
Integrating Mechanisms in Thrombotic Peripheral Arterial Disease
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Atherosclerosis
2.1. Mechanisms
2.2. Vascular Bed Specificity
2.3. Rupture vs. Erosion
3. Thrombo-Inflammation
3.1. Modulators of Platelet Function
3.2. Tissue Factor: Factor VII Pathway
3.3. Activators of the Factor XII-Dependent Intrinsic Coagulation Pathway
3.4. Cell–Cell Interactions
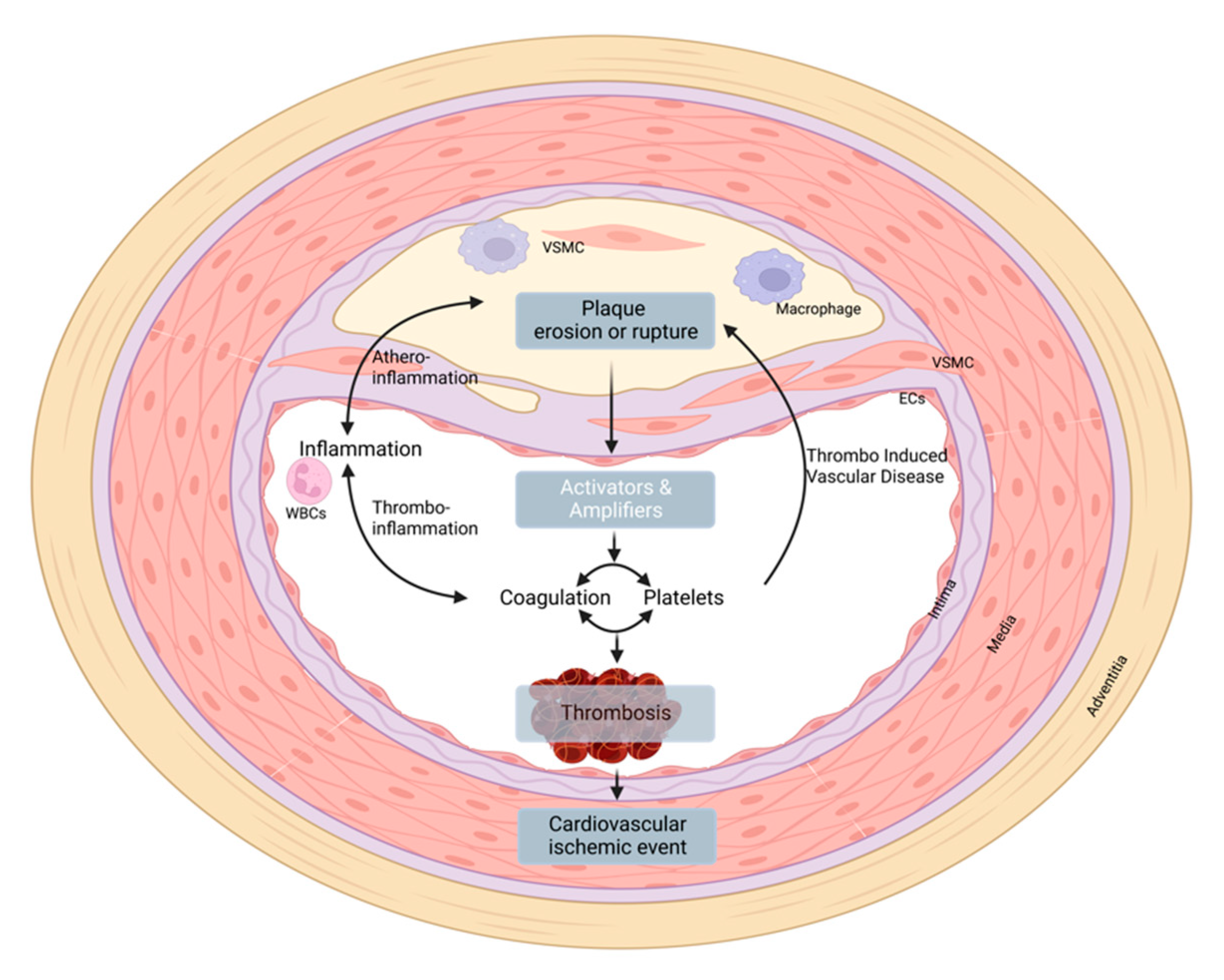
4. Thrombo-Induced Vascular Disease
5. Treatment Options
5.1. Current Antithrombotic Therapy in PAD
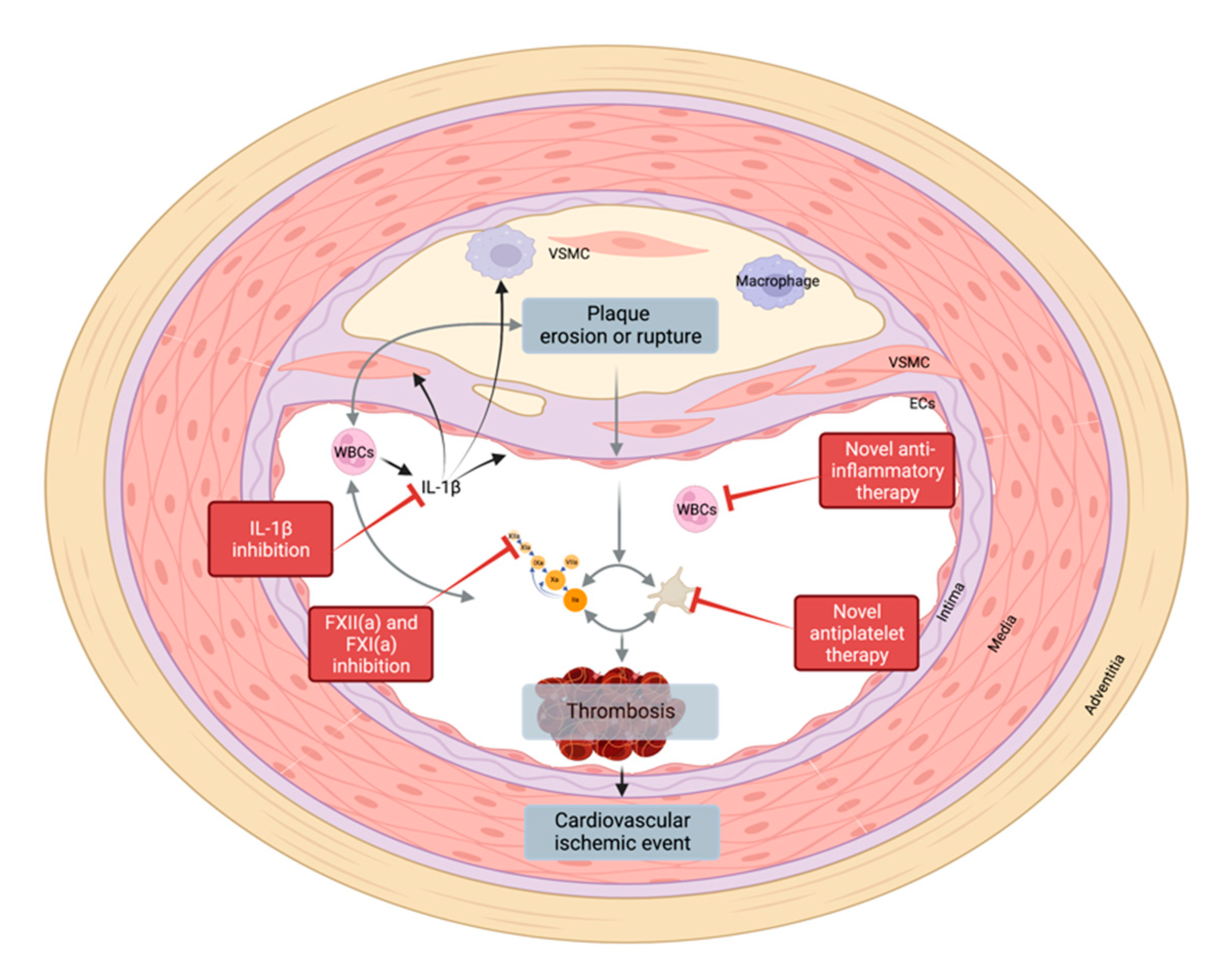
5.2. New Targets
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.R. TASC II Working Group Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67. [Google Scholar] [CrossRef] [PubMed]
- Del Río, C.V.; Mostaza, J.; Lahoz, C.; Sánchez-Arroyo, V.; Sabín, C.; López, S.; Patrón, P.; Fernández-García, P.; Fernández-Puntero, B.; Vicent, D.; et al. Prevalence of Peripheral Artery Disease (PAD) and Factors Associated: An Epidemiological Analysis from the Population-Based Screening PRE-Diabetes and Type 2 DIAbetes (SPREDIA-2) Study. PLoS ONE 2017, 12, e0186220. [Google Scholar]
- Selvin, E.; Erlinger, T.P. Prevalence of and Risk Factors for Peripheral Arterial Disease in the United States: Results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 2004, 110, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Kalbaugh, C.A.; Kucharska-Newton, A.; Wruck, L.; Lund, J.L.; Selvin, E.; Matsushita, K.; Bengtson, L.G.S.; Heiss, G.; Loehr, L. Peripheral Artery Disease Prevalence and Incidence Estimated from Both Outpatient and Inpatient Settings Among Medicare Fee-for-Service Beneficiaries in the Atherosclerosis Risk in Communities (ARIC) Study. J. Am. Heart Assoc. 2017, 6, e003796. [Google Scholar] [CrossRef] [PubMed]
- Kleinegris, M.-C.F.; ten Cate, H.; ten Cate-Hoek, A.J. D-Dimer as a Marker for Cardiovascular and Arterial Thrombotic Events in Patients with Peripheral Arterial Disease. A Systematic Review. Thromb. Haemost. 2013, 110, 233–243. [Google Scholar]
- Kremers, B.; Wübbeke, L.; Mees, B.; Ten Cate, H.; Spronk, H.; Ten Cate-Hoek, A. Plasma Biomarkers to Predict Cardiovascular Outcome in Patients with Peripheral Artery Disease: A Systematic Review and Meta-Analysis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2018–2032. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Spronk, H.M.H.; ten Cate, H. The Hemostatic System as a Modulator of Atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-Activated Receptors: Contribution to Physiology and Disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic Plaque Progression and Vulnerability to Rupture: Angiogenesis as a Source of Intraplaque Hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Howarth, S.P.S.; Tang, T.; Gillard, J.H. How Critical Is Fibrous Cap Thickness to Carotid Plaque Stability? Stroke 2006, 37, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Moreno, P.R.; Fayad, Z.A.; Corti, R.; Badimon, J.J. Atherothrombosis and High-Risk Plaque: Part I: Evolving Concepts. J. Am. Coll. Cardiol. 2005, 46, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jørgensen, H.F. Disease-Relevant Transcriptional Signatures Identified in Individual Smooth Muscle Cells from Healthy Mouse Vessels. Nat. Commun. 2018, 9, 4567. [Google Scholar] [CrossRef]
