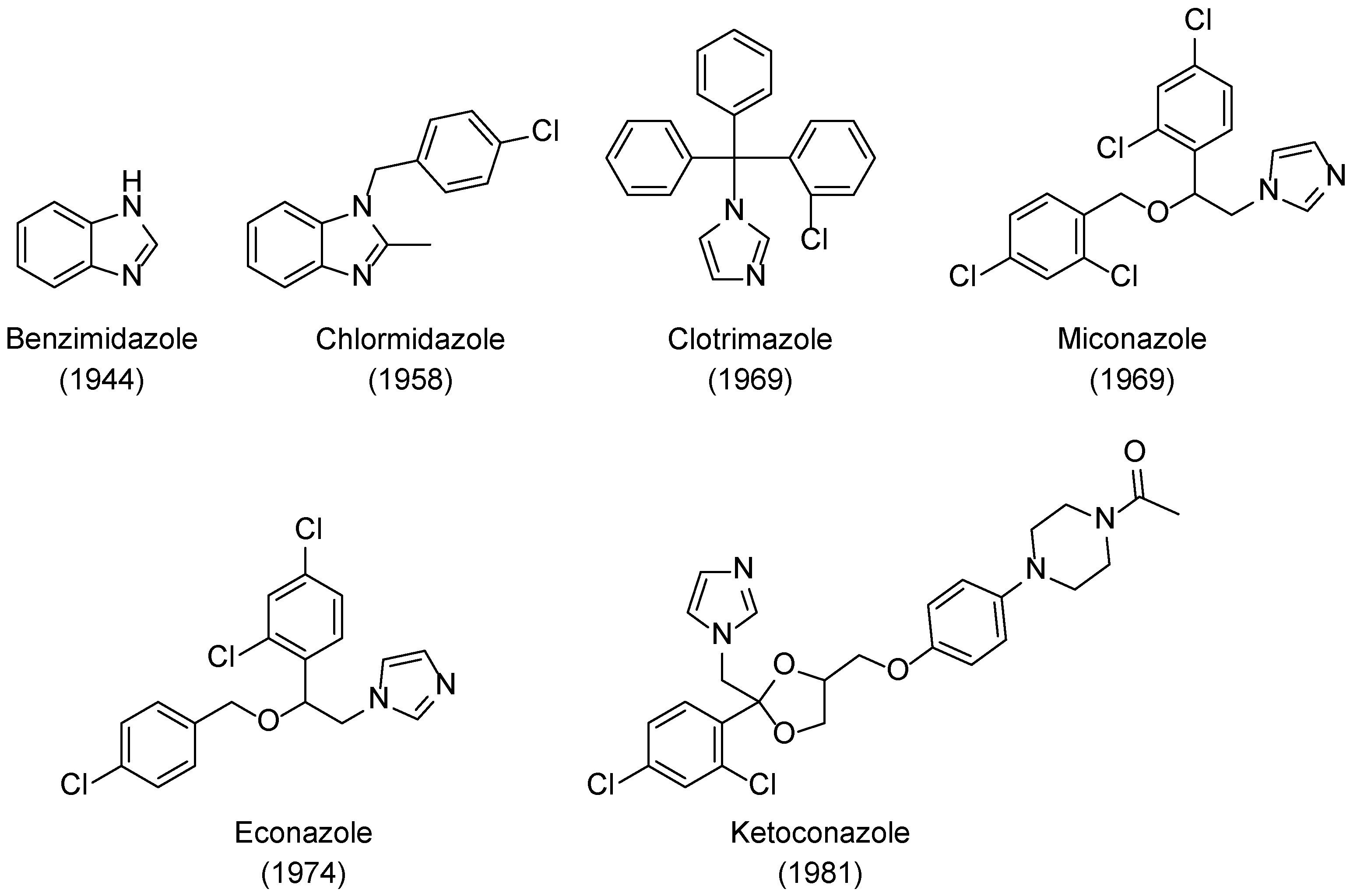
2. History and the Evolution of Azoles
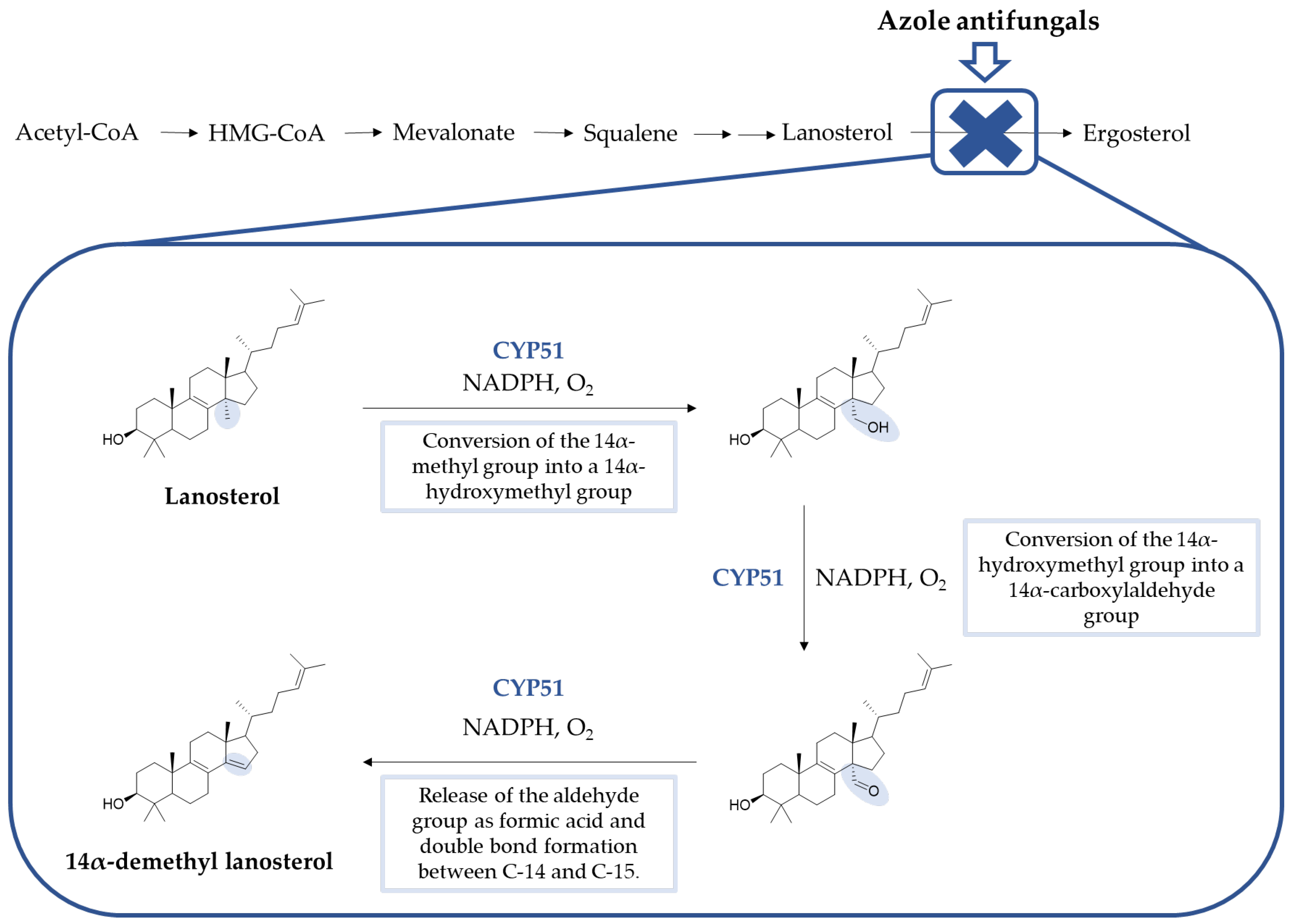
3. Mechanism of Action of Azole Antifungals
4. New Antifungal Azoles in Research and Development
4.1. Azoles in Clinical Trials
4.1.1. Luliconazole
4.1.2. Isavuconazole
4.1.3. SUBA-Itraconazole
4.1.4. Iodiconazole
4.1.5. Albaconazole
4.1.6. PC945
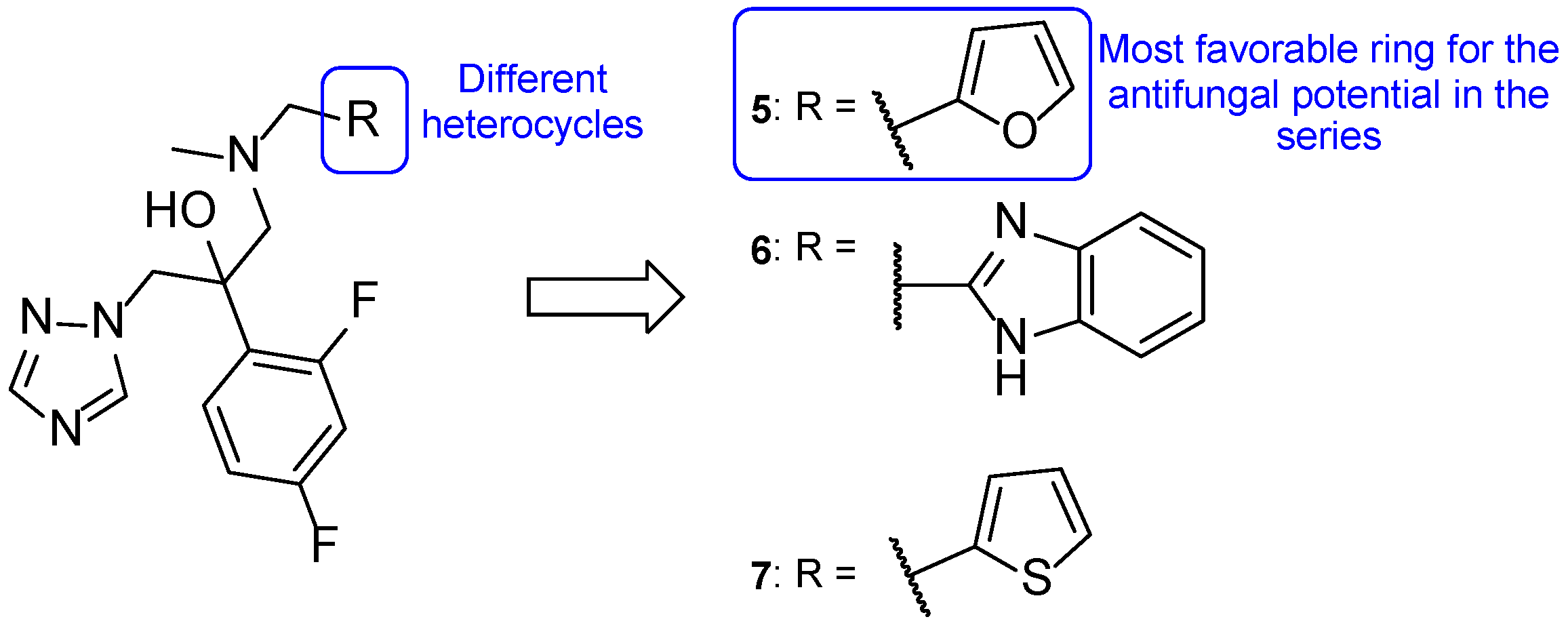
4.1.7. Tetrazole-Pyridine Hybrids
4.2. New Molecules with a Traditional Azole Pharmacophore
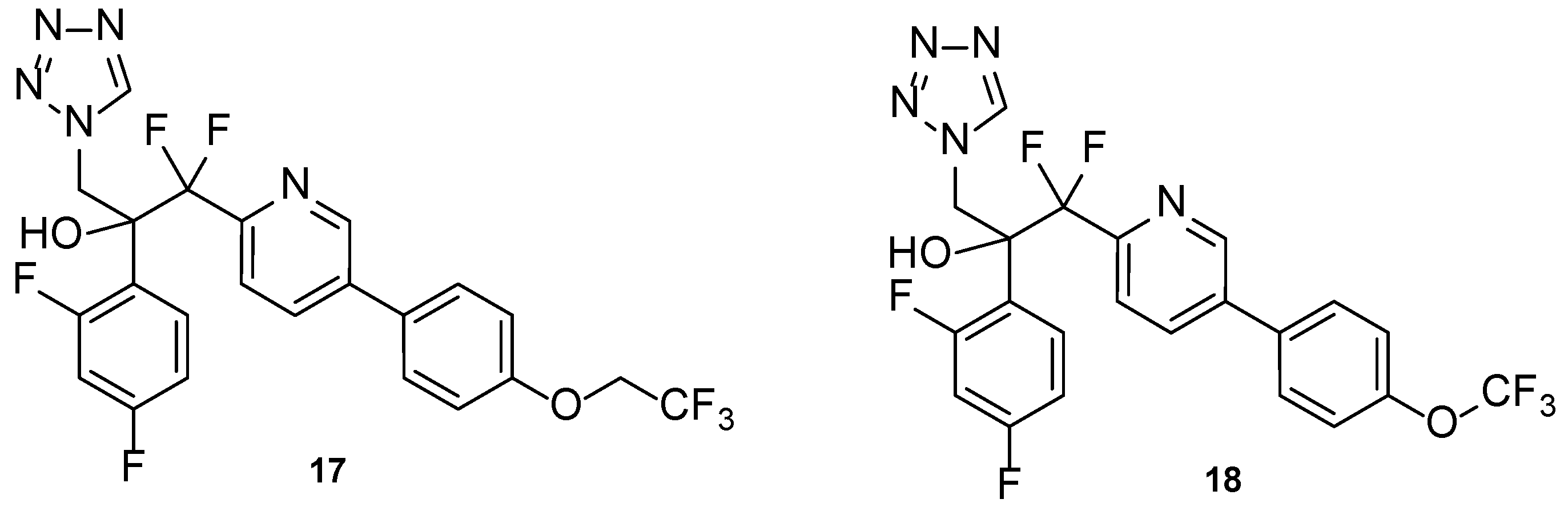
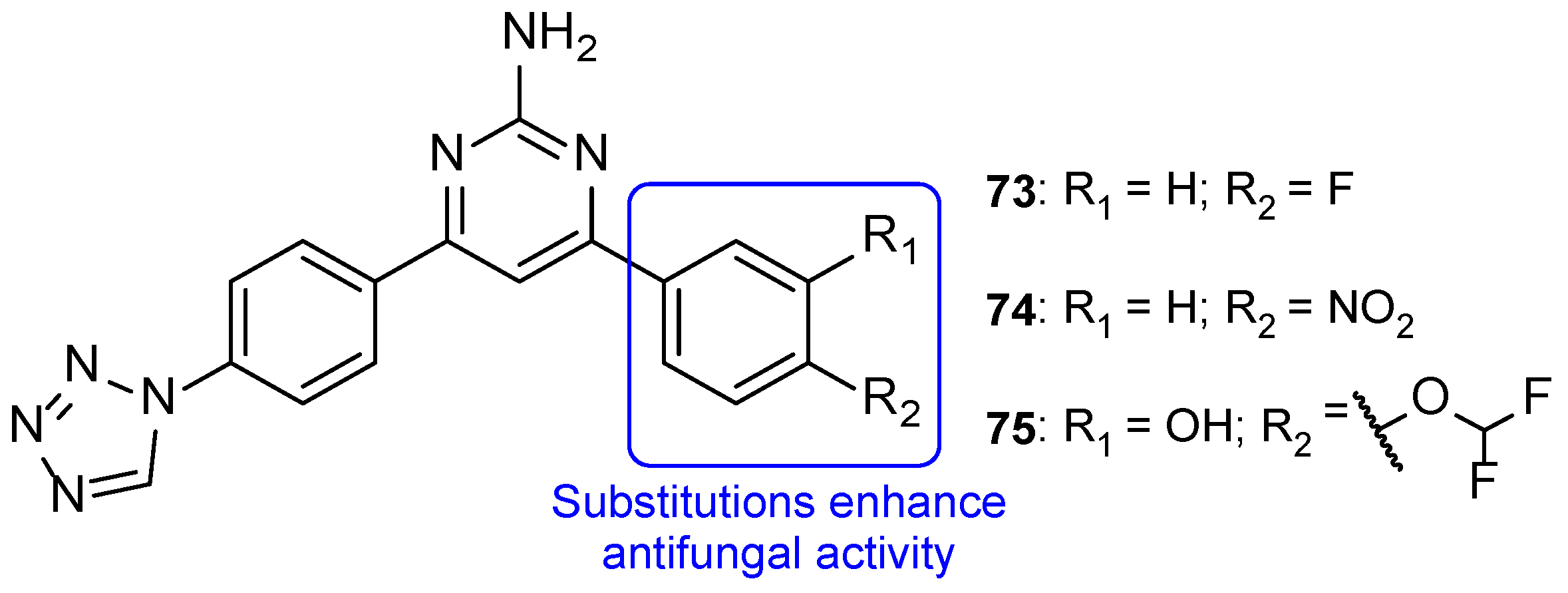
4.2.1. New Triazoles
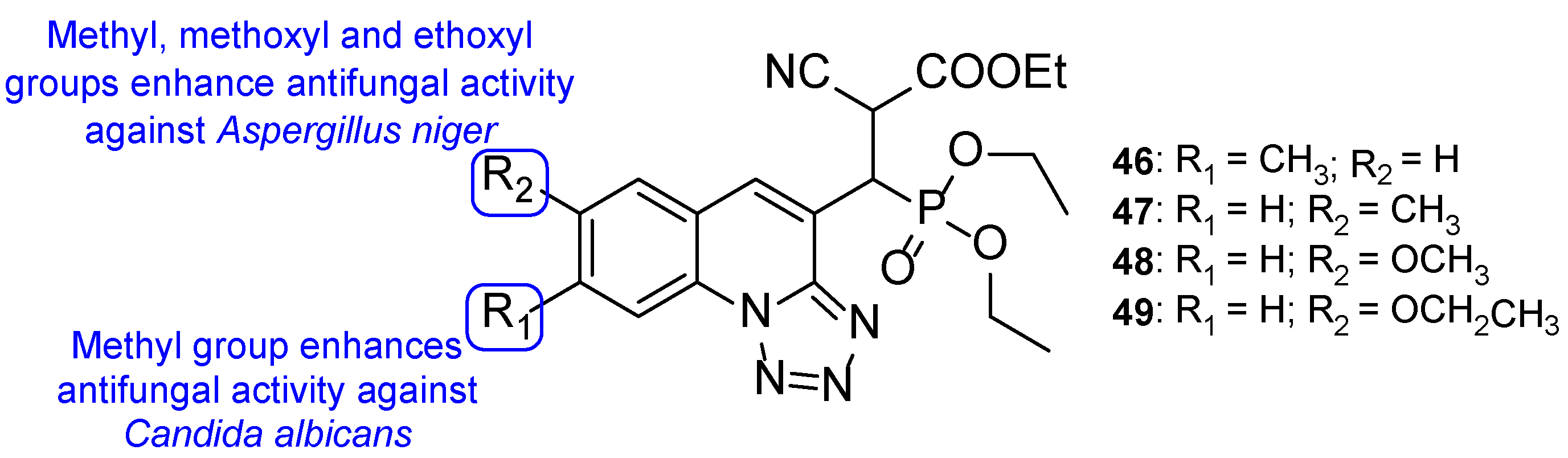
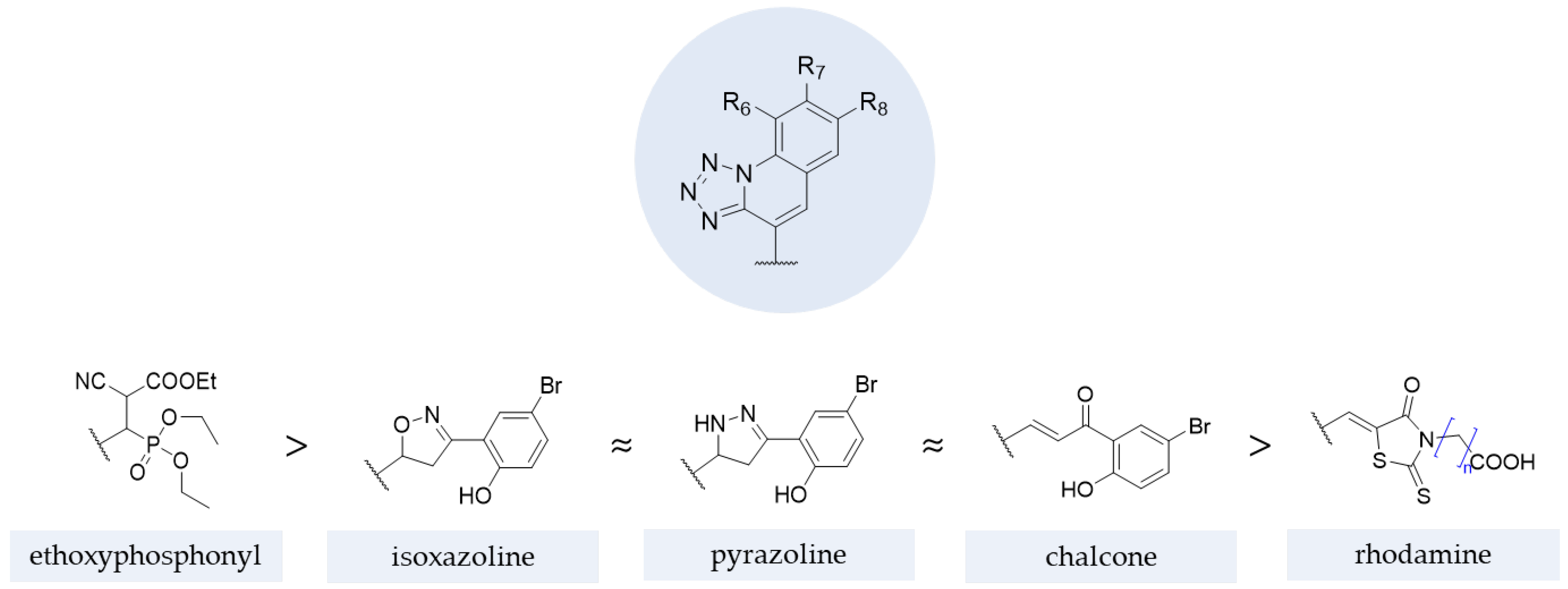
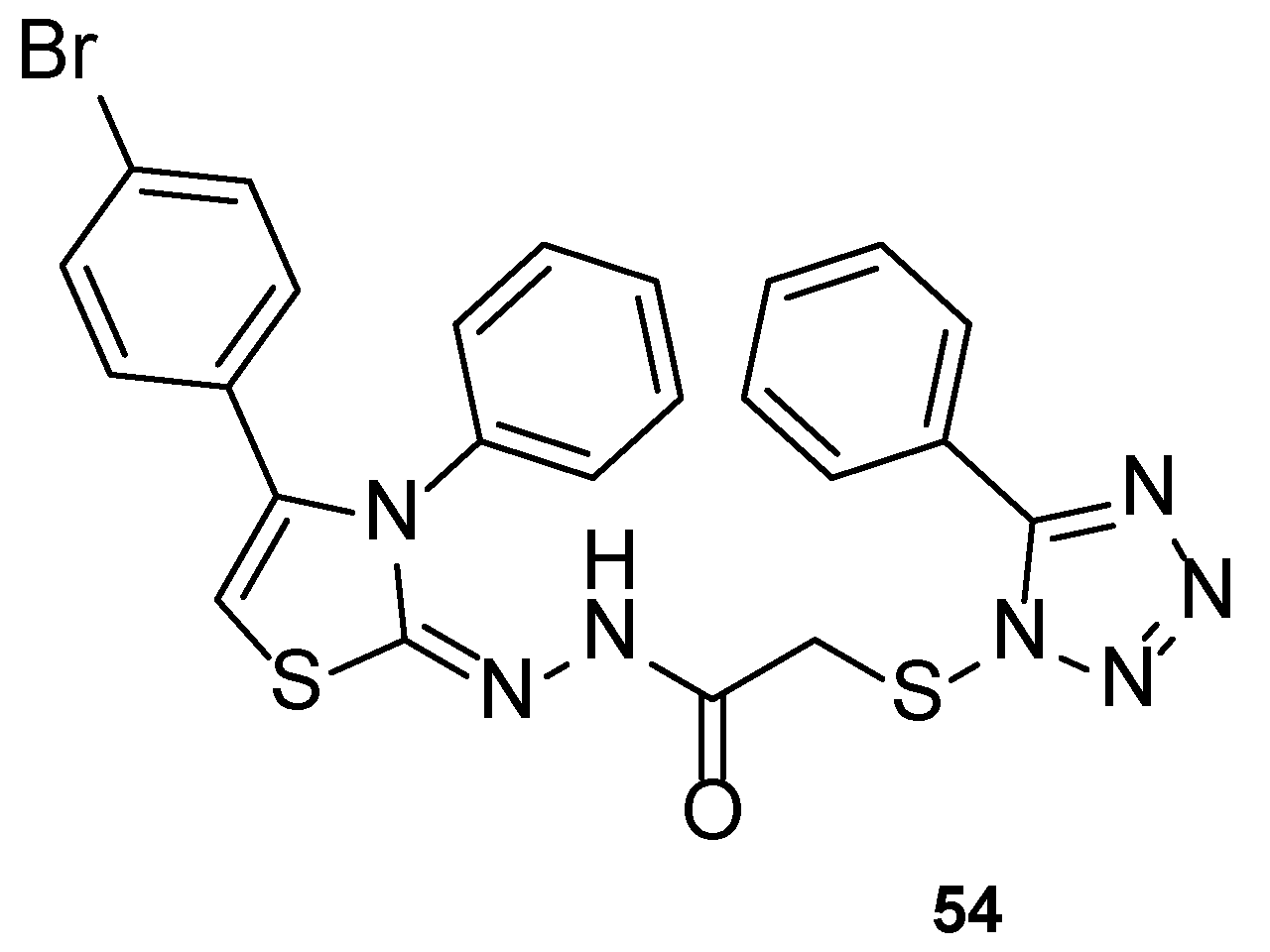
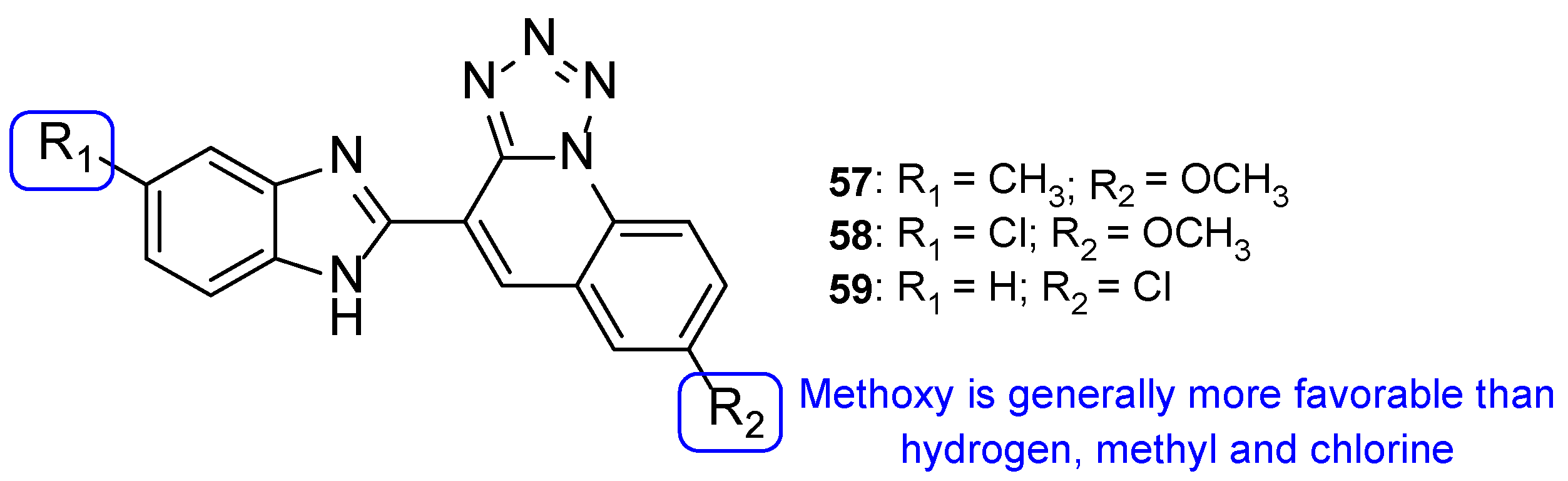
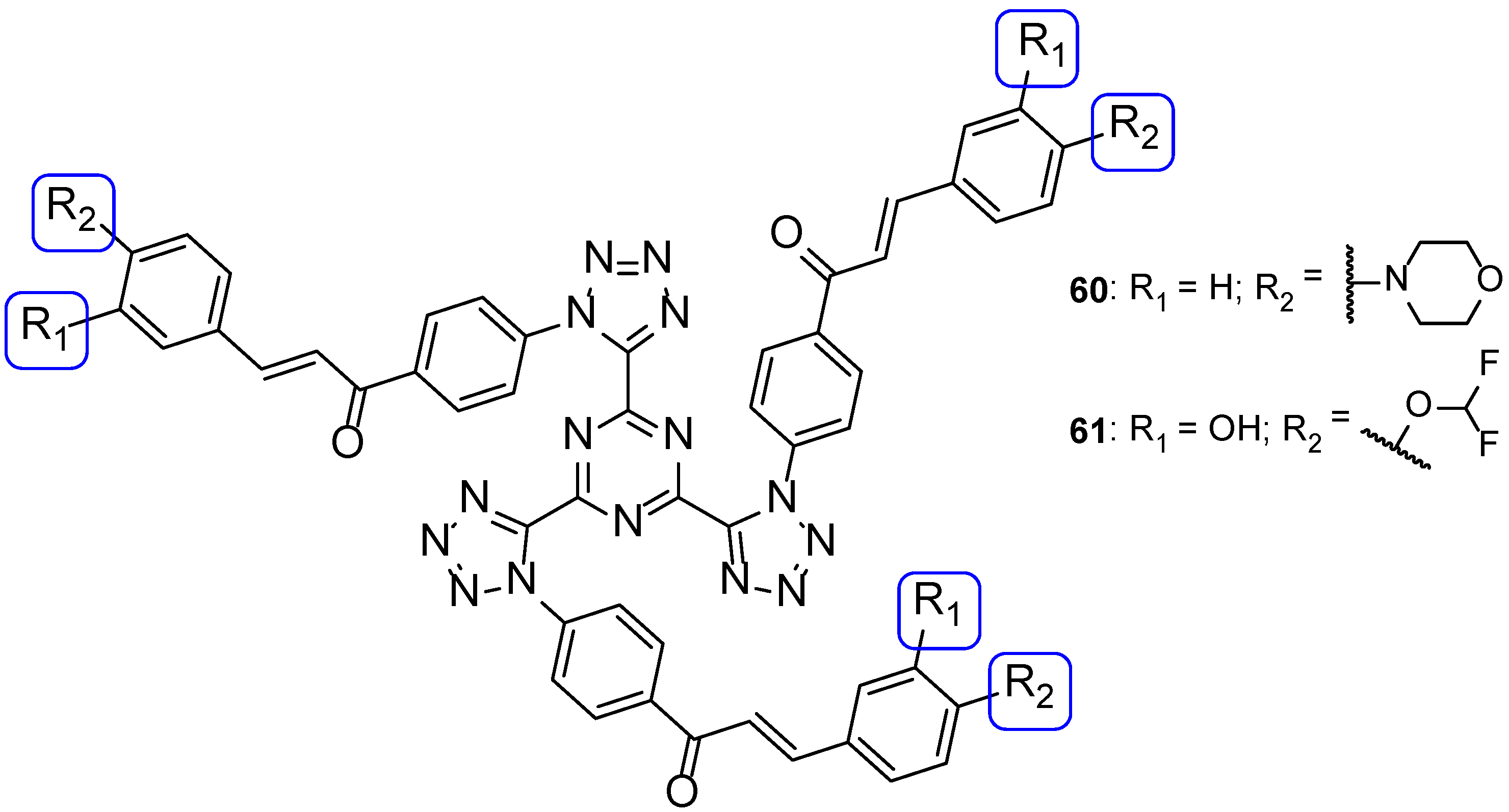
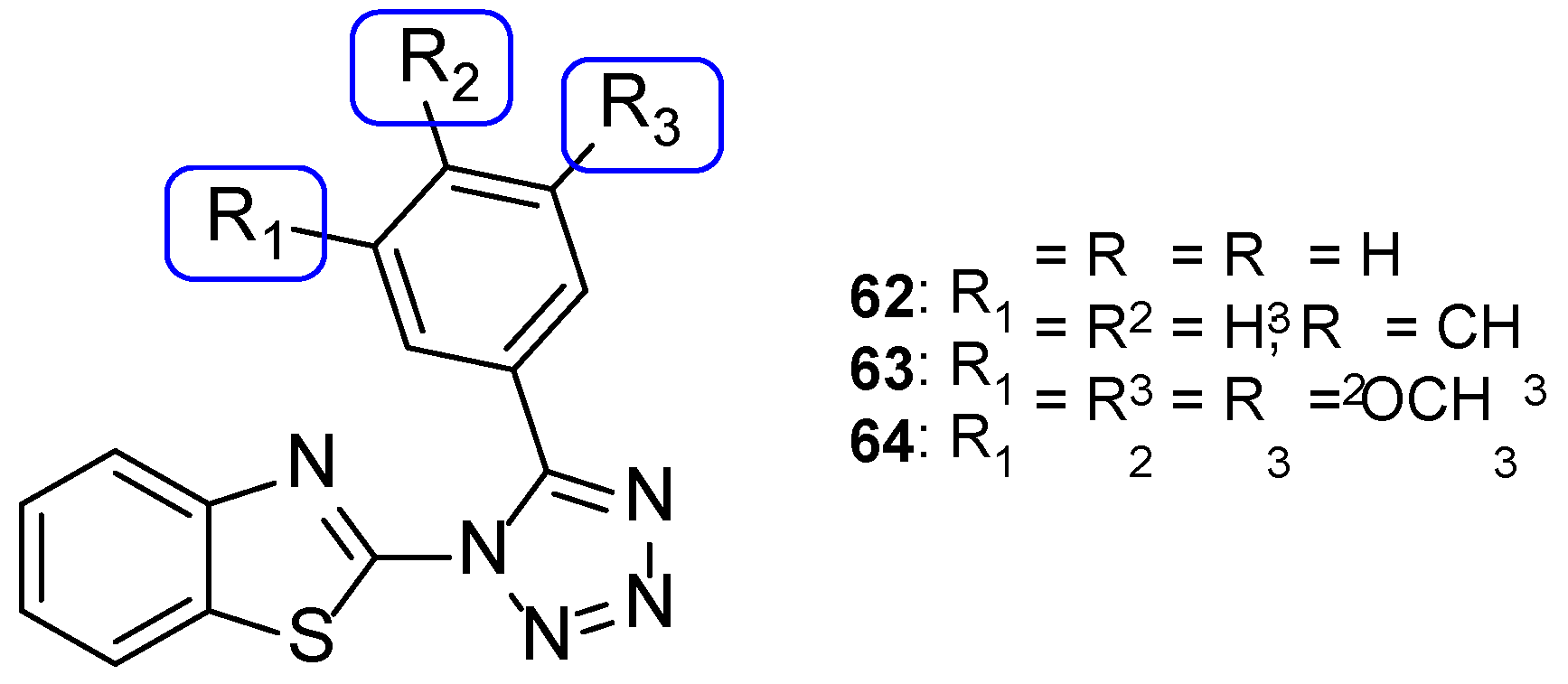
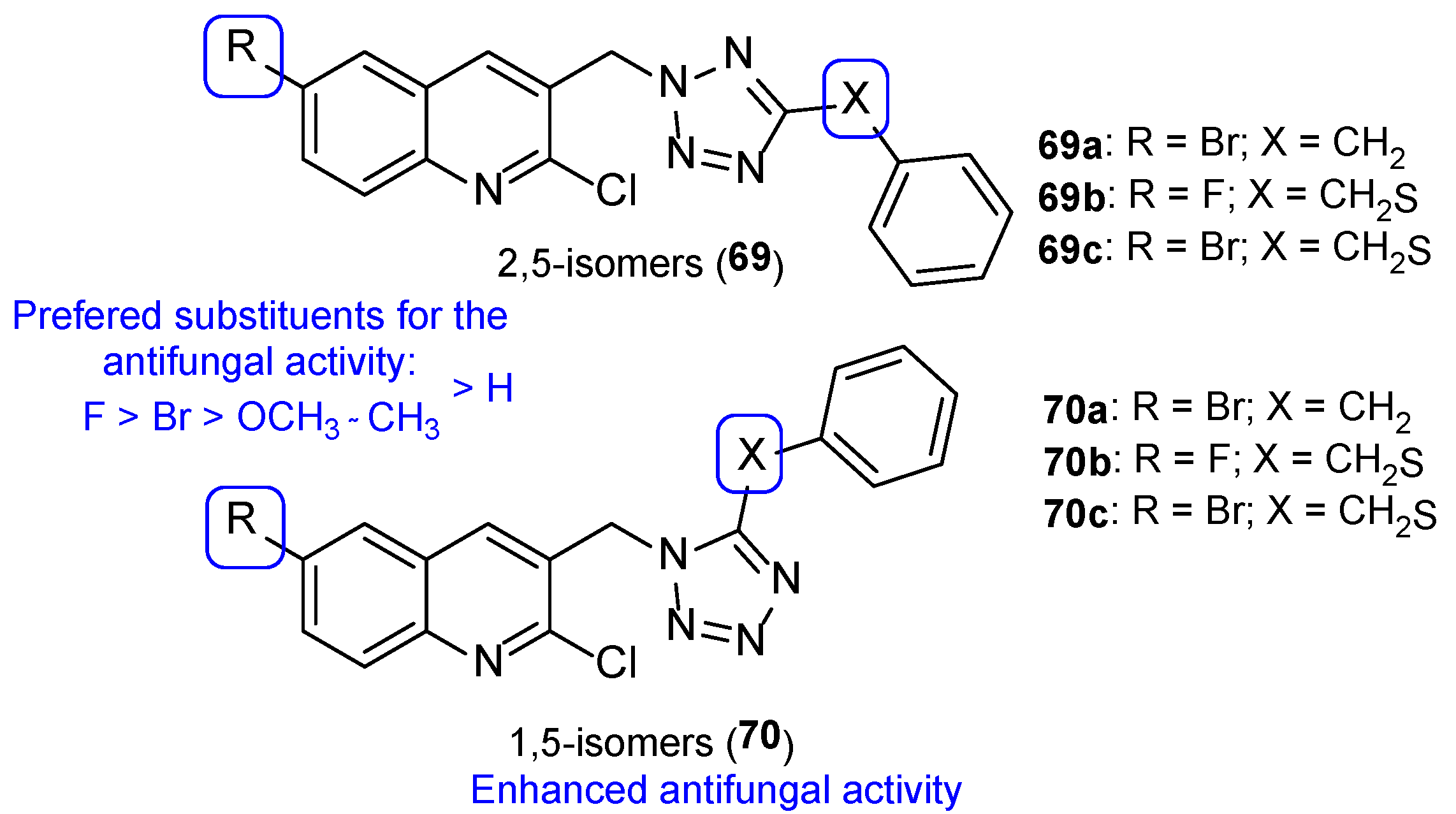
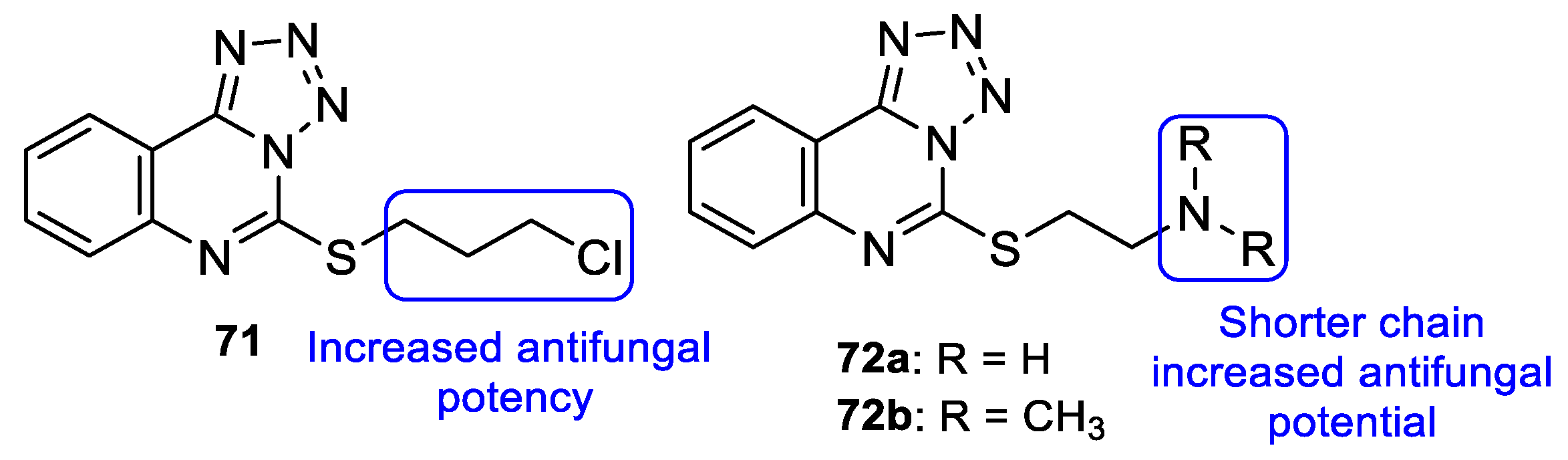
4.2.2. Tetrazoles
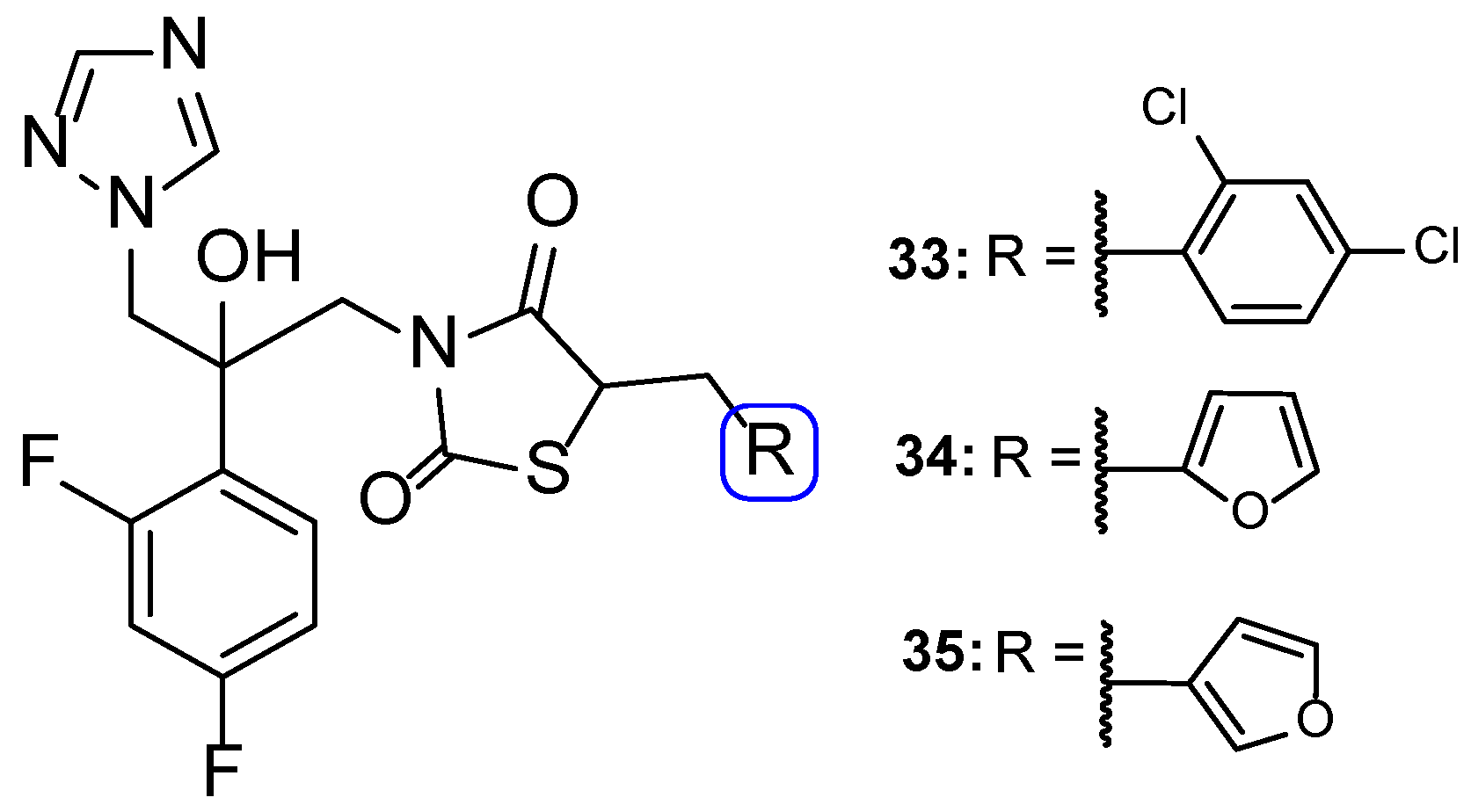
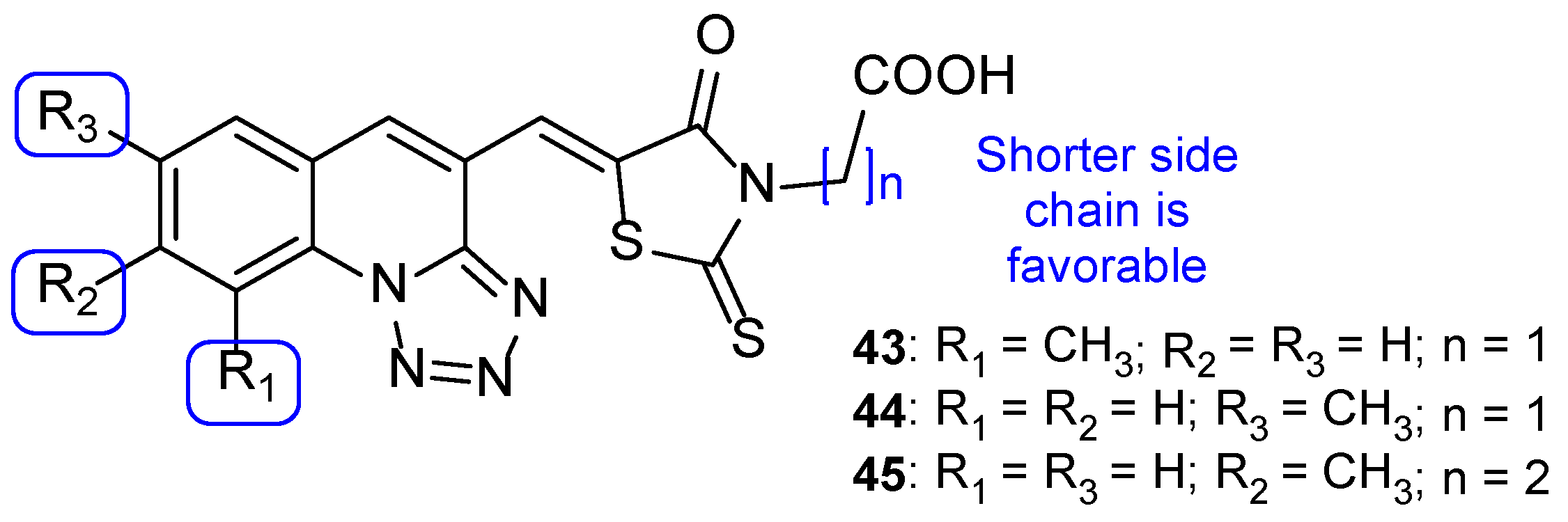
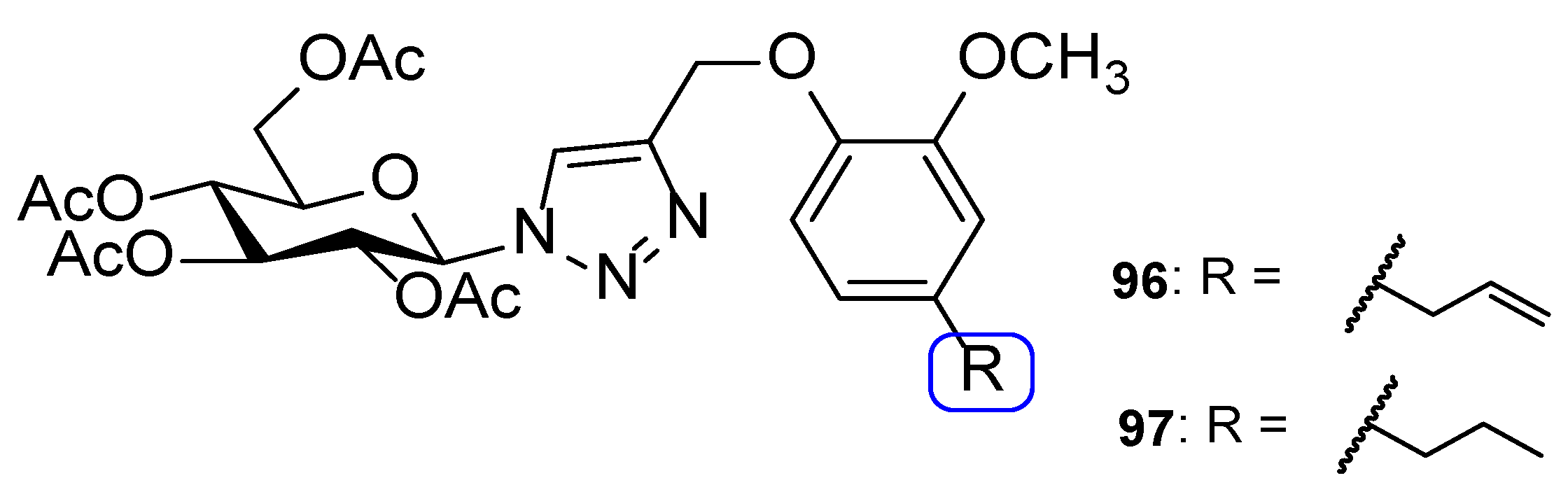
4.3. Novel Derivatives with Azole Moieties Aside the Traditional Pharmacophore
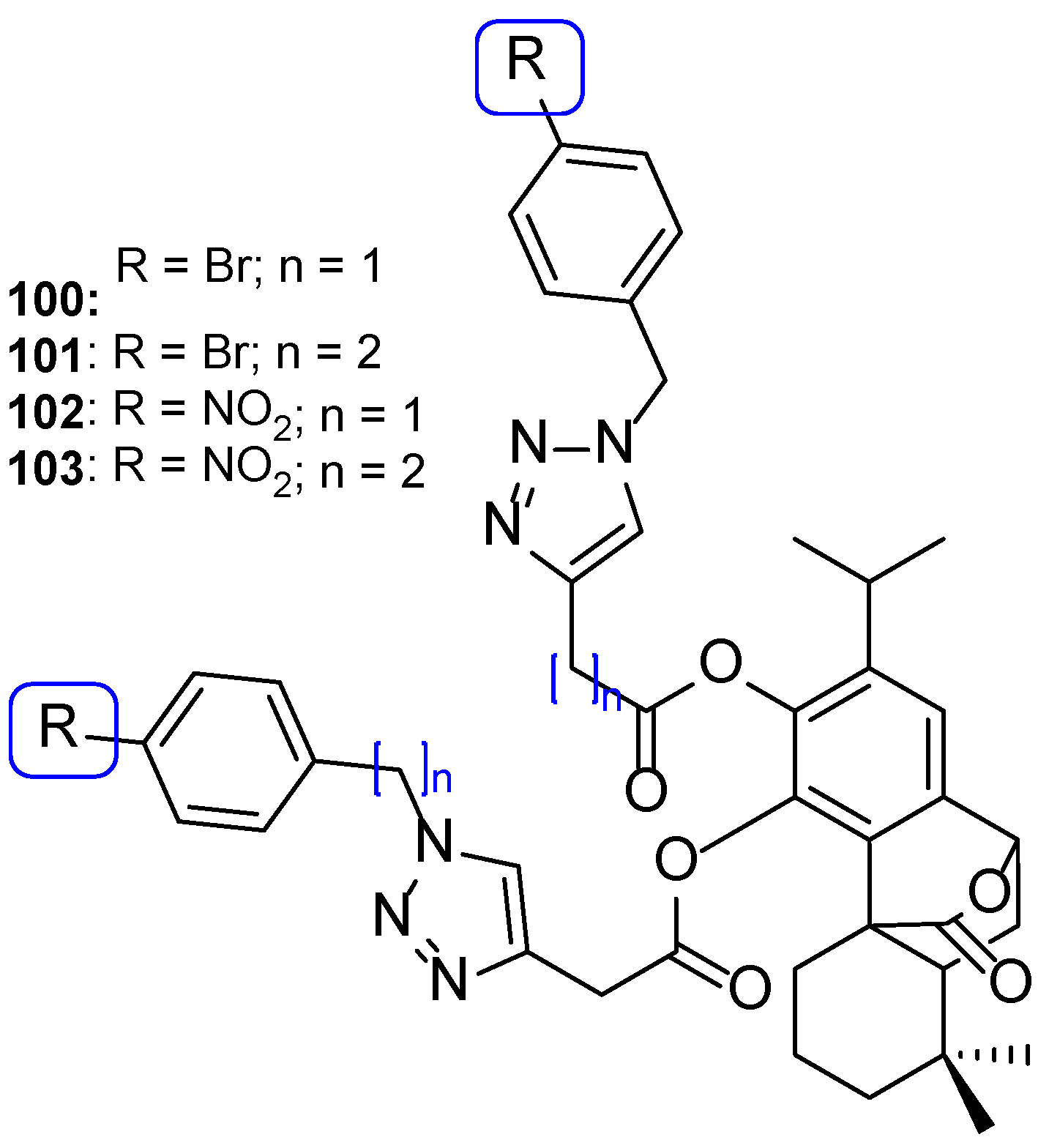
