
Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases

 ,
, 
 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
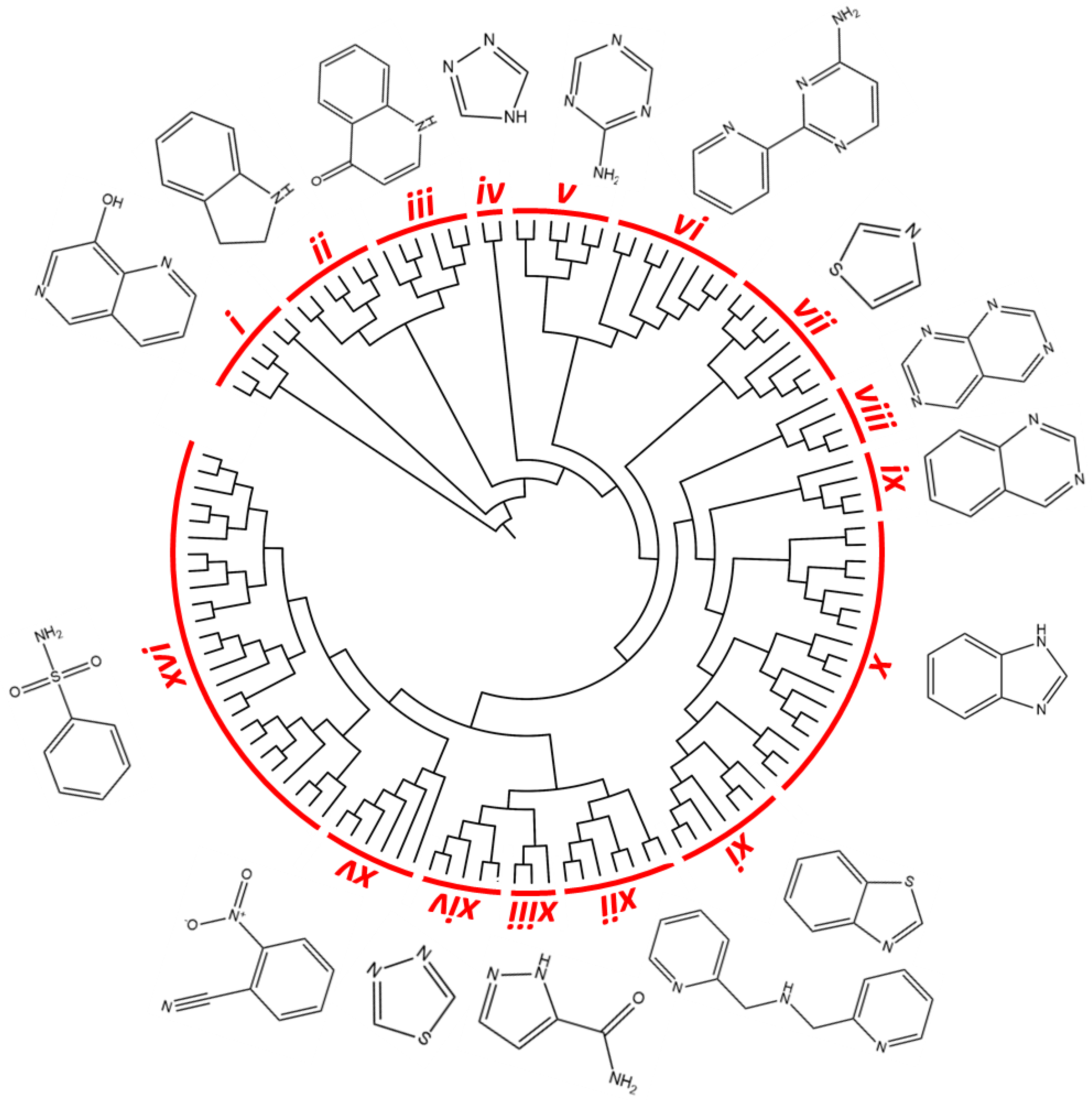
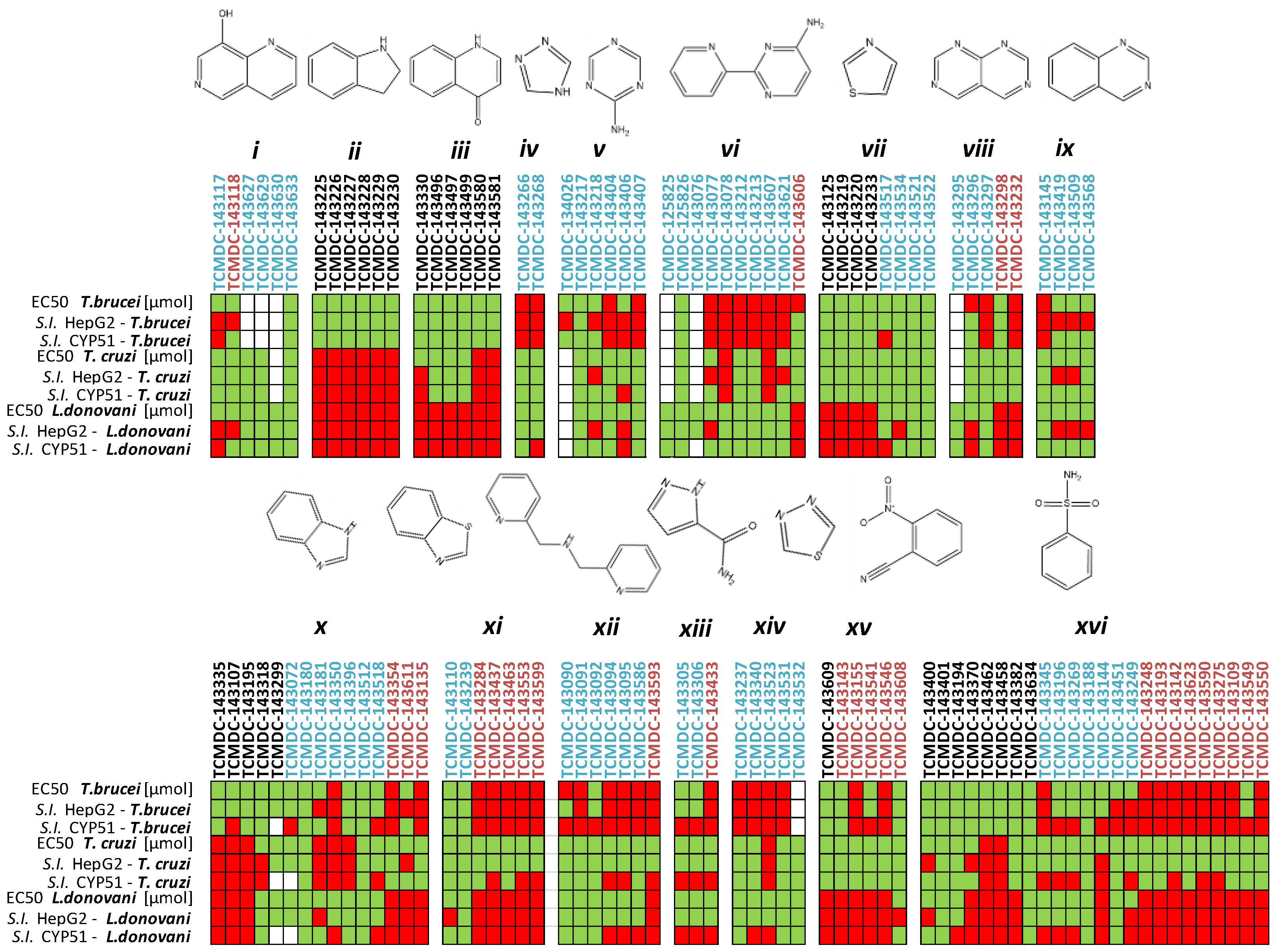
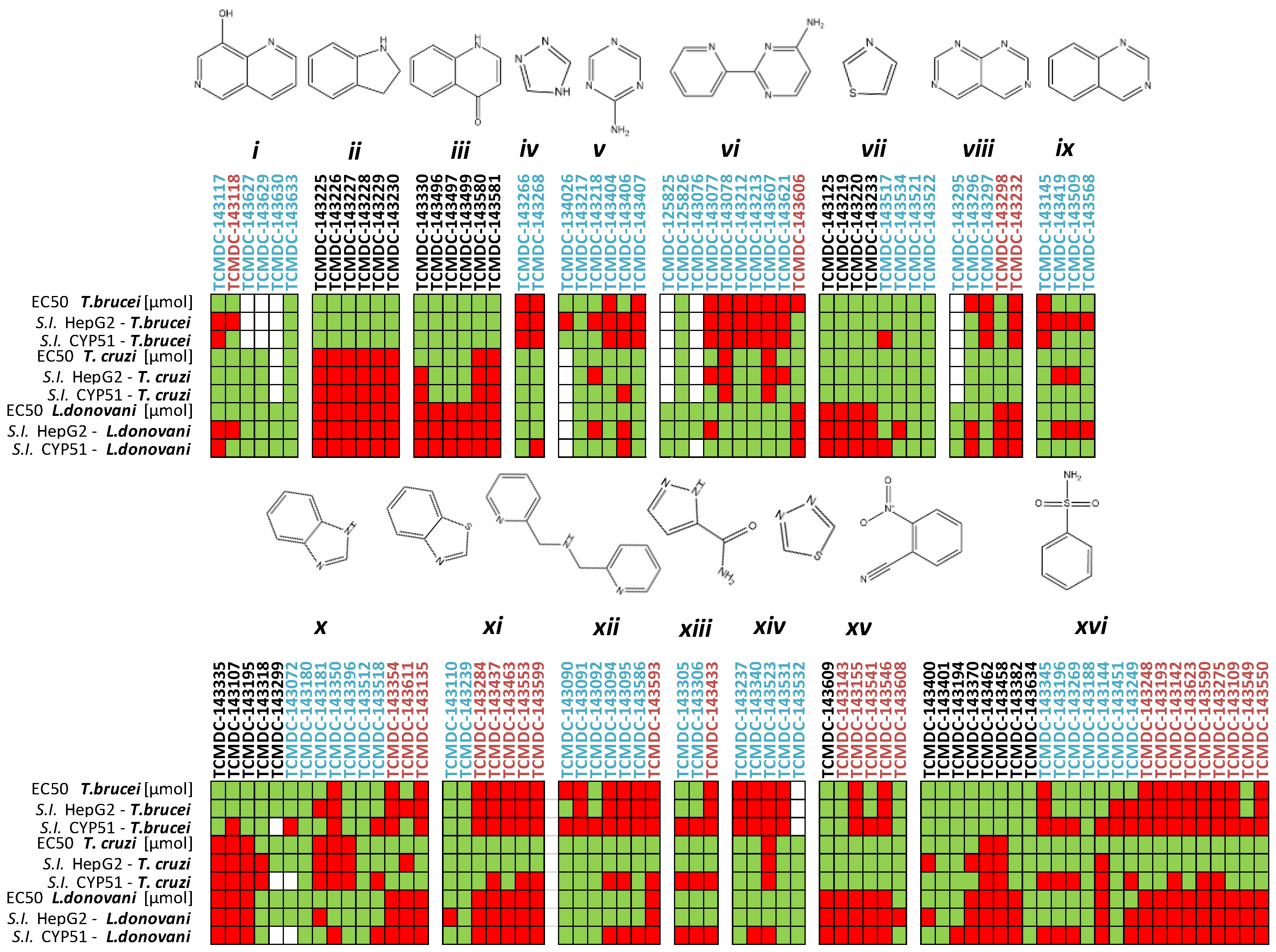
2.1. In Silico Evaluation of Drug-Likeness Properties and Hierarchical Clustering Analysis
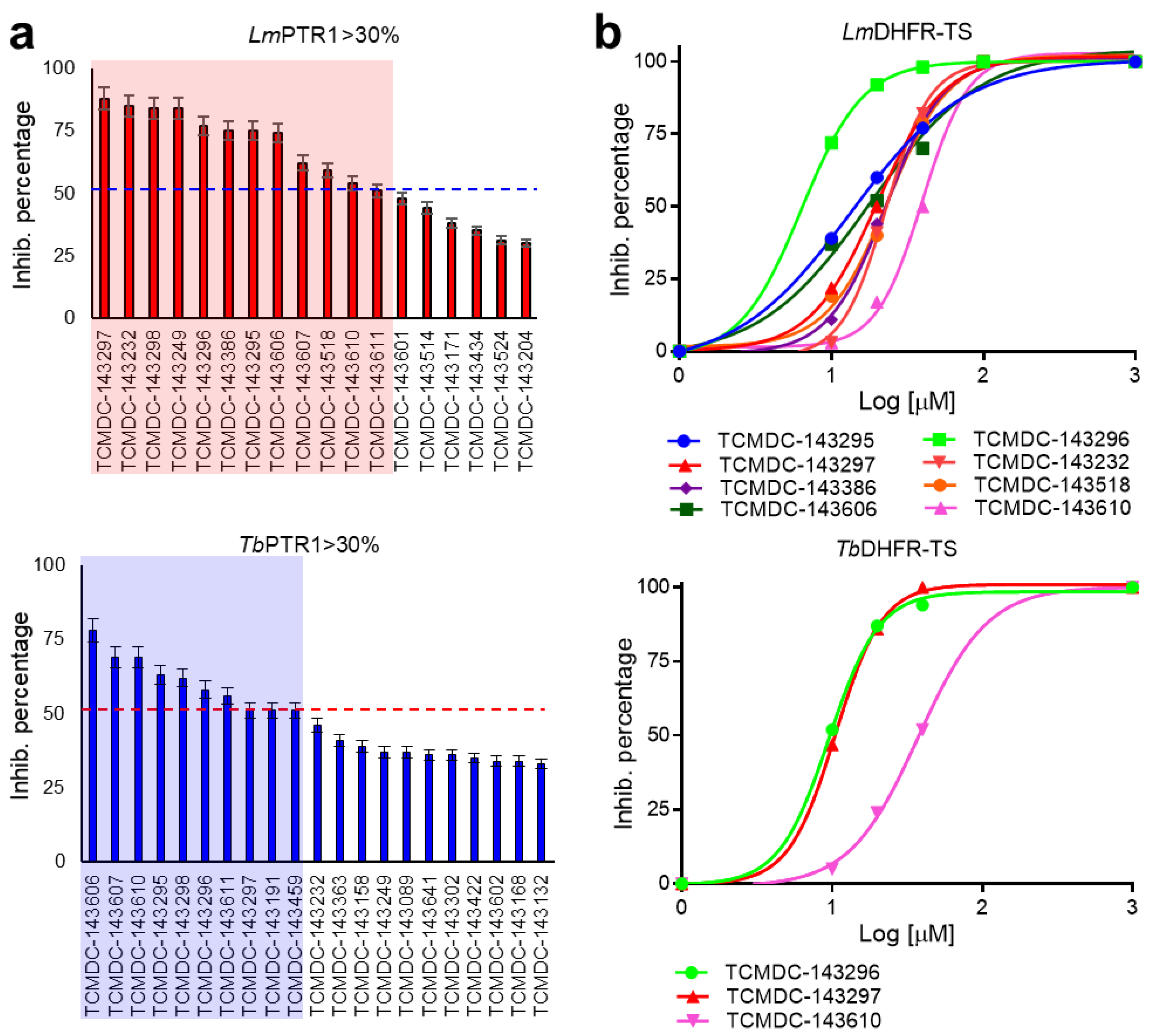
2.2. Inhibition of PTR1s and DHFRs
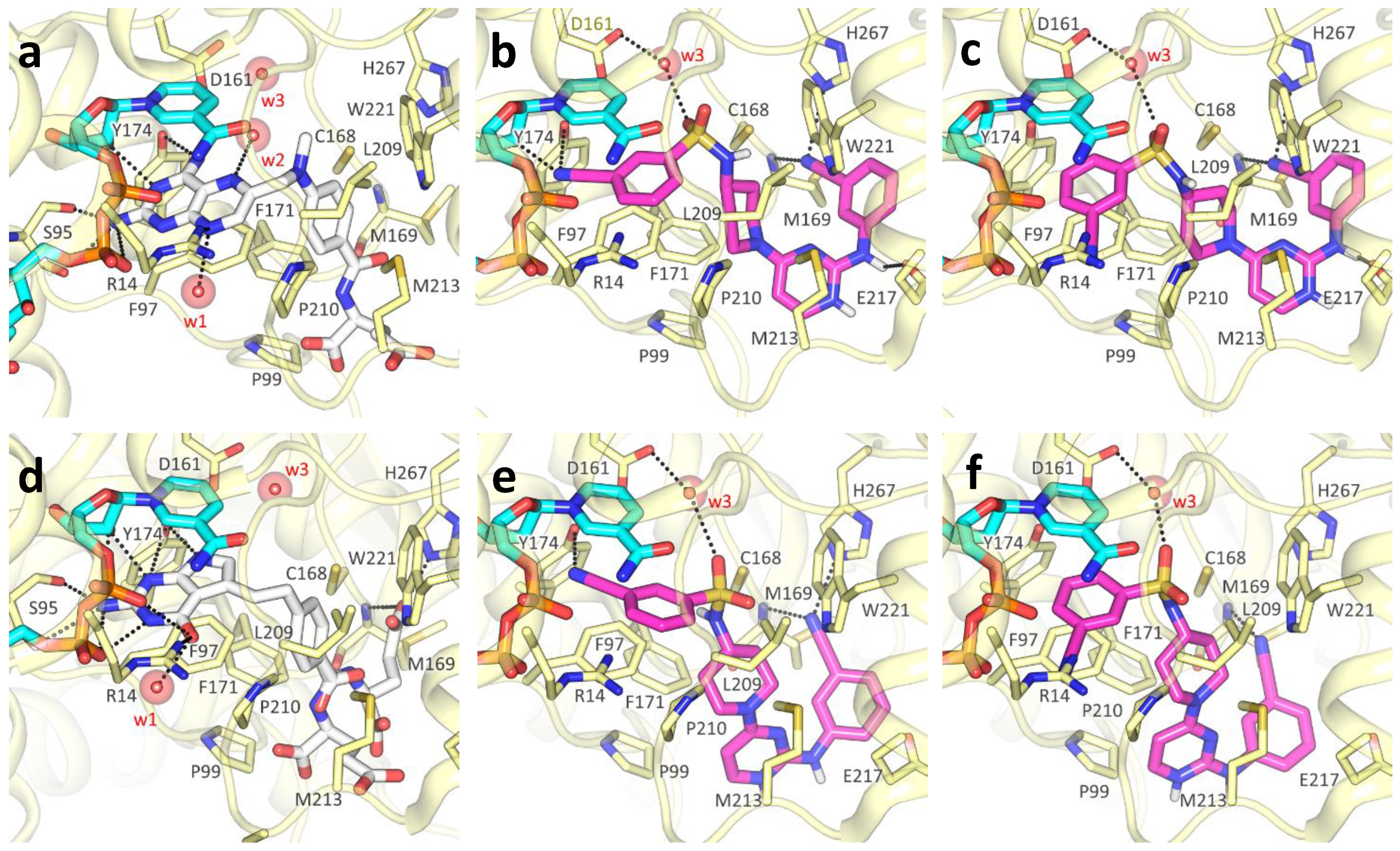
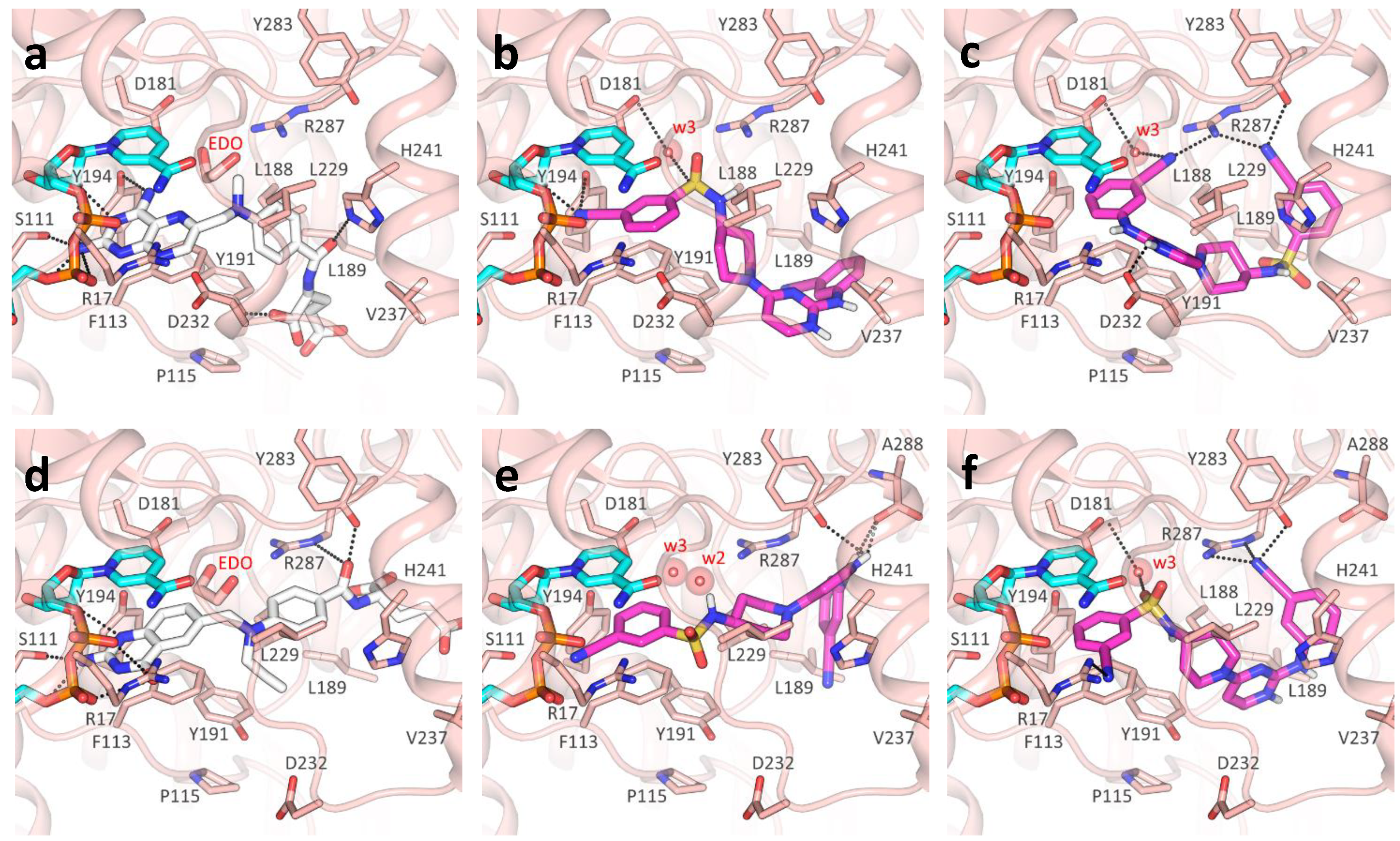
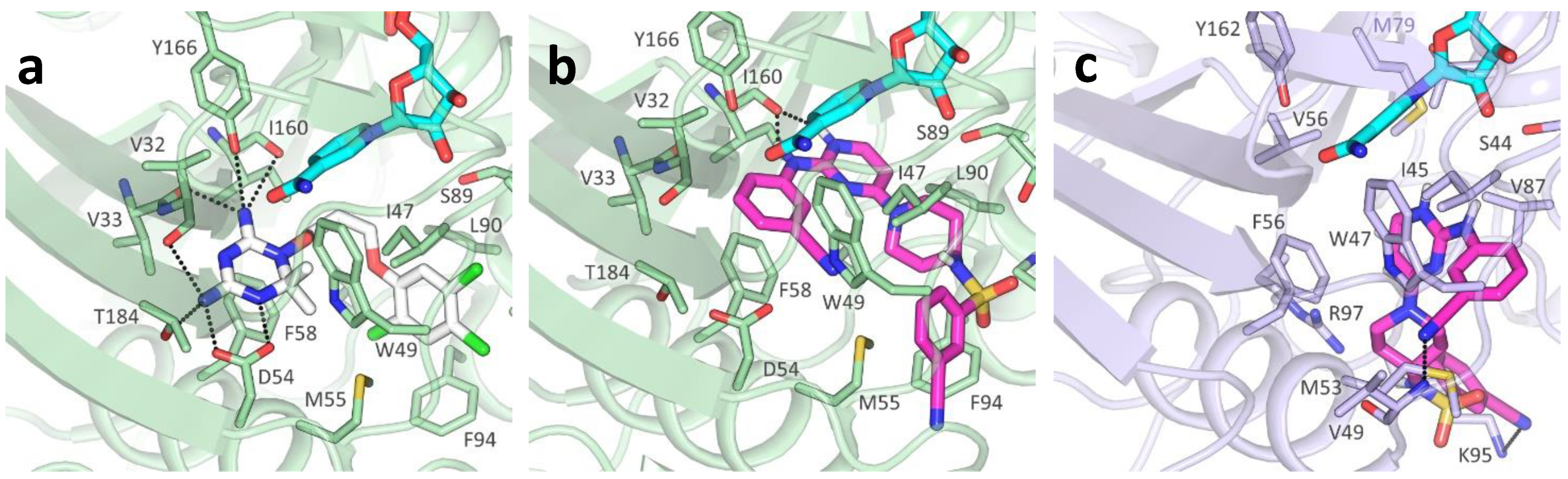
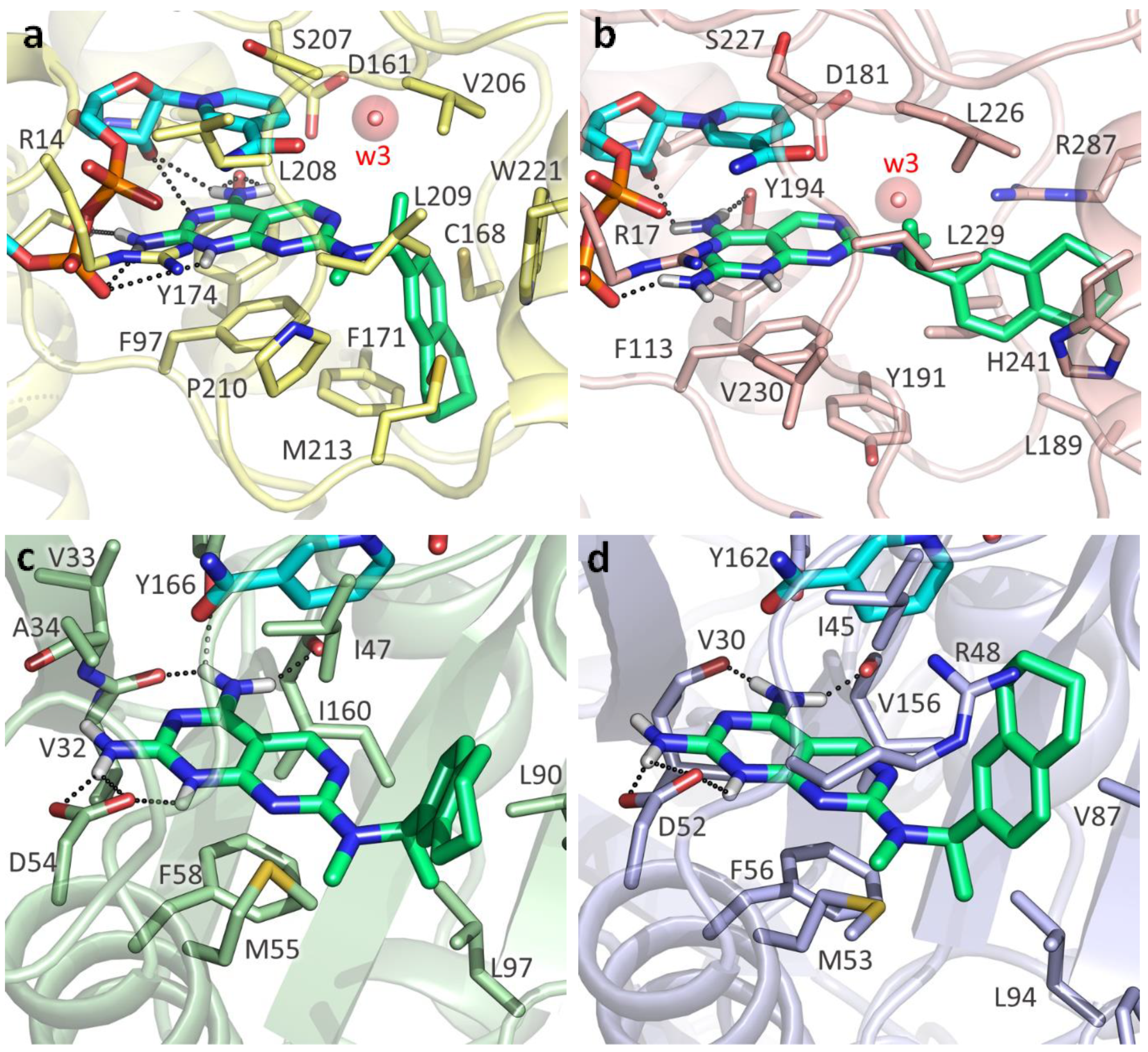
2.3. Molecular Docking
3. Materials and Methods
3.1. Reagents
3.2. In Silico Chemoinformatic and Clustering Analysis
3.3. Protein Purification
3.4. Anti-Kinetoplastid Chemical Boxes