- McClelland, R.L.; Jorgensen, N.W.; Budoff, M.; Blaha, M.J.; Post, W.S.; Kronmal, R.A.; Bild, D.E.; Shea, S.; Liu, K.; Watson, K.E.; et al. 10-Year Coronary Heart Disease Risk Prediction Using Coronary Artery Calcium and Traditional Risk Factors: Derivation in the MESA (Multi-Ethnic Study of Atherosclerosis) With Validation in the HNR (Heinz Nixdorf Recall) Study and the DHS (Dallas Heart Study). J. Am. Coll. Cardiol. 2015, 66, 1643–1653. [Google Scholar]
- Rennenberg, R.J.M.W.; Kessels, A.G.H.; Schurgers, L.J.; van Engelshoven, J.M.A.; de Leeuw, P.W.; Kroon, A.A. Vascular Calcifications as a Marker of Increased Cardiovascular Risk: A Meta-Analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G. Requiem for the “Vulnerable Plaque”. Eur. Heart J. 2015, 36, 2984–2987. [Google Scholar] [CrossRef]
- Libby, P.; Theroux, P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef]
- Partida, R.A.; Libby, P.; Crea, F.; Jang, I.-K. Plaque Erosion: A New in Vivo Diagnosis and a Potential Major Shift in the Management of Patients with Acute Coronary Syndromes. Eur. Heart J. 2018, 39, 2070–2076. [Google Scholar] [CrossRef]
- Bernardi, B.; Guidetti, G.F.; Campus, F.; Crittenden, J.R.; Graybiel, A.M.; Balduini, C.; Torti, M. The Small GTPase Rap1b Regulates the Cross Talk between Platelet Integrin alpha2beta1 and Integrin alphaIIbbeta3. Blood 2006, 107, 2728–2735. [Google Scholar] [CrossRef]
- Guidetti, G.; Bertoni, A.; Viola, M.; Tira, E.; Balduini, C.; Torti, M. The Small Proteoglycan Decorin Supports Adhesion and Activation of Human Platelets. Blood 2002, 100, 1707–1714. [Google Scholar] [CrossRef]
- Mazzucato, M.; Cozzi, M.R.; Pradella, P.; Perissinotto, D.; Malmstrom, A.; Morgelin, M.; Spessotto, P.; Colombatti, A.; De Marco, L.; Perris, R. Vascular PG-M/versican Variants Promote Platelet Adhesion at Low Shear Rates and Cooperate with Collagens to Induce Aggregation. FASEB J. 2002, 16, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Wight, T.N.; Virmani, R. Differential Accumulation of Proteoglycans and Hyaluronan in Culprit Lesions: Insights into Plaque Erosion. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Koshiishi, I.; Shizari, M.; Underhill, C.B. CD44 Can Mediate the Adhesion of Platelets to Hyaluronan. Blood 1994, 84, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, A.; Moura, R.; Hoylaerts, M.F. The Evolving Role of Thrombospondin-1 in Hemostasis and Vascular Biology. Cell. Mol. Life Sci. 2008, 65, 713–727. [Google Scholar] [CrossRef]
- Kuijpers, M.J.E.; de Witt, S.; Nergiz-Unal, R.; van Kruchten, R.; Korporaal, S.J.A.; Verhamme, P.; Febbraio, M.; Tjwa, M.; Voshol, P.J.; Hoylaerts, M.F.; et al. Supporting Roles of Platelet Thrombospondin-1 and CD36 in Thrombus Formation on Collagen. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1187–1192. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 Links Hyperlipidemia, Oxidant Stress and a Prothrombotic Phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- Nergiz-Unal, R.; Lamers, M.M.E.; Van Kruchten, R.; Luiken, J.J.; Cosemans, J.M.E.M.; Glatz, J.F.C.; Kuijpers, M.J.E.; Heemskerk, J.W.M. Signaling Role of CD36 in Platelet Activation and Thrombus Formation on Immobilized Thrombospondin or Oxidized Low-Density Lipoprotein. J. Thromb. Haemost. 2011, 9, 1835–1846. [Google Scholar] [CrossRef]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL Activates Blood Platelets through CD36/NOX2-Mediated Inhibition of the cGMP/protein Kinase G Signaling Cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef]