4.4. Use of Triazoles in the Preparation of New Antifungals from Natural Products
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kainz, K.; Bauer, M.A.; Madeo, F.; Carmona-Gutierrez, D. Fungal infections in humans: The silent crisis. Microb. Cell 2020, 7, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Kathiravan, M.K.; Salake, A.B.; Chothe, A.S.; Dudhe, P.B.; Watode, R.P.; Mukta, M.S.; Gadhwe, S. The biology and chemistry of antifungal agents: A review. Bioorg. Med. Chem. 2012, 20, 5678–5698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, A. Emerging Invasive Fungal Infections: Clinical Features and Controversies in Diagnosis and Treatment Processes. Infect. Drug Resist. 2020, 13, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Bouz, G.; Doležal, M. Advances in Antifungal Drug Development: An Up-To-Date Mini Review. Pharmaceuticals 2021, 14, 1312. [Google Scholar] [CrossRef]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef]
- Stewart, A.G.; Paterson, D.L. How urgent is the need for new antifungals? Expert Opin. Pharmacother. 2021, 22, 1857–1870. [Google Scholar] [CrossRef]
- Peyton, L.R.; Gallagher, S.; Hashemzadeh, M. Triazole antifungals: A review. Drugs Today 2015, 51, 705–718. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Kuznetsov, A. Introductory Chapter: Azoles, Their Importance, and Applications; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Wadhwa, P.; Singh, K. Multicomponent Reactions: A Sustainable Tool to 1,2- and 1,3-Azoles. Org. Biomol. Chem. 2018, 16, 9084–9116. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Jonnalagadda, S. A Review of Recent Advances in the Green Synthesis of Azole- and Pyran-based Fused Heterocycles Using MCRs and Sustainable Catalysts. Curr. Org. Chem. 2020, 24, 4–39. [Google Scholar] [CrossRef]
- Budeev, A.; Kantin, G.; Dar’in, D.; Krasavin, M. Diazocarbonyl and Related Compounds in the Synthesis of Azoles. Molecules 2021, 26, 2530. [Google Scholar] [CrossRef]
- Neha; Dwivedi, A.; Kumar, R.; Kumar, V. Recent Synthetic Strategies for Monocyclic Azole Nucleus and Its Role in Drug Discovery and Development. Curr. Org. Synth. 2017, 14, 321–340. [Google Scholar] [CrossRef]
- Siwach, A.; Verma, P.K. Synthesis and therapeutic potential of imidazole containing compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef]
- Woolley, D.W. Some biological effects produced by benzimidazole and their reversal by purines. J. Biol. Chem. 1944, 152, 225–232. [Google Scholar] [CrossRef]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef]
- Maertens, J.A. History of the development of azole derivatives. Clin. Microbiol. Infect. 2004, 10 (Suppl. 1), 1–10. [Google Scholar] [CrossRef]
- Chen, S.C.A.; Sorrell, T.C. Antifungal agents. Med. J. Aust. 2007, 187, 404–409. [Google Scholar] [CrossRef]
- Andes, D.; Dismukes, W. Azoles. In Essentials of Clinical Mycology; Springer: New York, NY, USA, 2011; pp. 61–93. [Google Scholar]
- Kane, A.; Carter, D.A. Augmenting Azoles with Drug Synergy to Expand the Antifungal Toolbox. Pharmaceuticals 2022, 15, 482. [Google Scholar] [CrossRef]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Delattin, N.; Cammue, B.P.; Thevissen, K. Reactive oxygen species-inducing antifungal agents and their activity against fungal biofilms. Future Med. Chem. 2014, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- S. cerevisiae CYP51 Complexed with Fluconazole in the Active Site. Available online: https://www.rcsb.org/structure/4WMZ (accessed on 4 July 2022).
