
Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis

 , and
, and 
Abstract
1. Introduction
1.1. HDACs: Functions, Classes, and Therapeutic Potential
1.2. Repurposing Anticancer HDACi as Antiparasitic Agents
1.3. Schistosomiasis—Key Facts
1.4. SmHDAC8, a Potential Drug Target in Schistosomes
2. Antischistosomal Effect of HDAC Inhibitors
2.1. Pan HDAC Inhibitors
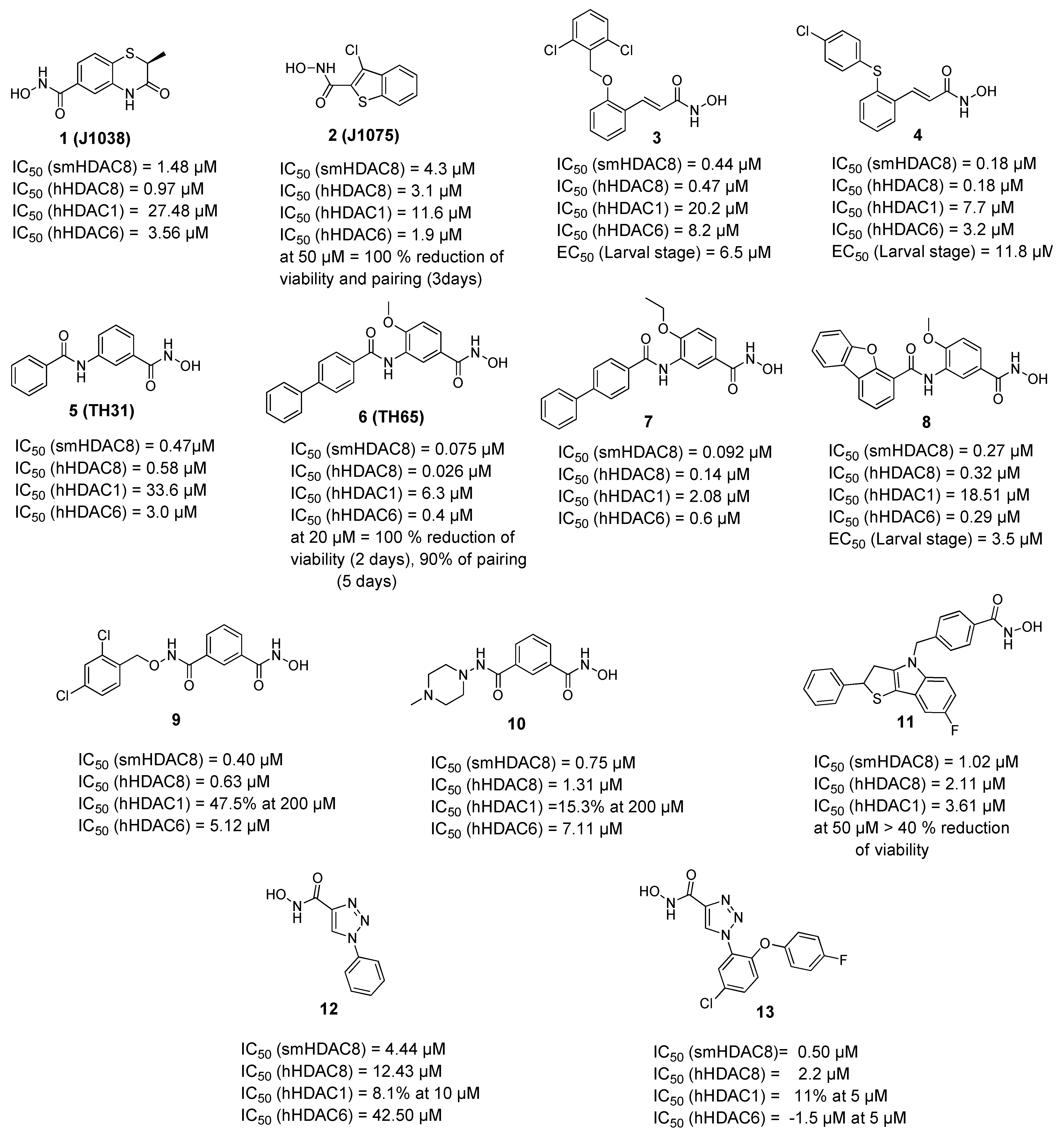
2.2. Selective smHDAC8 Inhibitors
2.2.1. Hydroxamic Acid Based Inhibitors
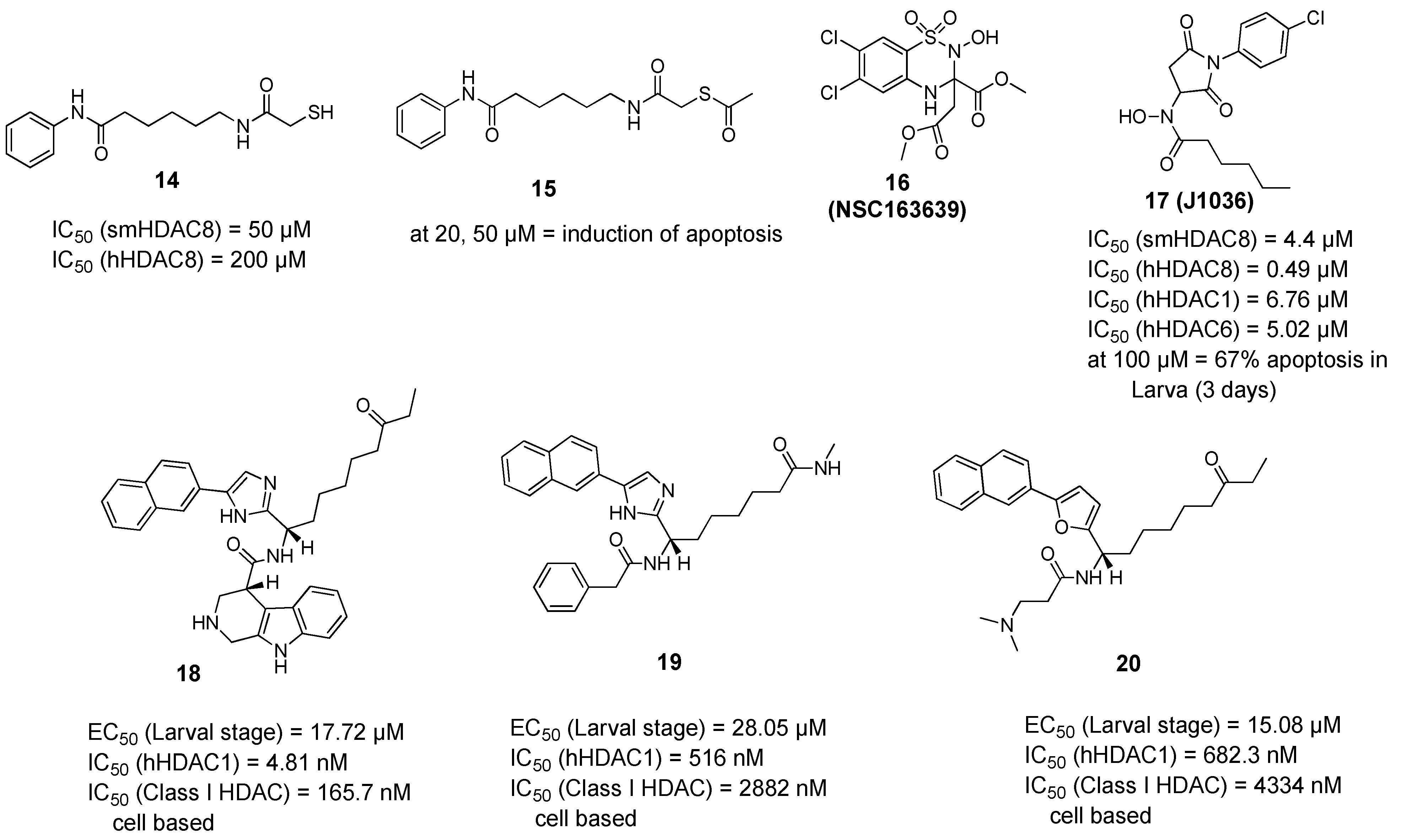
2.2.2. Non-Hydroxamic Acid Based Inhibitors
2.3. Molecular Pathways Affected by smHDAC8 Inhibitors
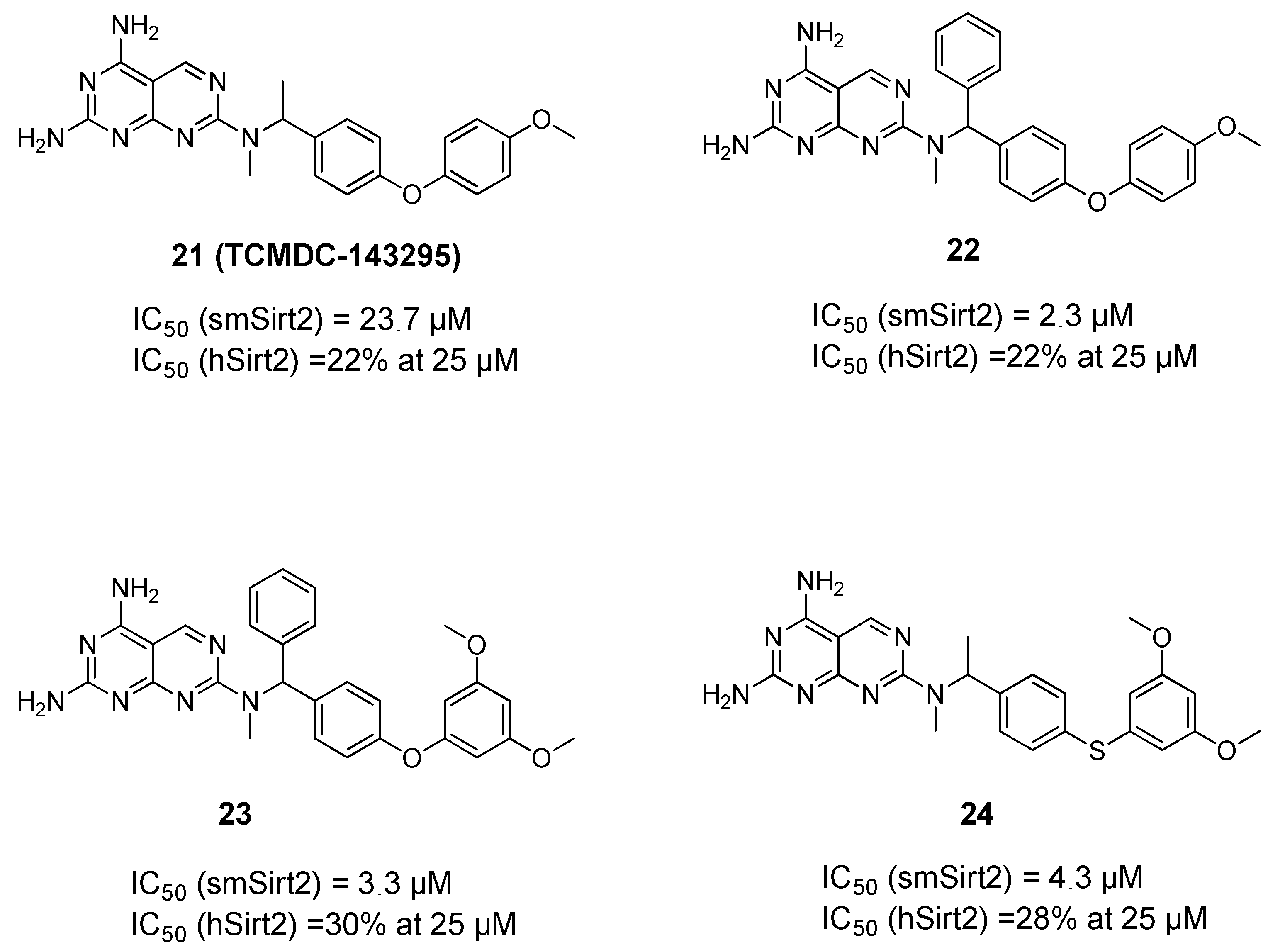
3. Schistosomal Sirtuins Are Potential Targets for Antischistosomal Therapy
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Jing, H.; Lin, H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Chen, B.; Zang, W.; Wang, J.; Huang, Y.; He, Y.; Yan, L.; Liu, J.; Zheng, W. The chemical biology of sirtuins. Chem. Soc. Rev. 2015, 44, 5246–5264. [Google Scholar] [CrossRef] [PubMed]
- Zwinderman, M.R.H.; De Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef]
- Mathias, R.; Guise, A.J.; Cristea, I.M. Post-translational Modifications Regulate Class IIa Histone Deacetylase (HDAC) Function in Health and Disease. Mol. Cell. Proteom. 2015, 14, 456–470. [Google Scholar] [CrossRef]
- Yao, Y.-L.; Yang, W.-M. Beyond Histone and Deacetylase: An Overview of Cytoplasmic Histone Deacetylases and Their Nonhistone Substrates. J. Biomed. Biotechnol. 2010, 2011, 1–15. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, C.-Y. Histone modifying enzymes: Novel disease biomarkers and assay development. Expert Rev. Mol. Diagn. 2016, 16, 297–306. [Google Scholar] [CrossRef]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The Substrate Specificity of Sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef] [PubMed]