- Choi, W.S.; Jeon, O.H.; Kim, D.S. CD40 Ligand Shedding Is Regulated by Interaction between Matrix Metalloproteinase-2 and Platelet Integrin αIIbβ3. J. Thromb. Haemost. 2010, 8, 1364–1371. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 Ligand on Activated Platelets Triggers an Inflammatory Reaction of Endothelial Cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Prasad, K.S.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L Stabilizes Arterial Thrombi by a beta3 Integrin--Dependent Mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.S.S.; Andre, P.; He, M.; Bao, M.; Manganello, J.; Phillips, D.R. Soluble CD40 Ligand Induces beta3 Integrin Tyrosine Phosphorylation and Triggers Platelet Activation by Outside-in Signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 12367–12371. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, D.; Hachem, A.; Théorêt, J.-F.; Gillis, M.-A.; Mourad, W.; Merhi, Y. Enhanced Levels of Soluble CD40 Ligand Exacerbate Platelet Aggregation and Thrombus Formation through a CD40-Dependent Tumor Necrosis Factor Receptor-Associated Factor-2/Rac1/p38 Mitogen-Activated Protein Kinase Signaling Pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.J.E.; Mattheij, N.J.A.; Cipolla, L.; van Geffen, J.P.; Lawrence, T.; Donners, M.M.P.C.; Boon, L.; Lievens, D.; Torti, M.; Noels, H.; et al. Platelet CD40L Modulates Thrombus Growth Via Phosphatidylinositol 3-Kinase β, and Not Via CD40 and IκB Kinase α. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1374–1381. [Google Scholar] [CrossRef]
- Lenti, M.; Falcinelli, E.; Pompili, M.; de Rango, P.; Conti, V.; Guglielmini, G.; Momi, S.; Corazzi, T.; Giordano, G.; Gresele, P. Matrix Metalloproteinase-2 of Human Carotid Atherosclerotic Plaques Promotes Platelet Activation. Correlation with ischaemic events. Thromb. Haemost. 2014, 111, 1089–1101. [Google Scholar] [CrossRef]
- Momi, S.; Falcinelli, E.; Giannini, S.; Ruggeri, L.; Cecchetti, L.; Corazzi, T.; Libert, C.; Gresele, P. Loss of Matrix Metalloproteinase 2 in Platelets Reduces Arterial Thrombosis in Vivo. J. Exp. Med. 2009, 206, 2365–2379. [Google Scholar] [CrossRef]
- Mastenbroek, T.G.; Feijge, M.A.H.; Kremers, R.M.W.; van den Bosch, M.T.J.; Swieringa, F.; De Groef, L.; Moons, L.; Bennett, C.; Ghevaert, C.; Johnson, J.L.; et al. Platelet-Associated Matrix Metalloproteinases Regulate Thrombus Formation and Exert Local Collagenolytic Activity. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2554–2561. [Google Scholar] [CrossRef]
- Soslau, G.; Mason, C.; Lynch, S.; Benjamin, J.; Ashak, D.; Prakash, J.M.; Moore, A.; Bagsiyao, P.; Albert, T.; Mathew, L.M.; et al. Intracellular Matrix Metalloproteinase-2 (MMP-2) Regulates Human Platelet Activation via Hydrolysis of Talin. Thromb. Haemost. 2014, 111, 140–153. [Google Scholar]
- Sebastiano, M.; Momi, S.; Falcinelli, E.; Bury, L.; Hoylaerts, M.F.; Gresele, P. A Novel Mechanism Regulating Human Platelet Activation by MMP-2-Mediated PAR1 Biased Signaling. Blood 2017, 129, 883–895. [Google Scholar] [CrossRef]
- Momi, S.; Falcinelli, E.; Petito, E.; Ciarrocca Taranta, G.; Ossoli, A.; Gresele, P. Matrix Metalloproteinase-2 on Activated Platelets Triggers Endothelial PAR-1 Initiating Atherosclerosis. Eur. Heart J. 2021, 43, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Geisler, T. Inflammatory Contribution of Platelets Revisited: New Players in the Arena of Inflammation. Semin. Thromb. Hemost. 2016, 42, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.G.; Harper, M.T.; Poole, A.W. SDF-1α Is a Novel Autocrine Activator of Platelets Operating through Its Receptor CXCR4. Cell. Signal. 2015, 27, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of Oxidized Platelet Lipidome: Implications for Coronary Artery Disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef]