- Crystal Structure of Sterol 14-Alpha Demethylase (CYP51) from Candida albicans in Complex with the Tetrazole-Based Antifungal Drug Candidate VT1161 (VT1). Available online: https://www.rcsb.org/structure/5TZ1 (accessed on 4 July 2022).
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 2007, 1770, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R.; Ghannoum, M. Emerging Issues in Antifungal Resistance. Infect. Dis. Clin. N. Am. 2020, 34, 921–943. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Bharti, S. Luliconazole for the treatment of fungal infections: An evidence-based review. Core Evid. 2014, 9, 113–124. [Google Scholar] [CrossRef]
- Niwano, Y.; Kuzuhara, N.; Kodama, H.; Yoshida, M.; Miyazaki, T.; Yamaguchi, H. In Vitro and In Vivo Antidermatophyte Activities of NND-502, a Novel Optically Active Imidazole Antimycotic Agent. Antimicrob. Agents Chemother. 1998, 42, 967–970. [Google Scholar] [CrossRef]
- Niwano, Y.; Kuzuhara, N.; Goto, Y.; Munechika, Y.; Kodama, H.; Kanai, K.; Yoshida, M.; Miyazaki, T.; Yamaguchi, H. Efficacy of NND-502, a novel imidazole antimycotic agent, in experimental models of Candida albicans and Aspergillus fumigatus infections. Int. J. Antimicrob. Agents 1999, 12, 221–228. [Google Scholar] [CrossRef]
- Study Evaluating the Drug Interaction Potential of Luliconazole Cream 1% in Participants with Tinea Pedis and Tinea Cruris. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02394340?term=luliconazole&draw=2&rank=1 (accessed on 28 June 2022).
- Maximal Use of Luliconazole Cream 1% in Pediatric Participants with Moderate to Severe Tinea Pedis or Tinea Cruris. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02767271?term=luliconazole&draw=2&rank=2 (accessed on 28 June 2022).
- Safety and Efficacy of Product 33525 (Luliconazole Cream 1%) in Pediatric Participants with Tinea Corporis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02767947?term=luliconazole&draw=2&rank=3 (accessed on 28 June 2022).
- Safety and Efficacy of Luliconazole Solution, 10% in Subjects with Mild to Moderate Onychomycosis (SOLUTION). Available online: https://www.clinicaltrials.gov/ct2/show/NCT01431820?term=luliconazole&draw=2&rank=6 (accessed on 28 June 2022).
- Multicenter Study of the Efficacy and Safety of Luliconazole Cream in Tinea Pedis (Athlete’s Foot). Available online: https://www.clinicaltrials.gov/ct2/show/NCT00869336?term=luliconazole&draw=2&rank=7 (accessed on 28 June 2022).
- Open-Label Pharmacokinetics/Safety Study of Luliconazole Solution, 10% in Distal Subungual Onychomycosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01044381?term=luliconazole&draw=2&rank=5 (accessed on 28 June 2022).
- Safety and Tolerability Study of SKX-16 (Luliconazole 10% Solution) in Subjects with Moderate to Severe Distal Subungual Onychomycosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05110638?term=luliconazole&draw=2&rank=4 (accessed on 28 June 2022).
- Ellsworth, M.; Ostrosky-Zeichner, L. Isavuconazole: Mechanism of Action, Clinical Efficacy, and Resistance. J. Fungi 2020, 6, 324. [Google Scholar] [CrossRef]
- Guinea, J.; Peláez, T.; Recio, S.; Torres-Narbona, M.; Bouza, E. In vitro antifungal activities of isavuconazole (BAL4815), voriconazole, and fluconazole against 1007 isolates of zygomycete, Candida, Aspergillus, Fusarium, and Scedosporium species. Antimicrob. Agents Chemother. 2008, 52, 1396–1400. [Google Scholar] [CrossRef]
- Datta, K.; Rhee, P.; Byrnes, E., 3rd; Garcia-Effron, G.; Perlin, D.S.; Staab, J.F.; Marr, K.A. Isavuconazole activity against Aspergillus lentulus, Neosartorya udagawae, and Cryptococcus gattii, emerging fungal pathogens with reduced azole susceptibility. J. Clin. Microbiol. 2013, 51, 3090–3093. [Google Scholar] [CrossRef]
- Seifert, H.; Aurbach, U.; Stefanik, D.; Cornely, O. In vitro activities of isavuconazole and other antifungal agents against Candida bloodstream isolates. Antimicrob. Agents Chemother. 2007, 51, 1818–1821. [Google Scholar] [CrossRef]
- Bongomin, F.; Maguire, N.; Moore, C.B.; Felton, T.; Rautemaa-Richardson, R. Isavuconazole and voriconazole for the treatment of chronic pulmonary aspergillosis: A retrospective comparison of rates of adverse events. Mycoses 2019, 62, 217–222. [Google Scholar] [CrossRef]
- Marty, F.M.; Ostrosky-Zeichner, L.; Cornely, O.A.; Mullane, K.M.; Perfect, J.R.; Thompson, G.R., 3rd; Alangaden, G.J.; Brown, J.M.; Fredricks, D.N.; Heinz, W.J.; et al. Isavuconazole treatment for mucormycosis: A single-arm open-label trial and case-control analysis. Lancet Infect. Dis. 2016, 16, 828–837. [Google Scholar] [CrossRef]
- Kullberg, B.J.; Viscoli, C.; Pappas, P.G.; Vazquez, J.; Ostrosky-Zeichner, L.; Rotstein, C.; Sobel, J.D.; Herbrecht, R.; Rahav, G.; Jaruratanasirikul, S.; et al. Isavuconazole Versus Caspofungin in the Treatment of Candidemia and Other Invasive Candida Infections: The ACTIVE Trial. Clin. Infect. Dis. 2019, 68, 1981–1989. [Google Scholar] [CrossRef]
- Pagano, L.; Cattaneo, C.; Quattrone, M.; Oberti, M.; Mazzitelli, M.; Trecarichi, E.M. Isavuconazole-Animal Data and Clinical Data. J. Fungi 2020, 6, 209. [Google Scholar] [CrossRef]
- Isavuconazole (BAL8557) for Primary Treatment of Invasive Aspergillosis. Available online: https://clinicaltrials.gov/ct2/show/NCT00412893 (accessed on 30 June 2022).
- Isavuconazole in the Treatment of Renally Impaired Aspergillosis and Rare Fungi (VITAL). Available online: https://www.clinicaltrials.gov/ct2/show/NCT00634049?term=NCT00634049&draw=2&rank=1 (accessed on 30 June 2022).
- Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections. Available online: https://clinicaltrials.gov/ct2/show/NCT00413218 (accessed on 30 June 2022).
- Abuhelwa, A.Y.; Foster, D.J.; Mudge, S.; Hayes, D.; Upton, R.N. Population pharmacokinetic modeling of itraconazole and hydroxyitraconazole for oral SUBA-itraconazole and sporanox capsule formulations in healthy subjects in fed and fasted states. Antimicrob. Agents Chemother. 2015, 59, 5681–5696. [Google Scholar] [CrossRef]
- Lindsay, J.; Sandaradura, I.; Wong, K.; Arthur, C.; Stevenson, W.; Kerridge, I.; Fay, K.; Coyle, L.; Greenwood, M. Serum levels, safety and tolerability of new formulation SUBA-itraconazole prophylaxis in patients with haematological malignancy or undergoing allogeneic stem cell transplantation. J. Antimicrob. Chemother. 2017, 72, 3414–3419. [Google Scholar] [CrossRef]
- Gintjee, T.J.; Donnelley, M.A.; Thompson, G.R., 3rd. Aspiring Antifungals: Review of Current Antifungal Pipeline Developments. J. Fungi 2020, 6, 28. [Google Scholar] [CrossRef]
- Endemic Mycoses Treatment with SUBA-Itraconazole vs Itraconazole (MSG15). Available online: https://www.clinicaltrials.gov/ct2/show/results/NCT03572049?term=NCT03572049&draw=2&rank=1 (accessed on 2 July 2022).
- Wu, L.; Zhou, K.; Zong, W.; Chen, Y.; Sheng, C. Single dose pharmacokinetics of topical iodiconazole creams in healthy Chinese volunteers. Xenobiotica 2021, 51, 427–433. [Google Scholar] [CrossRef]
- Sun, N.; Xie, Y.; Sheng, C.; Cao, Y.; Zhang, W.; Chen, H.; Fan, G. In vivo pharmacokinetics and in vitro antifungal activity of iodiconazole, a new triazole, determined by microdialysis sampling. Int. J. Antimicrob. Agents 2013, 41, 229–235. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, Y.; Wang, W.; Wang, S.; Xu, B.; Fan, G.; Dong, G.; Liu, Y.; Yao, J.; Miao, Z.; et al. Discovery of highly potent triazole antifungal derivatives by heterocycle-benzene bioisosteric replacement. Eur. J. Med. Chem. 2013, 64, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sigurgeirsson, B.; van Rossem, K.; Malahias, S.; Raterink, K. A phase II, randomized, double-blind, placebo-controlled, parallel group, dose-ranging study to investigate the efficacy and safety of 4 dose regimens of oral albaconazole in patients with distal subungual onychomycosis. J. Am. Acad. Dermatol. 2013, 69, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety Study of 4 Dose Regimens of Oral Albaconazole in Subjects with Distal Subungual Onychomycosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00730405?term=NCT00730405&draw=2&rank=1 (accessed on 1 July 2022).
- Ding, Z.; Ni, T.; Xie, F.; Hao, Y.; Yu, S.; Chai, X.; Jin, Y.; Wang, T.; Jiang, Y.; Zhang, D. Design, synthesis, and structure-activity relationship studies of novel triazole agents with strong antifungal activity against Aspergillus fumigatus. Bioorg. Med. Chem. Lett. 2020, 30, 126951. [Google Scholar] [CrossRef] [PubMed]
- Colley, T.; Alanio, A.; Kelly, S.L.; Sehra, G.; Kizawa, Y.; Warrilow, A.G.S.; Parker, J.E.; Kelly, D.E.; Kimura, G.; Anderson-Dring, L.; et al. In Vitro and In Vivo Antifungal Profile of a Novel and Long-Acting Inhaled Azole, PC945, on Aspergillus fumigatus Infection. Antimicrob. Agents Chemother. 2017, 61, e02280-16. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Zagaliotis, P.; Walsh, T. Novel antifungal agents in clinical trials version 2; peer review: 2 approved. F1000Research 2022, 10, 507. [Google Scholar] [CrossRef]
- Colley, T.; Sehra, G.; Daly, L.; Kimura, G.; Nakaoki, T.; Nishimoto, Y.; Kizawa, Y.; Strong, P.; Rapeport, G.; Ito, K. Antifungal synergy of a topical triazole, PC945, with a systemic triazole against respiratory Aspergillus fumigatus infection. Sci. Rep. 2019, 9, 9482. [Google Scholar] [CrossRef]
- Kimura, G.; Nakaoki, T.; Colley, T.; Rapeport, G.; Strong, P.; Ito, K.; Kizawa, Y. In Vivo Biomarker Analysis of the Effects of Intranasally Dosed PC945, a Novel Antifungal Triazole, on Aspergillus fumigatus Infection in Immunocompromised Mice. Antimicrob. Agents Chemother. 2017, 61, e00124-17. [Google Scholar] [CrossRef]
- Cass, L.; Murray, A.; Davis, A.; Woodward, K.; Albayaty, M.; Ito, K.; Strong, P.; Ayrton, J.; Brindley, C.; Prosser, J.; et al. Safety and nonclinical and clinical pharmacokinetics of PC945, a novel inhaled triazole antifungal agent. Pharmacol. Res. Perspect. 2021, 9, e00690. [Google Scholar] [CrossRef]
- Safety and Efficacy of PC945 in Combination with Other Antifungal Therapy for the Treatment of Refractory Invasive Pulmonary Aspergillosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05238116?term=PC-945&draw=2&rank=3 (accessed on 28 June 2022).