3.4.1. Primary Screening
3.4.2. Secondary Screening (Dose–Response Curve)
3.5. Molecular Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engels, D.; Zhou, X.-N. Neglected tropical diseases: An effective global response to local poverty-related disease priorities. Infect. Dis. Poverty 2020, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, K.; Honek, J.; Cadmus, C.C.; Efferth, T. Trypanosomatid Parasites Causing Neglected Diseases. Curr. Med. Chem. 2010, 17, 1594–1617. [Google Scholar] [CrossRef] [PubMed]
- Legros, D.; Ollivier, G.; Gastellu-Etchegorry, M.; Paquet, C.; Burri, C.; Jannin, J.; Büscher, P. Treatment of human African trypanosomia-sis—Present situation and needs for research and development. Lancet Infect. Dis. 2002, 2, 437–440. [Google Scholar] [CrossRef]
- Trouiller, P.; Olliaro, P.; Torreele, E.; Orbinski, J.; Laing, R.; Ford, N. Drug development for neglected diseases: A deficient market and a public-health policy failure. Lancet 2002, 359, 2188–2194. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G.E. The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol. 2008, 64, 116–126. [Google Scholar] [CrossRef]
- Santos, S.S.; de Araújo, R.V.; Giarolla, J.; Seoud, O.E.; Ferreira, E.I. Searching for drugs for Chagas disease, leishmaniasis and schisto-somiasis: A review. Int. J. Antimicrob. Agents 2020, 55, 105906. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; de Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, J.A.; deMecca, M.M.; Bartel, L.C. Toxic Side