- Wilcox, J.N.; Smith, K.M.; Schwartz, S.M.; Gordon, D. Localization of Tissue Factor in the Normal Vessel Wall and in the Atherosclerotic Plaque. Proc. Natl. Acad. Sci. USA 1989, 86, 2839–2843. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Heeneman, S.; Kilinç, E.; Kassák, P.; Van Oerle, R.; Winckers, K.; Govers-Riemslag, J.W.P.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.A.P.; et al. Early Atherosclerosis Exhibits an Enhanced Procoagulant State. Circulation 2010, 122, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Olsen, O.H. Allosteric Activation of Coagulation Factor VIIa. Front. Biosci. 2011, 16, 3156–3163. [Google Scholar] [CrossRef]
- Butenas, S.; Mann, K.G. Kinetics of Human Factor VII Activation. Biochemistry 1996, 35, 1904–1910. [Google Scholar] [CrossRef]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How It All Starts: Initiation of the Clotting Cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Munnix, I.C.A.; Auger, J.M.; Govers-Riemslag, J.W.P.; Cosemans, J.M.E.M.; Kuijpers, M.J.E.; Spronk, H.M.; Watson, S.P.; Renné, T.; Heemskerk, J.W.M. Dual Role of Collagen in Factor XII-Dependent Thrombus Formation. Blood 2009, 114, 881–890. [Google Scholar] [CrossRef]
- White-Adams, T.C.; Berny, M.A.; Patel, I.A. Laminin Promotes Coagulation and Thrombus Formation in a Factor XII-dependent Manner. J. Thromb. Haemost. 2010, 8, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.J.E.; van der Meijden, P.E.J.; Feijge, M.A.H.; Mattheij, N.J.A.; May, F.; Govers-Riemslag, J.; Meijers, J.C.M.; Heemskerk, J.W.M.; Renné, T.; Cosemans, J.M.E.M. Factor XII Regulates the Pathological Process of Thrombus Formation on Ruptured Plaques. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef]
- Naudin, C.; Burillo, E.; Blankenberg, S.; Butler, L.; Renné, T. Factor XII Contact Activation. Semin. Thromb. Hemost. 2017, 43, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meijden, P.E.J.; Van Schilfgaarde, M.; Van Oerle, R.; Renné, T.; ten Cate, H.; Spronk, H.M.H. Platelet- and Erythrocyte-Derived Microparticles Trigger Thrombin Generation via Factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Henderson, M.W.; Mooberry, M.; Ilich, A.; Ellsworth, P.; Piegore, M.; Skinner, S.C.; Pawlinski, R.; Welsby, I.; Renné, T.; et al. Red Blood Cell Microvesicles Activate the Contact System, Leading to Factor IX Activation via 2 Independent Pathways. Blood 2020, 135, 755–765. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Promote Thrombin Generation through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.-B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In Vitro Activation of Coagulation by Human Neutrophil DNA and Histone Proteins but Not Neutrophil Extracellular Traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular RNA Constitutes a Natural Procoagulant Cofactor in Blood Coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393. [Google Scholar] [CrossRef]