- The Effect of Early Treatment of PC945 on Aspergillus fumigatus Lung Infection in Lung Transplant Patients. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03905447?term=PC-945&draw=2&rank=2 (accessed on 28 June 2022).
- The effect of PC945 on Aspergillus fumigatus Lung Infection in Patients with Cystic Fibrosis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03870841?term=PC-945&draw=2&rank=1 (accessed on 28 June 2022).
- PC945 Prophylaxis or Pre-emptive Therapy against Pulmonary Aspergillosis in Lung Transplant Recipients. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05037851?term=PC-945&draw=2&rank=4 (accessed on 28 June 2022).
- The Effect of PC945 on Aspergillus or Candida Lung Infections in Patients with Asthma or Chronic Respiratory Diseases. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03745196?term=PC-945&draw=2&rank=5 (accessed on 28 June 2022).
- A Study to Investigate the Safety, Tolerability and Pharmacokinetics of Single and Repeat Doses of PC945. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02715570?term=PC-945&draw=2&rank=6 (accessed on 28 June 2022).
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Warrilow, A.G.; Hull, C.M.; Parker, J.E.; Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef]
- Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Sobel, J.D.; Lilly, E.A.; Fidel, P.L., Jr. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob. Agents Chemother. 2015, 59, 5567–5573. [Google Scholar] [CrossRef]
- Shubitz, L.F.; Trinh, H.T.; Galgiani, J.N.; Lewis, M.L.; Fothergill, A.W.; Wiederhold, N.P.; Barker, B.M.; Lewis, E.R.; Doyle, A.L.; Hoekstra, W.J.; et al. Evaluation of VT-1161 for Treatment of Coccidioidomycosis in Murine Infection Models. Antimicrob. Agents Chemother. 2015, 59, 7249–7254. [Google Scholar] [CrossRef]
- Sobel, J.D.; Nyirjesy, P. Oteseconazole: An advance in treatment of recurrent vulvovaginal candidiasis. Future Microbiol. 2021, 16, 1453–1461. [Google Scholar] [CrossRef]
- Brand, S.R.; Degenhardt, T.P.; Person, K.; Sobel, J.D.; Nyirjesy, P.; Schotzinger, R.J.; Tavakkol, A. A phase 2, randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the efficacy and safety of orally administered VT-1161 in the treatment of recurrent vulvovaginal candidiasis. Am. J. Obstet. Gynecol. 2018, 218, 624.e621–624.e629. [Google Scholar] [CrossRef]
- FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection). Available online: https://www.businesswire.com/news/home/20220428005301/en/FDA-Approves-Mycovia-Pharmaceuticals%E2%80%99-VIVJOA%E2%84%A2-oteseconazole-the-First-and-Only-FDA-Approved-Medication-for-Recurrent-Vulvovaginal-Candidiasis-Chronic-Yeast-Infection (accessed on 7 July 2022).
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=215888 (accessed on 7 July 2022).
- Elewski, B.; Brand, S.; Degenhardt, T.; Curelop, S.; Pollak, R.; Schotzinger, R.; Tavakkol, A. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the efficacy and safety of VT-1161 oral tablets in the treatment of patients with distal and lateral subungual onychomycosis of the toenail. Br. J. Dermatol. 2021, 184, 270–280. [Google Scholar] [CrossRef]
- Brand, S.R.; Sobel, J.D.; Nyirjesy, P.; Ghannoum, M.A.; Schotzinger, R.J.; Degenhardt, T.P. A Randomized Phase 2 Study of VT-1161 for the Treatment of Acute Vulvovaginal Candidiasis. Clin. Infect. Dis. 2021, 73, e1518–e1524. [Google Scholar] [CrossRef]
- Warrilow, A.G.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The Investigational Drug VT-1129 Is a Highly Potent Inhibitor of Cryptococcus Species CYP51 but Only Weakly Inhibits the Human Enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Fothergill, A.W.; Iqbal, N.; Bolden, C.B.; Grossman, N.T.; Garvey, E.P.; Brand, S.R.; Hoekstra, W.J.; Schotzinger, R.J.; Ottinger, E.; et al. The Investigational Fungal Cyp51 Inhibitor VT-1129 Demonstrates Potent In Vitro Activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrob. Agents Chemother. 2016, 60, 2528–2531. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Najvar, L.K.; Garvey, E.P.; Brand, S.R.; Xu, X.; Ottinger, E.A.; Alimardanov, A.; Cradock, J.; Behnke, M.; Hoekstra, W.J.; et al. The Fungal Cyp51 Inhibitor VT-1129 Is Efficacious in an Experimental Model of Cryptococcal Meningitis. Antimicrob. Agents Chemother. 2018, 62, e01071-18. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Xu, X.; Wang, A.; Najvar, L.K.; Garvey, E.P.; Ottinger, E.A.; Alimardanov, A.; Cradock, J.; Behnke, M.; Hoekstra, W.J.; et al. In Vivo Efficacy of VT-1129 against Experimental Cryptococcal Meningitis with the Use of a Loading Dose-Maintenance Dose Administration Strategy. Antimicrob. Agents Chemother. 2018, 62, e01315-18. [Google Scholar] [CrossRef]
- Gonzalez-Lara, M.F.; Sifuentes-Osornio, J.; Ostrosky-Zeichner, L. Drugs in Clinical Development for Fungal Infections. Drugs 2017, 77, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Sharp, A.D.; Warn, P.A.; Yates, C.M.; Atari, M.; Thomas, S.; Schotzinger, R.J. The novel fungal CYP51 inhibitor VT-1598 displays classic dose-dependent antifungal activity in murine models of invasive aspergillosis. Med. Mycol. 2020, 58, 505–513. [Google Scholar] [CrossRef] [PubMed]
- A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients with Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03561701?term=VT-1161&draw=2&rank=1 (accessed on 28 June 2022).
- A Study of Oral Oteseconazole for the Treatment of Patients with Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03562156?term=VT-1161&draw=2&rank=7 (accessed on 28 June 2022).
- Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients with Recurrent Yeast Infections (ultraVIOLET). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03840616?term=VT-1161&draw=2&rank=3 (accessed on 28 June 2022).
- A Study to Evaluate Oral VT-1161 in the Treatment of Patients with Recurrent Vaginal Candidiasis (Yeast Infection). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02267382?term=VT-1161&draw=2&rank=2 (accessed on 28 June 2022).
- A Study to Evaluate the Efficacy and Safety of Oral VT-1161 in Patients with Onychomycosis of the Toenail. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02267356?term=VT-1161&draw=2&rank=4 (accessed on 28 June 2022).
- A Study to Evaluate the Efficacy and Safety of Oral VT-1161 in Patients with Acute Vaginal Candidiasis (Yeast Infection). Available online: https://www.clinicaltrials.gov/ct2/show/NCT01891331?term=VT-1161&draw=2&rank=5 (accessed on 28 June 2022).
- A Study to Evaluate the Efficacy and Safety of Oral VT-1161 in Patients with Moderate—Severe Interdigital Tinea Pedis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01891305?term=VT-1161&draw=2&rank=6 (accessed on 28 June 2022).
- Safety and Pharmacokinetics of VT-1598. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04208321?term=VT1598&draw=2&rank=1 (accessed on 28 June 2022).
- Wang, Y.; Xu, K.; Bai, G.; Huang, L.; Wu, Q.; Pan, W.; Yu, S. Synthesis and antifungal activity of novel triazole compounds containing piperazine moiety. Molecules 2014, 19, 11333–11340. [Google Scholar] [CrossRef]
- Hashemi, S.; Badali, H.; Faramarzi, M.; Samadi, N.; Afsarian, M.; Irannejad, H.; Emami, S. Novel triazole alcohol antifungals derived from fluconazole: Design, synthesis, and biological activity. Mol. Divers. 2014, 19, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Gu, J.; Wang, C.; Wang, S.; Liu, N.; Jiang, Y.; Dong, G.; Wang, Y.; Liu, Y.; Yao, J.; et al. Design, synthesis and antifungal activity of novel triazole derivatives containing substituted 1,2,3-triazole-piperdine side chains. Eur. J. Med. Chem. 2014, 82, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, Y.; Badali, H.; Hashemi, S.M.; Ansari, M.; Fakhim, H.; Fallah, M.; Shokrzadeh, M.; Emami, S. New potent antifungal triazole alcohols containing N-benzylpiperazine carbodithioate moiety: Synthesis, in vitro evaluation and in silico study. Bioorg. Chem. 2019, 90, 103060. [Google Scholar] [CrossRef]
- Wu, S.; He, X.; Che, X.; Wang, S.; Liu, Y.; Jiang, Y.; Liu, N.; Dong, G.; Yao, J.; Miao, Z.; et al. Inside Cover: From Antidiabetic to Antifungal: Discovery of Highly Potent Triazole-Thiazolidinedione Hybrids as Novel Antifungal Agents. ChemMedChem 2014, 9, 2639–2646. [Google Scholar] [CrossRef]