- Osborne, B.; Bentley, N.L.; Montgomery, M.K.; Turner, N. The role of mitochondrial sirtuins in health and disease. Free Radic. Biol. Med. 2016, 100, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Danaher, R.J.; Zhang, L.; Donley, C.J.; Laungani, N.A.; Hui, S.E.; Miller, C.S.; Westlund, K.N. Histone deacetylase inhibitors prevent persistent hypersensitivity in an orofacial neuropathic pain model. Mol. Pain 2018, 14, 1744806918796763. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.J.; Advani, A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 2630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Q.; Yang, Q.; Tang, J.; Xu, C.; Gai, D.; Chen, X.; Chen, J. Histone Deacetylase 3 Inhibitor Suppresses Hepatitis C Virus Replication by Regulating Apo-A1 and LEAP-1 Expression. Virol. Sin. 2018, 33, 418–428. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Zhang, H.; Gatta, E.; Glover, E.J.; Pandey, S.C.; Lasek, A.W. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 2019, 78, 79–87. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kee, H.J.; Sun, S.; Seok, Y.M.; Ryu, Y.; Kim, G.R.; Kee, S.-J.; Pflieger, M.; Kurz, T.; Kassack, M.U.; et al. Histone deacetylase inhibitor LMK235 attenuates vascular constriction and aortic remodelling in hypertension. J. Cell. Mol. Med. 2019, 23, 2801–2812. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Ryu, Y.; Seok, Y.M.; Choi, S.Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens. 2019, 25, 13. [Google Scholar] [CrossRef] [PubMed]
- Romeiro, L.A.S.; Nunes, J.L.D.C.; Miranda, C.D.O.; Cardoso, G.S.H.R.; de Oliveira, A.S.; Gandini, A.; Kobrlova, T.; Soukup, O.; Rossi, M.; Senger, J.; et al. Novel Sustainable-by-Design HDAC Inhibitors for the Treatment of Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10, 671–676.e7. [Google Scholar] [CrossRef]