- Oehmcke, S.; Mörgelin, M.; Herwald, H. Activation of the Human Contact System on Neutrophil Extracellular Traps. J. Innate Immun. 2009, 1, 225–230. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.C.; Weitz, J.I.; Liaw, P.C. Neutrophil Extracellular Traps Promote Thrombin Generation through Platelet-Dependent and Platelet-Independent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil Extracellular Traps License Macrophages for Cytokine Production in Atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of Functional Tissue Factor by Neutrophil Extracellular Traps in Culprit Artery of Acute Myocardial Infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R. The Prowess of Platelets in Immunity and Inflammation. Thromb Haemost 2016, 116, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M.E.M. Platelet Interaction with Activated Endothelium: Mechanistic Insights from Microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Filippi, M.-D. Mechanism of Diapedesis: Importance of the Transcellular Route. Adv. Immunol. 2016, 129, 25–53. [Google Scholar]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Schmitt, M.M.N. Platelets and Their Chemokines in Atherosclerosis-Clinical Applications. Front. Physiol. 2014, 5, 294. [Google Scholar] [CrossRef]
- Swystun, L.L.; Liaw, P.C. The Role of Leukocytes in Thrombosis. Blood Am. Soc. Hematol. 2016, 128, 753–762. [Google Scholar] [CrossRef]
- Hartwig, H.; Silvestre Roig, C.; Daemen, M.; Lutgens, E.; Soehnlein, O. Neutrophils in Atherosclerosis. A Brief Overview. Hamostaseologie 2015, 35, 121–127. [Google Scholar]
- Bowley, S.R.; Fang, C.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Protein Disulfide Isomerase Secretion Following Vascular Injury Initiates a Regulatory Pathway for Thrombus Formation. Nat. Commun. 2017, 8, 14151. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal Coupling of Coagulation and Innate Immunity via Neutrophil Serine Proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-Associated Molecular Pattern-Activated Neutrophil Extracellular Trap Exacerbates Sterile Inflammatory Liver Injury. Hepatology 2015, 62, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-Derived HMGB1 Is a Critical Mediator of Thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular Histones Are Major Mediators of Death in Sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones Induce Rapid and Profound Thrombocytopenia in Mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef]
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice in Vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Boudoulas, H. The Role of Red Blood Cells in the Progression and Instability of Atherosclerotic Plaque. Int. J. Cardiol. 2010, 142, 2–7. [Google Scholar] [CrossRef]
- Michel, J.-B.; Martin-Ventura, J.L. Red Blood Cells and Hemoglobin in Human Atherosclerosis and Related Arterial Diseases. Int. J. Mol. Sci. 2020, 21, 6756. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Cortese-Krott, M.M.; Kelm, M.; Li, N.; Pernow, J. Novel Perspectives on Redox Signaling in Red Blood Cells and Platelets in Cardiovascular Disease. Free Radic. Biol. Med. 2021, 168, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Turpin, C.; Catan, A.; Meilhac, O.; Bourdon, E.; Canonne-Hergaux, F.; Rondeau, P. Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 5843. [Google Scholar] [CrossRef]