Effects of Drugs Used to Treat Chagas’ Disease (American Trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Kedzierski, L.; Sakthianandeswaren, A.; Curtis, J.; Andrews, P.; Junk, P.; Kedzierska, K. Leishmaniasis: Current Treatment and Prospects for New Drugs and Vaccines. Curr. Med. Chem. 2009, 16, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Panecka-Hofman, J.; Pöhner, I.; Spyrakis, F.; Zeppelin, T.; Di Pisa, F.; Dello Iacono, L.; Bonucci, A.; Quotadamo, A.; Venturelli, A.; Mangani, S.; et al. Comparative mapping of on-targets and off-targets for the discovery of anti-trypanosomatid folate pathway inhibitors. Biochim. Biophys. Acta BBA—Gen. Subj. 2017, 1861, 3215–3230. [Google Scholar] [CrossRef]
- Di Pisa, F.; Landi, G.; Dello Iacono, L.; Pozzi, C.; Borsari, C.; Ferrari, S.; Santucci, M.; Santarem, N.; Cordeiro-da-Silva, A.; Moraes, C.B.; et al. Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules 2017, 22, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linciano, P.; Cullia, G.; Borsari, C.; Santucci, M.; Ferrari, S.; Witt, G.; Gul, S.; Kuzikov, M.; Ellinger, B.; Santarém, N.; et al. Identification of a 2,4-diaminopyrimidine scaffold targeting Trypanosoma brucei pteridine reductase 1 from the LIBRA compound library screening campaign. Eur. J. Med. Chem. 2020, 189, 112047. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef]
- Ubeda, J.-M.; Légaré, D.; Raymond, F.; Ouameur, A.A.; Boisvert, S.; Rigault, P.; Corbeil, J.; Tremblay, M.J.; Olivier, M.; Papadopoulou, B.; et