- Steelant, B.; Wawrzyniak, P.; Martens, K.; Jonckheere, A.-C.; Pugin, B.; Schrijvers, R.; Bullens, D.M.A.; Vanoirbeek, J.; Krawczyk, K.; Dreher, A.; et al. Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2019, 144, 1242–1253. [Google Scholar] [CrossRef]
- Sangwan, R.; Rajan, R.; Mandal, P.K. HDAC as onco target: Reviewing the synthetic approaches with SAR study of their inhibitors. Eur. J. Med. Chem. 2018, 158, 620–706. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, R.; Mautone, N.; Rovere, A.; Rotili, D.; Mai, A. Targeting histone acetylation/deacetylation in parasites: An update (2017–2020). Curr. Opin. Chem. Biol. 2020, 57, 65–74. [Google Scholar] [CrossRef]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.; Ng, S.S.; Fairlie, D.P.; Skinner-Adams, T.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef]
- Oliveira, G. Cancer and parasitic infections: Similarities and opportunities for the development of new control tools. Rev. da Soc. Bras. Med. Trop. 2014, 47, 1–2. [Google Scholar] [CrossRef]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine Deacetylase Inhibitors in Parasites: Past, Present, and Future Perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef]
- Hansen, F.K.; Sumanadasa, S.D.; Stenzel, K.; Duffy, S.; Meister, S.; Marek, L.; Schmetter, R.; Kuna, K.; Hamacher, A.; Mordmüller, B.; et al. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014, 82, 204–213. [Google Scholar] [CrossRef]
- Ontoria, J.M.; Paonessa, G.; Ponzi, S.; Ferrigno, F.; Nizi, E.; Biancofiore, I.; Malancona, S.; Graziani, R.; Roberts, D.; Willis, P.; et al. Discovery of a Selective Series of Inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. [Google Scholar] [CrossRef] [PubMed]