- Dann, R.; Hadi, T.; Montenont, E.; Boytard, L.; Alebrahim, D.; Feinstein, J.; Allen, N.; Simon, R.; Barone, K.; Uryu, K.; et al. Platelet-Derived MRP-14 Induces Monocyte Activation in Patients with Symptomatic Peripheral Artery Disease. J. Am. Coll. Cardiol. 2018, 71, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.D.; Cornwell, M.G.; Zhou, H.; Rockman, C.; Heguy, A.; Suarez, Y.; Cheng, H.S.; Feinberg, M.W.; Hochman, J.S.; Ruggles, K.V.; et al. Gene Expression Signature in Patients with Symptomatic Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Schlegel, M.; Zhou, F.; Gorenchtein, M.; Bolstorff, J.; Moore, K.J.; Fisher, E.A.; Berger, J.S. Platelet Regulation of Myeloid Suppressor of Cytokine Signaling 3 Accelerates Atherosclerosis. Sci. Transl. Med. 2019, 11, eaax0481. [Google Scholar] [CrossRef]
- Loeffen, R.; Spronk, H.M.H.; ten Cate, H. The Impact of Blood Coagulability on Atherosclerosis and Cardiovascular Disease. J. Thromb. Haemost. 2012, 10, 1207–1216. [Google Scholar] [CrossRef]
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. Genome-Wide Association Study of Peripheral Artery Disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef]
- Spronk, H.M.H.; De Jong, A.M.; Verheule, S.; De Boer, H.C.; Maass, A.H.; Lau, D.H.; Rienstra, M.; van Hunnik, A.; Kuiper, M.; Lumeij, S.; et al. Hypercoagulability Causes Atrial Fibrosis and Promotes Atrial Fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar] [CrossRef]
- de Moerloose, P.; Boehlen, F. Inherited Thrombophilia in Arterial Disease: A Selective Review. Semin. Hematol. 2007, 44, 106–113. [Google Scholar] [CrossRef]
- Houbballah, R.; LaMuraglia, G.M. Clotting Problems: Diagnosis and Management of Underlying Coagulopathies. Semin. Vasc. Surg. 2010, 23, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Fukuda, D.; Tanaka, K.; Higashikuni, Y.; Hirata, Y.; Nishimoto, S.; Yagi, S.; Yamada, H.; Soeki, T.; Wakatsuki, T.; et al. Rivaroxaban, a Novel Oral Anticoagulant, Attenuates Atherosclerotic Plaque Progression and Destabilization in ApoE-Deficient Mice. Atherosclerosis 2015, 242, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Bea, F.; Kreuzer, J.; Preusch, M.; Schaab, S.; Isermann, B.; Rosenfeld, M.E.; Katus, H.; Blessing, E. Melagatran Reduces Advanced Atherosclerotic Lesion Size and May Promote Plaque Stability in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-O.; Kratz, M.T.; Schirmer, S.H.; Baumhäkel, M.; Böhm, M. The Effects of Direct Thrombin Inhibition with Dabigatran on Plaque Formation and Endothelial Function in Apolipoprotein E-Deficient Mice. J. Pharmacol. Exp. Ther. 2012, 343, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Otten, J.J.T.; Heeneman, S.; Leenders, P.; van Oerle, R.; Soehnlein, O.; Loubele, S.T.B.G.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.A.P.; et al. Genetic and Pharmacological Modifications of Thrombin Formation in Apolipoprotein E-Deficient Mice Determine Atherosclerosis Severity and Atherothrombosis Onset in a Neutrophil-Dependent Manner. PLoS ONE 2013, 8, e55784. [Google Scholar] [CrossRef]
- Zhou, Q.; Bea, F.; Preusch, M.; Wang, H.; Isermann, B.; Shahzad, K.; Katus, H.A.; Blessing, E. Evaluation of Plaque Stability of Advanced Atherosclerotic Lesions in Apo E-Deficient Mice after Treatment with the Oral Factor Xa Inhibitor Rivaroxaban. Mediat. Inflamm. 2011, 2011, 432080. [Google Scholar] [CrossRef]
- Posthuma, J.J.; Posma, J.J.N.; van Oerle, R.; Leenders, P.; van Gorp, R.H.; Jaminon, A.M.G.; Mackman, N.; Heitmeier, S.; Schurgers, L.J.; Ten Cate, H.; et al. Targeting Coagulation Factor Xa Promotes Regression of Advanced Atherosclerosis in Apolipoprotein-E Deficient Mice. Sci. Rep. 2019, 9, 3909. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin Signalling and Protease-Activated Receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Coughlin, S.R. Protease-Activated Receptors in Hemostasis, Thrombosis and Vascular Biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef]