- Fang, X.F.; Li, D.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.W.; Zhou, C.H. Novel potentially antifungal hybrids of 5-flucytosine and fluconazole: Design, synthesis and bioactive evaluation. Bioorg. Med. Chem. Lett. 2017, 27, 4964–4969. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically active antifungal azoles: Synthesis and antifungal activity of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-[2-[4-aryl-piperazin-1-yl]-ethyl]-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem. 2004, 12, 2225–2238. [Google Scholar] [CrossRef]
- Qian, A.; Zheng, Y.; Wang, R.; Wei, J.; Cui, Y.; Cao, X.; Yang, Y. Design, synthesis, and structure-activity relationship studies of novel tetrazole antifungal agents with potent activity, broad antifungal spectrum and high selectivity. Bioorg. Med. Chem. Lett. 2018, 28, 344–350. [Google Scholar] [CrossRef]
- Subhedar, D.D.; Shaikh, M.H.; Nawale, L.; Yeware, A.; Sarkar, D.; Khan, F.A.; Sangshetti, J.N.; Shingate, B.B. Novel tetrazoloquinoline-rhodanine conjugates: Highly efficient synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2016, 26, 2278–2283. [Google Scholar] [CrossRef]
- Kategaonkar, A.H.; Sadaphal, S.A.; Shelke, K.F.; Kategaonkar, A.H.; Shingate, B.B.; Shingare, M.S. Synthesis and in vitro Antimicrobial Activity of New Ethyl 2-(Ethoxyphosphono)-1-cyano-2-(substituted tetrazolo [1,5-a]quinolin-4-yl)ethanoate Derivatives. Chin. J. Chem. 2010, 28, 243–249. [Google Scholar] [CrossRef]
- Nikam, M.D.; Mahajan, P.S.; Damale, M.G.; Sangshetti, J.N.; Dabhade, S.K.; Shinde, D.W.; Gill, C.H. Synthesis, molecular docking and biological evaluation of some novel tetrazolo [1,5-a]quinoline incorporated pyrazoline and isoxazoline derivatives. Med. Chem. Res. 2015, 24, 3372–3386. [Google Scholar] [CrossRef]
- Wang, S.Q.; Wang, Y.F.; Xu, Z. Tetrazole hybrids and their antifungal activities. Eur. J. Med. Chem. 2019, 170, 225–234. [Google Scholar] [CrossRef]
- Dhayanithi, V.; Syed, S.S.; Kumaran, K.; Reguraman, K.; Sankar, J.; Ragavan, R.; Goud, S.K.; Kumari, N.S.; Pati, H.N. Synthesis of selected 5-thio-substituted tetrazole derivatives and evaluation of their antibacterial and antifungal activities. J. Serb. Chem. Soc. 2011, 76, 165–175. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Kaplancıklı, Z.A.; Ciftçi, G.A.; Demirel, R. Synthesis and biological evaluation of thiazoline derivatives as new antimicrobial and anticancer agents. Eur. J. Med. Chem. 2014, 74, 264–277. [Google Scholar] [CrossRef]
- Ozkay, Y.; Tunalı, Y.; Karaca, H.; Işıkdağ, I. Antimicrobial activity of a new combination system of benzimidazole and various azoles. Arch. Pharm. 2011, 344, 264–271. [Google Scholar] [CrossRef]
- Mungra, D.C.; Patel, M.P.; Patel, R.G. Microwave-assisted synthesis of some new tetrazolo [1,5-a]quinoline-based benzimidazoles catalyzed by p-TsOH and investigation of their antimicrobial activity. Med. Chem. Res. 2011, 20, 782–789. [Google Scholar] [CrossRef]
- Vembu, S.; Pazhamalai, S.; Gopalakrishnan, M. Synthesis, spectral characterization, and effective antifungal evaluation of 1H-tetrazole containing 1,3,5-triazine dendrimers. Med. Chem. Res. 2016, 25, 1916–1924. [Google Scholar] [CrossRef]
- Shanmugapandiyan, P.; Atmakuru, R. Synthesis and antimicrobial activity of 1-(benzothiazol-2′-yl)-5- phenyl-tetrazole. Asian J. Chem. 2008, 20, 992–998. [Google Scholar]
- Łukowska-Chojnacka, E.; Mierzejewska, J.; Milner-Krawczyk, M.; Bondaryk, M.; Staniszewska, M. Synthesis of novel tetrazole derivatives and evaluation of their antifungal activity. Bioorg. Med. Chem. 2016, 24, 6058–6065. [Google Scholar] [CrossRef] [PubMed]
- Kanakaraju, S.; Suresh, L. Design, synthesis, in vitro antimicrobial and cytotoxic evaluation of novel 1,2,3-selena/thiadiazolyltetrazole derivatives. RSC Adv. 2015, 5, 29325–29334. [Google Scholar] [CrossRef]
- Shaikh, S.K.J.; Kamble, R.R.; Somagond, S.M.; Devarajegowda, H.C.; Dixit, S.R.; Joshi, S.D. Tetrazolylmethyl quinolines: Design, docking studies, synthesis, anticancer and antifungal analyses. Eur. J. Med. Chem. 2017, 128, 258–273. [Google Scholar] [CrossRef]
- Antypenko, L.M.; Kovalenko, S.I.; Antypenko, O.M.; Katsev, A.M.; Achkasova, O.M. Design and Evaluation of Novel Antimicrobial and Anticancer Agents among Tetrazolo [1,5-c]quinazoline-5-thione S-Derivatives. Sci. Pharm. 2013, 81, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Vembu, S.; Pavadai, P.; Gopalakrishnan, M. Synthesis, in vitro antifungal and antitubercular evaluation of novel amino pyrimidines based tetrazole derivatives. J. Pharm. Res. 2014, 88, 1552–1558. [Google Scholar]
- Vembu, S.; Pavadai, P.; Gopalakrishnan, M. Design, in silico molecular docking studies, synthesis, spectral characterization and in vitro antifungal evaluation of 1-(4-(1H-tetrazole-1-yl) phenyl)-3-arylprop-2-en-1-ones. Der Pharma Chem. 2014, 6, 35–44. [Google Scholar]
- Dofe, V.S.; Sarkate, A.P.; Kathwate, S.H.; Gill, C.H. Synthesis, antimicrobial activity and anti-biofilm activity of novel tetrazole derivatives. Heterocycl. Commun. 2017, 23, 325–330. [Google Scholar] [CrossRef]
- Nandha, B.; Hazra, K.; Chandra, J.N.; Nargund, L.V.G.; Pp, R.; Harish, M.S.; Puranik, D. Synthesis of some new substituted fluoro benzimidazoles and their antimicrobial screening. Der Pharma Chem. 2013, 5, 287–295. [Google Scholar]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Al-Ghamdi, A.R.; Ghabbour, H.A.; Al-Agamy, M.H.; Attia, M.I. Synthesis, structure elucidation, and antifungal potential of certain new benzodioxole-imidazole molecular hybrids bearing ester functionalities. Drug Des. Devel. Ther. 2019, 13, 775–789. [Google Scholar] [CrossRef]
- Kumar, L.; Lal, N.; Kumar, V.; Sarswat, A.; Jangir, S.; Bala, V.; Kumar, L.; Kushwaha, B.; Pandey, A.K.; Siddiqi, M.I.; et al. Azole-carbodithioate hybrids as vaginal anti-Candida contraceptive agents: Design, synthesis and docking studies. Eur. J. Med. Chem. 2013, 70, 68–77. [Google Scholar] [CrossRef]
- Malūkaitė, D.; Grybaitė, B.; Vaickelionienė, R.; Vaickelionis, G.; Sapijanskaitė-Banevič, B.; Kavaliauskas, P.; Mickevičius, V. Synthesis of Novel Thiazole Derivatives Bearing β-Amino Acid and Aromatic Moieties as Promising Scaffolds for the Development of New Antibacterial and Antifungal Candidates Targeting Multidrug-Resistant Pathogens. Molecules 2022, 27, 74. [Google Scholar]
- Guo, H.-Y.; Chen, Z.-A.; Shen, Q.-K.; Quan, Z.-S. Application of triazoles in the structural modification of natural products. J. Enzym. Inhib. Med. Chem. 2021, 36, 1115–1144. [Google Scholar] [CrossRef]
- Junior, E.F.C.; Guimarães, C.; Franco, L.L.; Alves, R.J.; Kato, K.C.; Martins, H.R.; de Souza Filho, J.D.; Bemquerer, M.P.; Munhoz, V.H.O.; Resende, J.M.; et al. Glycotriazole-peptides derived from the peptide HSP1: Synergistic effect of triazole and saccharide rings on the antifungal activity. Amino Acids 2017, 49, 1389–1400. [Google Scholar] [CrossRef]
- de Souza, T.B.; Orlandi, M.; Coelho, L.F.L.; Malaquias, L.C.C.; Dias, A.L.T.; de Carvalho, R.R.; Silva, N.C.; Carvalho, D.T. Synthesis and in vitro evaluation of antifungal and cytotoxic activities of eugenol glycosides. Med. Chem. Res. 2014, 23, 496–502. [Google Scholar] [CrossRef]
- de Souza, T.B.; de Oliveira Brito, K.M.; Silva, N.C.; Rocha, R.P.; de Sousa, G.F.; Duarte, L.P.; Coelho, L.F.L.; Dias, A.L.T.; Veloso, M.P.; Carvalho, D.T.; et al. New Eugenol Glucoside-based Derivative Shows Fungistatic and Fungicidal Activity against Opportunistic Candida glabrata. Chem. Biol. Drug Des. 2016, 87, 83–90. [Google Scholar] [CrossRef]
- Hipólito, T.M.M.; Bastos, G.T.L.; Barbosa, T.W.L.; de Souza, T.B.; Coelho, L.F.L.; Dias, A.L.T.; Rodríguez, I.C.; dos Santos, M.H.; Dias, D.F.; Franco, L.L.; et al. Synthesis, activity, and docking studies of eugenol-based glucosides as new agents against Candida sp. Chem. Biol. Drug Des. 2018, 92, 1514–1524. [Google Scholar] [CrossRef]
- Magalhães, L.S.; Reis, A.C.C.; Nakao, I.A.; Péret, V.A.C.; Reis, R.; Silva, N.C.; Dias, A.L.T.; Carvalho, D.T.; Dias, D.F.; Brandão, G.C.; et al. Glucosyl-1,2,3-triazoles derived from eugenol and analogues: Synthesis, anti-Candida activity, and molecular modeling studies in CYP-51. Chem. Biol. Drug Des. 2021, 98, 903–913. [Google Scholar] [CrossRef]
- Goswami, L.; Gupta, L.; Paul, S.; Vermani, M.; Vijayaraghavan, P.; Bhattacharya, A.K. Design and synthesis of eugenol/isoeugenol glycoconjugates and other analogues as antifungal agents against Aspergillus fumigatus. RSC Med. Chem. 2022, 13, 955–962. [Google Scholar] [CrossRef]