- De Vreese, R.; De Kock, C.; Smith, P.J.; Chibale, K.; D’Hooghe, M. Exploration of thiaheterocyclic hHDAC6 inhibitors as potential antiplasmodial agents. Futur. Med. Chem. 2017, 9, 357–364. [Google Scholar] [CrossRef]
- Diedrich, D.; Stenzel, K.; Hesping, E.; Antonova-Koch, Y.; Gebru, T.; Duffy, S.; Fisher, G.; Schöler, A.; Meister, S.; Kurz, T.; et al. One-pot, multi-component synthesis and structure-activity relationships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur. J. Med. Chem. 2018, 158, 801–813. [Google Scholar] [CrossRef]
- Bouchut, A.; Rotili, D.; Pierrot, C.; Valente, S.; Lafitte, S.; Schultz, J.; Hoglund, U.; Mazzone, R.; Lucidi, A.; Fabrizi, G.; et al. Identification of novel quinazoline derivatives as potent antiplasmodial agents. Eur. J. Med. Chem. 2019, 161, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Mackwitz, M.K.W.; Hesping, E.; Antonova-Koch, Y.; Diedrich, D.; Woldearegai, T.G.; Skinner-Adams, A.P.D.T.; Clarke, M.; Schöler, A.; Limbach, L.; Kurz, T.; et al. Structure–Activity and Structure–Toxicity Relationships of Peptoid-Based Histone Deacetylase Inhibitors with Dual-Stage Antiplasmodial Activity. ChemMedChem 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Zuma, A.A.; de Souza, W. Histone deacetylases as targets for antitrypanosomal drugs. Futur. Sci. OA 2018, 4, FSO325. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Taylor, M.C.; Horn, D.; Loza, E.; Kalvinsh, I.; Björkling, F. Inhibitors of human histone deacetylase with potent activity against the African trypanosome Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2012, 22, 1886–1890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carrillo, A.K.; Guiguemde, W.A.; Guy, R.K. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorg. Med. Chem. 2015, 23, 5151–5155. [Google Scholar] [CrossRef]
- Loeuillet, C.; Touquet, B.; Guichou, J.F.; Labesse, G.; Sereno, D. A Tiny Change Makes a Big Difference in the Anti-Parasitic Activities of an HDAC Inhibitor. Int. J. Mol. Sci. 2019, 20, 2973. [Google Scholar] [CrossRef]