- Posma, J.J.N.; Posthuma, J.J.; Spronk, H.M.H. Coagulation and Non-Coagulation Effects of Thrombin. J. Thromb. Haemost. 2016, 14, 1908–1916. [Google Scholar] [CrossRef]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Activated Protein C: Biased for Translation. Blood 2015, 125, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Mann, A.; Conrad, K.; Saum, K.; Hall, D.E.; McKinney, L.M.; Robbins, N.; Thompson, J.; Peairs, A.D.; Camerer, E.; et al. PAR2 (Protease-Activated Receptor 2) Deficiency Attenuates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Zuo, Z.; Zheng, Y.; Wang, X.; Zhou, Q.; Chen, L.; Ma, G. Protease-Activated Receptor-2 Deficiency Attenuates Atherosclerotic Lesion Progression and Instability in Apolipoprotein E-Deficient Mice. Front. Pharmacol. 2017, 8, 647. [Google Scholar] [CrossRef]
- Hara, T.; Phuong, P.T.; Fukuda, D.; Yamaguchi, K.; Murata, C.; Nishimoto, S.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; et al. Protease-Activated Receptor-2 Plays a Critical Role in Vascular Inflammation and Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation 2018, 138, 1706–1719. [Google Scholar] [CrossRef] [PubMed]
- Plank, F.; Beyer, C.; Friedrich, G.; Stühlinger, M.; Hintringer, F.; Dichtl, W.; Wildauer, M.; Feuchtner, G. Influence of Vitamin K Antagonists and Direct Oral Anticoagulation on Coronary Artery Disease: A CTA Analysis. Int. J. Cardiol. 2018, 260, 11–15. [Google Scholar] [CrossRef]
- CAPRIE Steering Committee. A Randomised, Blinded, Trial of Clopidogrel versus Aspirin in Patients at Risk of Ischaemic Events (CAPRIE). Lancet 1996, 348, 1329–1339. [Google Scholar] [CrossRef]
- Birkeland, K.; Parra, D.; Rosenstein, R. Antiplatelet Therapy in Acute Coronary Syndromes: Focus on Ticagrelor. J. Blood Med. 2010, 1, 197–219. [Google Scholar]
- Galimzhanov, A.M.; Azizov, B.S. Ticagrelor for Asian Patients with Acute Coronary Syndrome in Real-World Practice: A Systematic Review and Meta-Analysis of Observational Studies. Indian Heart J. 2019, 71, 15–24. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Kapadia, S.R.; Bajzer, C.T.; Chew, D.P.; Ziada, K.M.; Mukherjee, D.; Roffi, M.; Topol, E.J.; Yadav, J.S. Dual Antiplatelet Therapy with Clopidogrel and Aspirin after Carotid Artery Stenting. J. Invasive Cardiol. 2001, 13, 767–771. [Google Scholar]
- Bhatt, D.L.; Fox, K.A.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen, E.A.; Creager, M.A.; Easton, J.D.; et al. Clopidogrel and Aspirin versus Aspirin Alone for the Prevention of Atherothrombotic Events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef]
- Mega, J.L.; Braunwald, E.; Wiviott, S.D.; Bassand, J.-P.; Bhatt, D.L.; Bode, C.; Burton, P.; Cohen, M.; Cook-Bruns, N.; Fox, K.A.A.; et al. Rivaroxaban in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 366, 9–19. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Bauersachs, R.M.; Anand, S.S.; Debus, E.S.; Nehler, M.R.; Patel, M.R.; Fanelli, F.; Capell, W.H.; Diao, L.; Jaeger, N.; et al. Rivaroxaban in Peripheral Artery Disease after Revascularization. N. Engl. J. Med. 2020, 382, 1994–2004. [Google Scholar] [CrossRef]