- Pyta, K.; Blecha, M.; Janas, A.; Klich, K.; Pecyna, P.; Gajecka, M.; Przybylski, P. Synthesis, structure and antimicrobial evaluation of a new gossypol triazole conjugates functionalized with aliphatic chains and benzyloxy groups. Bioorg. Med. Chem. Lett. 2016, 26, 4322–4326. [Google Scholar] [CrossRef]
- Pertino, M.W.; Theoduloz, C.; Butassi, E.; Zacchino, S.; Schmeda-Hirschmann, G. Synthesis, Antiproliferative and Antifungal Activities of 1,2,3-Triazole-Substituted Carnosic Acid and Carnosol Derivatives. Molecules 2015, 20, 8666–8686. [Google Scholar] [CrossRef] [PubMed]
- Irfan, M.; Aneja, B.; Yadava, U.; Khan, S.I.; Manzoor, N.; Daniliuc, C.G.; Abid, M. Synthesis, QSAR and anticandidal evaluation of 1,2,3-triazoles derived from naturally bioactive scaffolds. Eur. J. Med. Chem. 2015, 93, 246–254. [Google Scholar] [CrossRef] [PubMed]



















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
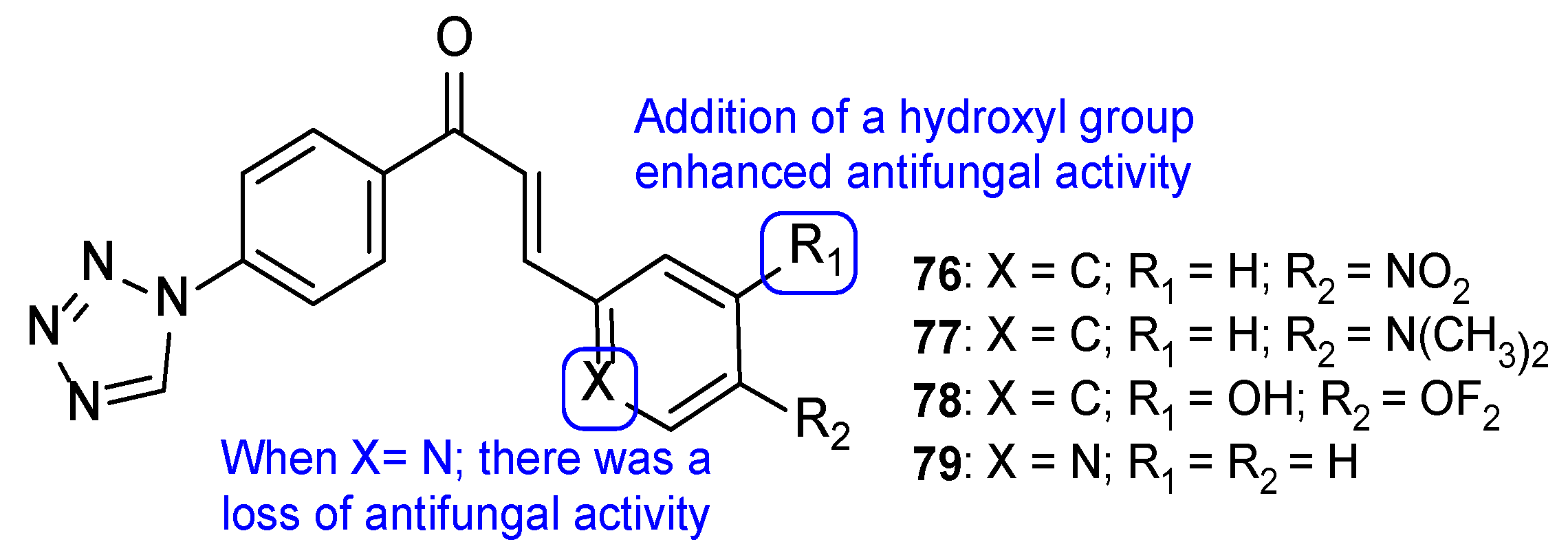{kind=link}
{kind=link}
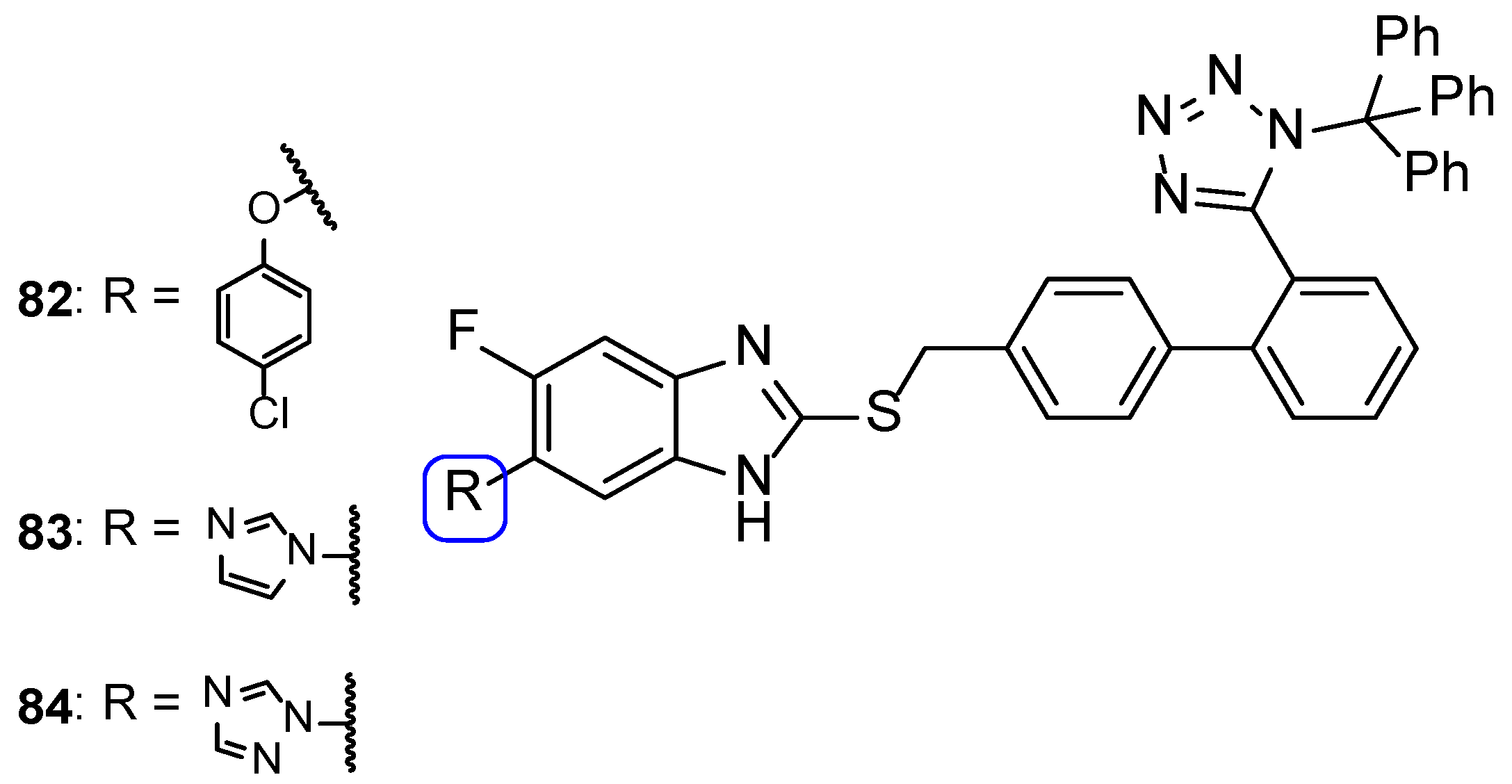{kind=link}
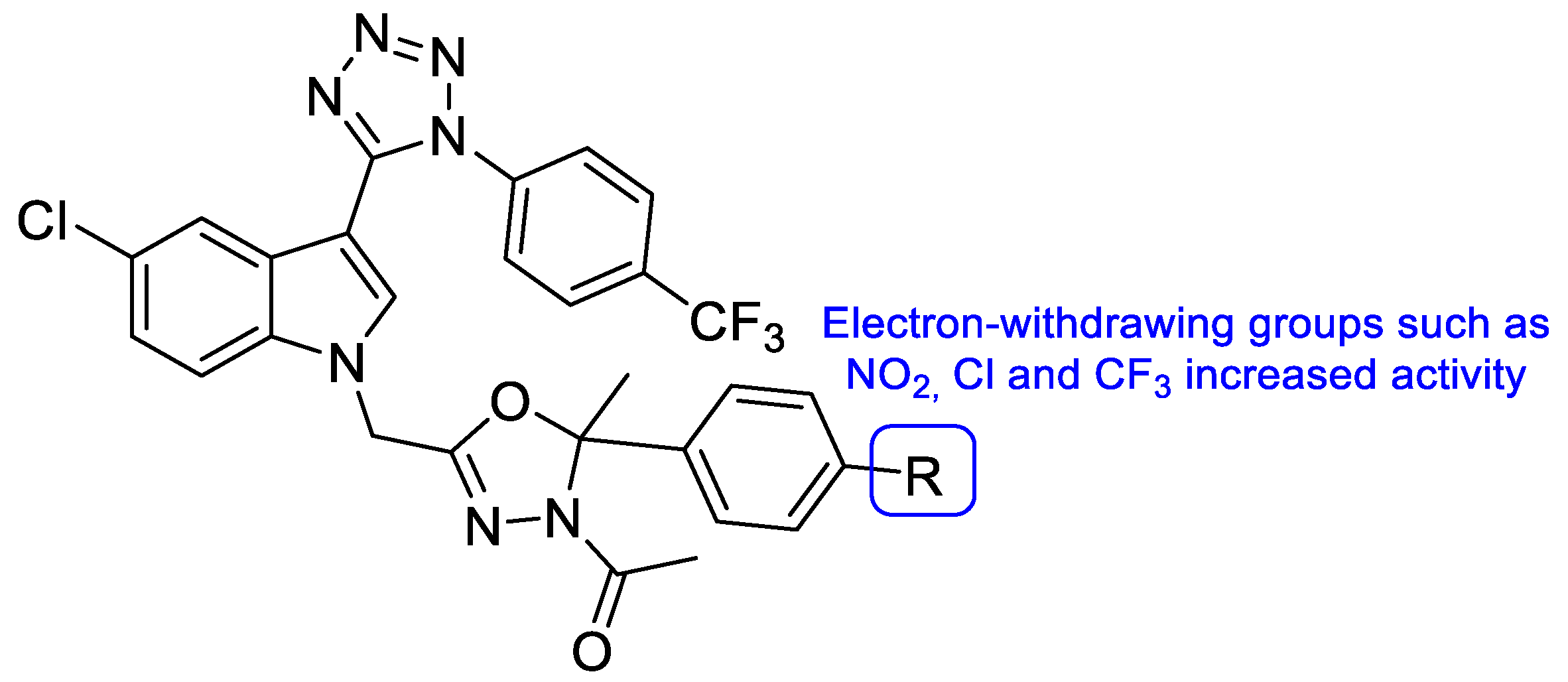{kind=link}
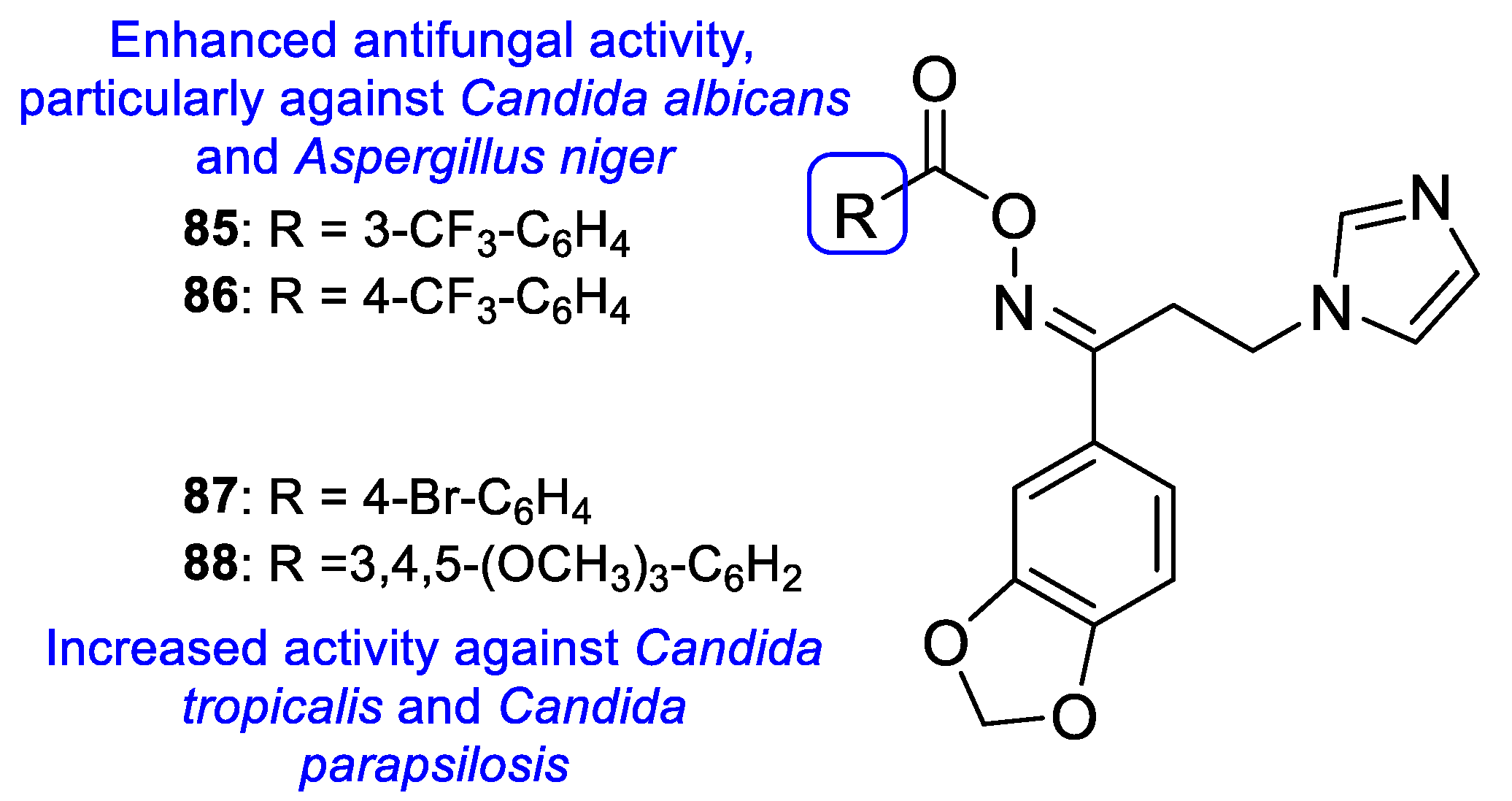{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|
| 4 | NCT02394340 | Study evaluating the drug interaction potential of luliconazole cream 1% in participants with tinea pedis and tinea cruris | Tinea pedis Tinea cruris | Completed | [33] |
| NCT02767271 | Maximal use of luliconazole cream 1% in pediatric participants with moderate to severe tinea pedis or tinea cruris | Tinea pedis Tinea cruris | Completed | [34] | |
| 4 | NCT02767947 | Safety and efficacy of product 33525 (luliconazole cream 1%) in pediatric participants with tinea corporis | Tinea corporis | Completed | [35] |
| 2, 3 | NCT01431820 | Safety and efficacy of luliconazole solution, 10% in subjects with mild to moderate onychomycosis (solution) | Distal and lateral subungual onychomycosis | Completed | [36] |
| 2 | NCT00869336 | Multicenter study of the efficacy and safety of luliconazole cream in tinea pedis (athlete’s foot) | Tinea pedis | Completed | [37] |
| 1, 2 | NCT01044381 | Open-label pharmacokinetics/safety study of luliconazole solution, 10% in distal subungual onychomycosis | Onychomycosis | Completed | [38] |
| 1 | NCT05110638 | Safety and tolerability study of SKX-16 (luliconazole 10% solution) in subjects with moderate to severe distal subungual onychomycosis | Onychomycosis of toenail | Active, not recruiting | [39] |
| Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|
| 3 | NCT00412893 | Isavuconazole (BAL8557) for primary treatment of invasive aspergillosis | Aspergillosis Invasive fungal infections | Completed | [48] |
| NCT00634049 | Isavuconazole in the treatment of renally impaired aspergillosis and rare fungi (vital) | Aspergillosis Invasive fungal infections | Completed | [49] | |
| NCT00413218 | Isavuconazole (BAL8557) in the treatment of candidemia and other invasive Candida infections | Candidiasis, invasive Candidemia Mycoses | Completed | [50] |
| Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|
| 3 | NCT03572049 | Endemic mycoses treatment with SUBA-itraconazole vs itraconazole (MSG15) | Invasive fungal infections | Completed | [54] |
| Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|
| 2 | NCT00730405 | Efficacy and safety study of 4 dose regimens of oral albaconazole in subjects with distal subungual onychomycosis | Onychomycosis | Completed | [59] |
| Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|
| 3 | NCT05238116 | Safety and efficacy of PC945 in combination with other antifungal therapy for the treatment of refractory invasive pulmonary aspergillosis | Refractory IPA | Recruiting | [66] |
| 2 | NCT03905447 | The effect of early treatment of PC945 on Aspergillus fumigatus lung infection in lung transplant patients | Aspergillosis Lung transplant infection | Terminated | [67] |
| NCT03870841 | The effect of PC945 on Aspergillus fumigatus lung infection in patients with cystic fibrosis | Aspergillosis Cystic fibrosis | Terminated | [68] | |
| NCT05037851 | PC945 prophylaxis or pre-emptive therapy against pulmonary aspergillosis in lung transplant recipients | Pulmonary aspergillosis | Recruiting | [69] | |
| NCT03745196 | The effect of PC945 on Aspergillus or Candida lung infections in patients with asthma or chronic respiratory diseases | Asthma Respiratory candidiasis Respiratory aspergillosis COPD Bronchiectasis | Terminated | [70] | |
| 1 | NCT02715570 | A study to investigate the safety, tolerability and pharmacokinetics of single and repeat doses of PC945 | Aspergillosis | Completed | [71] |
| Drug | Phase | Identifier | Title | Condition(s) | Status | Ref. |
|---|---|---|---|---|---|---|
| Oteseconazole (17) | 3 | NCT03562156; NCT03561701 | A study of oral oteseconazole for the treatment of patients with recurrent vaginal candidiasis (yeast infection) (violet) | Recurrent Vaginal Candidiasis | Completed | [88,89] |
| NCT03840616 | Study of oral oteseconazole (VT-1161) for acute yeast infections in patients with recurrent yeast infections (ultraviolet) | Recurrent vaginal candidiasis | Completed | [90] | ||
| 2 | NCT02267382 | A study to evaluate oral VT-1161 in the treatment of patients with recurrent vaginal candidiasis (yeast infection) | Recurrent vaginal candidiasis | Completed | [77,91] | |
| NCT02267356 | A study to evaluate the efficacy and safety of oral VT-1161 in patients with onychomycosis of the toenail | Onychomycosis | Completed | [92] | ||
| NCT01891331 | A study to evaluate the efficacy and safety of oral VT-1161 in patients with acute vaginal candidiasis (yeast infection) | Candidiasis, vulvovaginal | Completed | [93] | ||
| NCT01891305 | A study to evaluate the efficacy and safety of oral VT-1161 in patients with moderate—severe interdigital tinea pedis | Tinea pedis | Completed | [94] | ||
| VT-1598 (19) | 1 | NCT04208321 | Safety and pharmacokinetics of VT-1598 | Coccidioidomycosis | Completed | [95] |
 | |||
|---|---|---|---|
| X | Y | R | SAR |
Triazole  | F |  | R1 = Halogens, methyl, cyanide, and nitro groups led to promising antifungal potential against Candida albicans, Candida parapsilosis, Cryptococcus neoformans, and Nannizzia gypsea |
| F or Cl |  | R1 = Di-chloro-phenyl, paired with Y = Cl, led to the highest antifungal potential of the tested series Y = Cl increased antifungal activity against C. albicans, C. parapsilosis, C. neoformans, Epidermophyton floccosum and Trichophyton mentagrophytes | |
| F |  | R1 = butyrate and butyric acid increased antifungal potential against all tested strains; 4-acetyl or 4-trifluoromethoxy-phenyl groups increased activity for C. albicans; 2-methyl-phenyl group increased activity against C. parapsilosis and Candida glabrata | |
| F or Cl |  | R1 = 4-Chloro showed high antifungal activity against C. albicans, C. glabrata, C. parapsilosis, Candida krusei and Candida tropicalis. Y = F analogs presented higher antifungal potency | |
| F |  | R1 = Di-chloro-phenyl, furan ring presented promising results against C. albicans | |
| F | 1 | 1: R1 = alkyl chains were not beneficial for antifungal activity 2: R1–3 = one fluor substituent; two chlorine substituents: increased antifungal activity against C. albicans | |
2 | |||
| F | 1 | 1 and 2: positional isomers R1 = trifluoromethyl led to high antifungal activity against C. albicans, C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, C. neoformans, Aspergillus fumigatus, and Aspergillus niger | |
2 | |||
Tetrazole  | F |  | R1 = Alicyclic side chains up to six-members enhance the antifungal potency against C. albicans, C. parapsilosis, C. glabrata, C. neoformans, and A. fumigatus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, M.M.; Carvalho, D.T.; Sousa, E.; Pinto, E. New Antifungal Agents with Azole Moieties. Pharmaceuticals 2022, 15, 1427. https://doi.org/10.3390/ph15111427
Teixeira MM, Carvalho DT, Sousa E, Pinto E. New Antifungal Agents with Azole Moieties. Pharmaceuticals. 2022; 15(11):1427. https://doi.org/10.3390/ph15111427
Chicago/Turabian StyleTeixeira, Melissa Martins, Diogo Teixeira Carvalho, Emília Sousa, and Eugénia Pinto. 2022. "New Antifungal Agents with Azole Moieties" Pharmaceuticals 15, no. 11: 1427. https://doi.org/10.3390/ph15111427
APA StyleTeixeira, M. M., Carvalho, D. T., Sousa, E., & Pinto, E. (2022). New Antifungal Agents with Azole Moieties. Pharmaceuticals, 15(11), 1427. https://doi.org/10.3390/ph15111427