- Araujo-Silva, C.A.; De Souza, W.; Martins-Duarte, E.S.; Vommaro, R.C. HDAC inhibitors Tubastatin A and SAHA affect parasite cell division and are potential anti-Toxoplasma gondii chemotherapeutics. Int. J. Parasitol. Drugs Drug Resist. 2021, 15, 25–35. [Google Scholar] [CrossRef]
- Corpas-López, V.; Díaz-Gavilán, M.; Franco-Montalbán, F.; Merino-Espinosa, G.; López-Viota, M.; López-Viota, J.; Belmonte-Reche, E.; Palacio, J.P.-D.; de Pedro, N.; Gómez-Vidal, J.A.; et al. A nanodelivered Vorinostat derivative is a promising oral compound for the treatment of visceral leishmaniasis. Pharmacol. Res. 2019, 139, 375–383. [Google Scholar] [CrossRef]
- Vaca, H.R.; Celentano, A.M.; Macchiaroli, N.; Kamenetzky, L.; Camicia, F.; Rosenzvit, M.C. Histone deacetylase enzymes as potential drug targets of Neglected Tropical Diseases caused by cestodes. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 120–132. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.Â.; Silva, E.B.M.; Agripino, J.D.M.; Onofre, T.S.; Calla, L.F.A.; Heimburg, T.; Ghazy, E.; Bayer, T.; da Silva, V.H.F.; Ribeiro, P.D.; et al. Histone deacetylases inhibitors as new potential drugs against Leishmania braziliensis, the main causative agent of new world tegumentary leishmaniasis. Biochem. Pharmacol. 2020, 180, 114191. [Google Scholar] [CrossRef]
- Vaca, H.R.; Celentano, A.M.; Toscanini, M.A.; Heimburg, T.; Ghazy, E.; Zeyen, P.; Hauser, A.-T.; Oliveira, G.; Elissondo, M.C.; Jung, M.; et al. The potential for histone deacetylase (HDAC) inhibitors as cestocidal drugs. PLoS Negl. Trop. Dis. 2021, 15, e0009226. [Google Scholar] [CrossRef]
- World Health Organization. Schistosomiasis. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 19 July 2021).