- Hess, C.N.; Szarek, M.; Anand, S.S.; Bauersachs, R.M.; Patel, M.R.; Debus, E.S.; Nehler, M.R.; Capell, W.H.; Beckman, J.A.; Piazza, G.; et al. Rivaroxaban and Risk of Venous Thromboembolism in Patients with Symptomatic Peripheral Artery Disease After Lower Extremity Revascularization. JAMA Netw. Open 2022, 5, e2215580. [Google Scholar] [CrossRef]
- Willems, L.H.; Maas, D.P.M.S.M.; Kramers, K.; Reijnen, M.M.P.J.; Riksen, N.P.; Ten Cate, H.; van der Vijver-Coppen, R.J.; de Borst, G.J.; Mees, B.M.E.; Zeebregts, C.J.; et al. Antithrombotic Therapy for Symptomatic Peripheral Arterial Disease: A Systematic Review and Network Meta-Analysis. Drugs 2022, 82, 1287–1302. [Google Scholar] [CrossRef]
- Russell, K.S.; Yates, D.P.; Kramer, C.M.; Feller, A.; Mahling, P.; Colin, L.; Clough, T.; Wang, T.; LaPerna, L.; Patel, A.; et al. A Randomized, Placebo-Controlled Trial of Canakinumab in Patients with Peripheral Artery Disease. Vasc. Med. 2019, 24, 414–421. [Google Scholar] [CrossRef]
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 Stimulates the Release of Procoagulant Microvesicles from Human Monocytes and Differentially Increases Tissue Factor in Human Monocyte Subsets. Thromb. Haemost. 2017, 117, 1379–1390. [Google Scholar] [CrossRef]
- Altara, R.; Ghali, R.; Mallat, Z.; Cataliotti, A.; Booz, G.W.; Zouein, F.A. Conflicting Vascular and Metabolic Impact of the IL-33/sST2 Axis. Cardiovasc. Res. 2018, 114, 1578–1594. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Conover, C.A.; Overgaard, M.T.; Bailey, K.R.; Christiansen, M.; Holmes, D.R., Jr.; Virmani, R.; Oxvig, C.; Schwartz, R.S. Pregnancy-Associated Plasma Protein A as a Marker of Acute Coronary Syndromes. N. Engl. J. Med. 2001, 345, 1022–1029. [Google Scholar] [CrossRef]
- Cirillo, P.; Conte, S.; Pellegrino, G.; Ziviello, F.; Barra, G.; De Palma, R.; Leonardi, A.; Trimarco, B. Pregnancy-Associated Plasma Protein-A Promotes TF Procoagulant Activity in Human Endothelial Cells by Akt-NF-κB Axis. J. Thromb. Thrombolysis 2016, 42, 225–232. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grade | Symptoms |
|---|---|
| Stage I | Asymptomatic, incomplete blood vessel obstruction |
| Stage II | Mild claudication pain in limb |
| Stage IIA | Claudication at a distance > 200 m |
| Stage IIB | Claudication at a distance < 200 m |
| Stage III | Rest pain, mostly in the feet |
| Stage IV | Necrosis and/or gangrene of the limb |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, M.; van der Meijden, P.E.J.; Glunz, J.; Schurgers, L.; Lutgens, E.; ten Cate, H.; Heitmeier, S.; Spronk, H.M.H. Integrating Mechanisms in Thrombotic Peripheral Arterial Disease. Pharmaceuticals 2022, 15, 1428. https://doi.org/10.3390/ph15111428
Nagy M, van der Meijden PEJ, Glunz J, Schurgers L, Lutgens E, ten Cate H, Heitmeier S, Spronk HMH. Integrating Mechanisms in Thrombotic Peripheral Arterial Disease. Pharmaceuticals. 2022; 15(11):1428. https://doi.org/10.3390/ph15111428
Chicago/Turabian StyleNagy, Magdolna, Paola E. J. van der Meijden, Julia Glunz, Leon Schurgers, Esther Lutgens, Hugo ten Cate, Stefan Heitmeier, and Henri M. H. Spronk. 2022. "Integrating Mechanisms in Thrombotic Peripheral Arterial Disease" Pharmaceuticals 15, no. 11: 1428. https://doi.org/10.3390/ph15111428
APA StyleNagy, M., van der Meijden, P. E. J., Glunz, J., Schurgers, L., Lutgens, E., ten Cate, H., Heitmeier, S., & Spronk, H. M. H. (2022). Integrating Mechanisms in Thrombotic Peripheral Arterial Disease. Pharmaceuticals, 15(11), 1428. https://doi.org/10.3390/ph15111428