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gärtner, F.; Costa, J.M.C.d. Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob. Agents Chemother. 2017, 61, e02582-16. [Google Scholar] [CrossRef]
- Anderson, L.; Gomes, M.R.; DaSilva, L.F.; Pereira, A.D.S.A.; Mourão, M.M.; Romier, C.; Pierce, R.; Verjovski-Almeida, S. Histone deacetylase inhibition modulates histone acetylation at gene promoter regions and affects genome-wide gene transcription in Schistosoma mansoni. PLoS Negl. Trop. Dis. 2017, 11, e0005539. [Google Scholar] [CrossRef] [PubMed]
- Scholte, L.L.; Mourão, M.M.; Pais, F.; Melesina, J.; Robaa, D.; Volpini, A.C.; Sippl, W.; Pierce, R.J.; Oliveira, G.; Nahum, L.A. Evolutionary relationships among protein lysine deacetylases of parasites causing neglected diseases. Infect. Genet. Evol. 2017, 53, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Lancelot, J.; Caby, S.; Dubois-Abdesselem, F.; Vanderstraete, M.; Trolet, J.; Oliveira, G.; Bracher, F.; Jung, M.; Pierce, R.J. Schistosoma mansoni Sirtuins: Characterization and Potential as Chemotherapeutic Targets. PLoS Negl. Trop. Dis. 2013, 7, e2428. [Google Scholar] [CrossRef]
- Oger, F.; Dubois, F.; Caby, S.; Noël, C.; Cornette, J.; Bertin, B.; Capron, M.; Pierce, R.J. The class I histone deacetylases of the platyhelminth parasite Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2008, 377, 1079–1084. [Google Scholar] [CrossRef]
- Pierce, R. Targeting Schistosome Histone Modifying Enzymes for Drug Development. Curr. Pharm. Des. 2012, 18, 3567–3578. [Google Scholar] [CrossRef]
- Marek, M.; Kannan, S.; Hauser, A.-T.; Mourão, M.M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Morales, E.R.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef][Green Version]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef]
- Chua, M.J.; Arnold, M.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.; Skinner-Adams, T.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Melesina, J.; Hauser, A.-T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C.; et al. Discovery of Inhibitors of Schistosoma mansoni HDAC8 by Combining Homology Modeling, Virtual Screening, and in Vitro Validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.; Chakrabarti, A.; Lancelot, J.; Shaik, T.B.; Hausmann, K.; Melesina, J.; Schmidtkunz, K.; Marek, M.; Erdmann, F.; Schmidt, M.; et al. Synthesis, Crystallization Studies, and in vitro Characterization of Cinnamic Acid Derivatives as Sm HDAC8 Inhibitors for the Treatment of Schistosomiasis. ChemMedChem 2018, 13, 1517–1529. [Google Scholar] [CrossRef]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.-T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef]
- Panic, G.; Flores, D.; Ingram-Sieber, K.; Keiser, J. Fluorescence/luminescence-based markers for the assessment of Schistosoma mansoni schistosomula drug assays. Parasites Vectors 2015, 8, 1–12. [Google Scholar] [CrossRef]
- Ghazy, E.; Heimburg, T.; Lancelot, J.; Zeyen, P.; Schmidtkunz, K.; Truhn, A.; Darwish, S.; Simoben, C.V.; Shaik, T.B.; Erdmann, F.; et al. Synthesis, structure-activity relationships, cocrystallization and cellular characterization of novel smHDAC8 inhibitors for the treatment of schistosomiasis. Eur. J. Med. Chem. 2021, 225, 113745. [Google Scholar] [CrossRef]
- Simoben, C.; Ghazy, E.; Zeyen, P.; Darwish, S.; Schmidt, M.; Romier, C.; Robaa, D.; Sippl, W. Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors. Molecules 2021, 26, 2584. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, K.; Chakrabarti, A.; Melesina, J.; Hansen, F.K.; Lancelot, J.; Herkenhöhner, S.; Lungerich, B.; Marek, M.; Romier, C.; Pierce, R.J.; et al. Isophthalic Acid-Based HDAC Inhibitors as Potent Inhibitors of HDAC8 fromSchistosoma mansoni. Arch. Der. Pharm. 2017, 350, 1700096. [Google Scholar] [CrossRef] [PubMed]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone Deacetylase (HDAC) Inhibitors with a Novel Connecting Unit Linker Region Reveal a Selectivity Profile for HDAC4 and HDAC5 with Improved Activity against Chemoresistant Cancer Cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Saccoccia, F.; Brindisi, M.; Gimmelli, R.; Relitti, N.; Guidi, A.; Saraswati, A.P.; Cavella, C.; Brogi, S.; Chemi, G.; Butini, S.; et al. Screening and Phenotypical Characterization of Schistosoma mansoni Histone Deacetylase 8 (SmHDAC8) Inhibitors as Multistage Antischistosomal Agents. ACS Infect. Dis. 2020, 6, 100–113. [Google Scholar] [CrossRef]
- Kalinin, D.V.; Jana, S.K.; Pfafenrot, M.; Chakrabarti, A.; Melesina, J.; Shaik, T.B.; Lancelot, J.; Pierce, R.J.; Sippl, W.; Romier, C.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Triazole-Based smHDAC8 Inhibitors. ChemMedChem 2019, 15, 571–584. [Google Scholar] [CrossRef]
- Stolfa, D.A.; Marek, M.; Lancelot, J.; Hauser, A.-T.; Walter, A.; Leproult, E.; Melesina, J.; Rumpf, T.; Wurtz, J.-M.; Cavarelli, J.; et al. Molecular Basis for the Antiparasitic Activity of a Mercaptoacetamide Derivative That Inhibits Histone Deacetylase 8 (HDAC8) from the Human Pathogen Schistosoma mansoni. J. Mol. Biol. 2014, 426, 3442–3453. [Google Scholar] [CrossRef]
- Ballante, F.; Reddy, D.R.; Zhou, N.J.; Marshall, G.R. Structural insights of SmKDAC8 inhibitors: Targeting Schistosoma epigenetics through a combined structure-based 3D QSAR, in vitro and synthesis strategy. Bioorg. Med. Chem. 2017, 25, 2105–2132. [Google Scholar] [CrossRef]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef]
- Guidi, A.; Saccoccia, F.; Gennari, N.; Gimmelli, R.; Nizi, E.; Lalli, C.; Paonessa, G.; Papoff, G.; Bresciani, A.; Ruberti, G. Identification of novel multi-stage histone deacetylase (HDAC) inhibitors that impair Schistosoma mansoni viability and egg production. Parasites Vectors 2018, 11, 668. [Google Scholar] [CrossRef]
- Caby, S.; Pagliazzo, L.; Lancelot, J.; Saliou, J.-M.; Bertheaume, N.; Pierce, R.J.; Roger, E. Analysis of the interactome of Schistosoma mansoni histone deacetylase 8. PLoS Negl. Trop. Dis. 2017, 11, e0006089. [Google Scholar] [CrossRef]
- Pagliazzo, L.; Caby, S.; Lancelot, J.; Salomé-Desnoulez, S.; Saliou, J.-M.; Heimburg, T.; Chassat, T.; Cailliau, K.; Sippl, W.; Vicogne, J.; et al. Histone deacetylase 8 interacts with the GTPase SmRho1 in Schistosoma mansoni. PLoS Negl. Trop. Dis. 2021, 15, e0009503. [Google Scholar] [CrossRef]
- Monaldi, D.; Rotili, D.; Lancelot, J.; Marek, M.; Wössner, N.; Lucidi, A.; Tomaselli, D.; Ramos-Morales, E.; Romier, C.; Pierce, R.J.; et al. Structure–Reactivity Relationships on Substrates and Inhibitors of the Lysine Deacylase Sirtuin 2 from Schistosoma mansoni (SmSirt2). J. Med. Chem. 2019, 62, 8733–8759. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Marek, M.; Lancelot, J.; Karaman, B.; Almlöf, I.; Schultz, J.; Sippl, W.; Pierce, R.J.; Romier, C.; Jung, M. Fluorescence-Based Screening Assays for the NAD+-Dependent Histone Deacetylase smSirt2 from Schistosoma mansoni. J. Biomol. Screen. 2014, 20, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Ramos-Morales, E.; Picchi-Constante, G.F.; Bayer, T.; Norström, C.; Herp, D.; Sales-Junior, P.A.; Guerra-Slompo, E.P.; Hausmann, K.; Chakrabarti, A.; et al. Species-selective targeting of pathogens revealed by the atypical structure and active site of Trypanosoma cruzi histone deacetylase DAC2. Cell Rep. 2021, 37. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
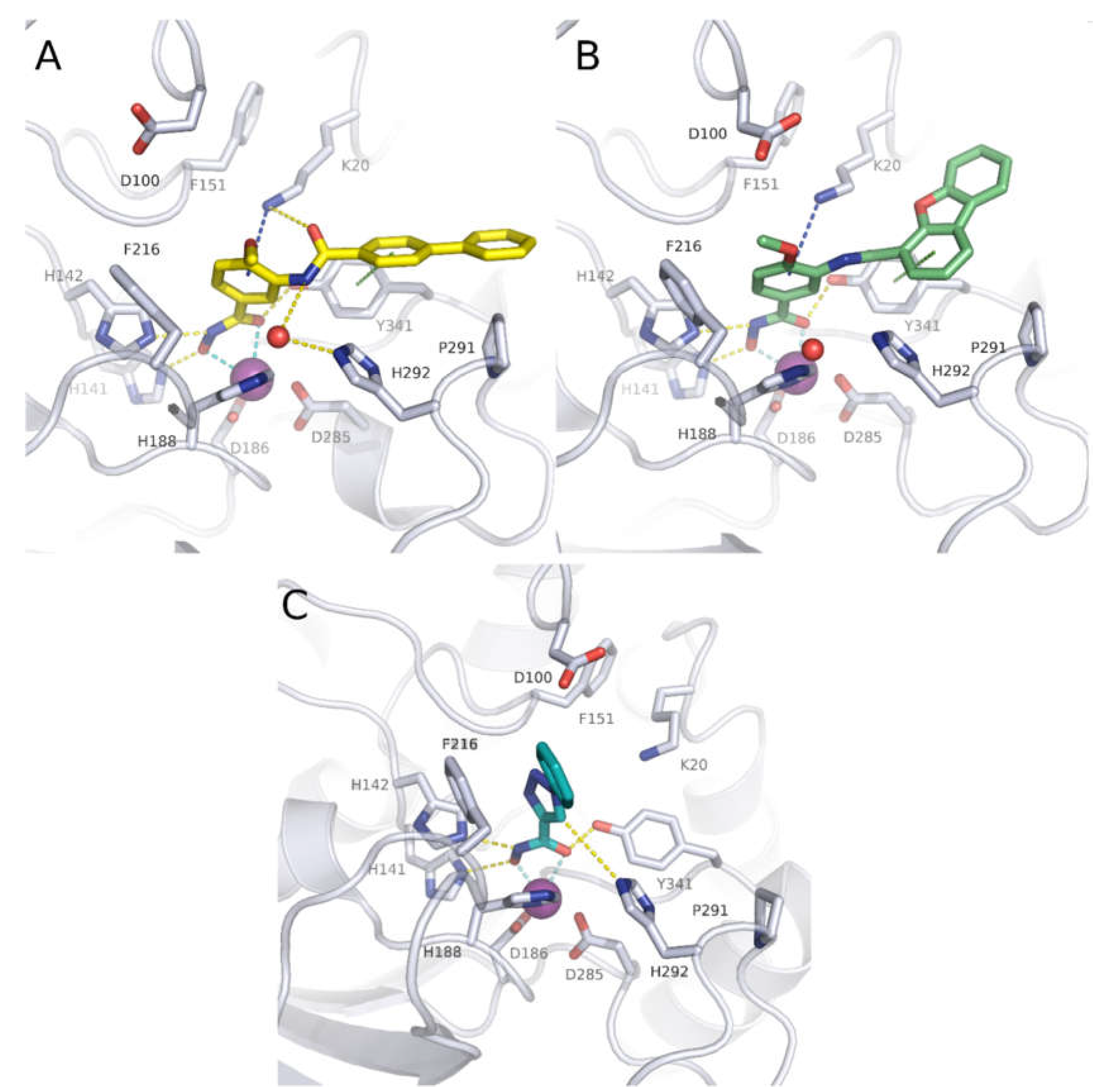
| Cpd | PDB ID | Zn2+-chelation * | H-bond triad ** | F216 (π-π) | F151 (π-π) | K20 (cation-π) | K20 (H-bond) | H292 | Y341 (π-π) | P291 (vdW) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 (J1038) | 4BZ8 | X | X | X | X (H-bond) | |||||
| 2 (J1075) | 4BZ9 | X | X | X | ||||||
| 3 | 6GXW | X | X | X | X (vdW) | X | ||||
| 4 | 6GXU | X | X | X | X (π-π) | |||||
| 5 (TH31) | 5FUE | X | X | X | X | X (H-bond) | X | |||
| 6 (TH65) | 6HTH | X | X | X | X | X (H-bond) | X | X | ||
| 8 | 7P3S | X | X | X | X (π-π) | X | ||||
| 12 | 6TLD | X | X | X (H-bond) | ||||||
| 13 | 6HU3 | X | X | X (vdW) | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghazy, E.; Abdelsalam, M.; Robaa, D.; Pierce, R.J.; Sippl, W. Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals 2022, 15, 80. https://doi.org/10.3390/ph15010080
Ghazy E, Abdelsalam M, Robaa D, Pierce RJ, Sippl W. Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals. 2022; 15(1):80. https://doi.org/10.3390/ph15010080
Chicago/Turabian StyleGhazy, Ehab, Mohamed Abdelsalam, Dina Robaa, Raymond J. Pierce, and Wolfgang Sippl. 2022. "Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis" Pharmaceuticals 15, no. 1: 80. https://doi.org/10.3390/ph15010080
APA StyleGhazy, E., Abdelsalam, M., Robaa, D., Pierce, R. J., & Sippl, W. (2022). Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals, 15(1), 80. https://doi.org/10.3390/ph15010080