al. Modulation of gene expression in drug resistant Leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol. 2008, 9, R115. [Google Scholar] [CrossRef] [Green Version]
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice: Essentiality of pteridine reductase 1. Mol. Microbiol. 2010, 77, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Landi, G.; Linciano, P.; Borsari, C.; Bertolacini, C.P.; Moraes, C.B.; Cordeiro-da-Silva, A.; Gul, S.; Witt, G.; Kuzikov, M.; Costi, M.P.; et al. Structural Insights into the Development of Cycloguanil Derivatives as Trypanosoma brucei Pteridine-Reductase-1 Inhibitors. ACS Infect. Dis. 2019, 5, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Tassone, G.; Landi, G.; Linciano, P.; Francesconi, V.; Tonelli, M.; Tagliazucchi, L.; Costi, M.; Mangani, S.; Pozzi, C. Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase. Pharmaceuticals 2021, 14, 636. [Google Scholar] [CrossRef] [PubMed]
- Landi, G.; Linciano, P.; Tassone, G.; Costi, M.P.; Mangani, S.; Pozzi, C. High-resolution crystal structure of Trypanosoma brucei pteridine reductase 1 in complex with an innovative tricyclic-based inhibitor. Acta Crystallogr. Sect. Struct. Biol. 2020, 76, 558–564. [Google Scholar] [CrossRef]
- Linciano, P.; Dawson, A.; Pöhner, I.; Costa, D.M.; Sá, M.S.; Cordeiro-Da-Silva, A.; Luciani, R.; Gul, S.; Witt, G.; Ellinger, B.; et al. Exploiting the 2-Amino-1,3,4-thiadiazole Scaffold to Inhibit Trypanosoma brucei Pteridine Reductase in Support of Early-Stage Drug Discovery. ACS Omega 2017, 2, 5666–5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, I.; Pilar Manzano, M.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New Compound Sets Identified from High Throughput Phenotypic Screening Against Three Kinetoplastid Parasites: An Open Resource. Sci. Rep. 2015, 5, srep08771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipandula, W.; Young, S.A.; MacNeill, S.A.; Smith, T.K. Screening of the MMV and GSK open access chemical boxes using a viability assay developed against the kinetoplastid Crithidia fasciculata. Mol. Biochem. Parasitol. 2018, 222, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez de Iturrate, P.; Sebastián-Pérez, V.; Nácher-Vázquez, M.; Tremper, C.S.; Smirlis, D.; Martín, J.; Campillo, N.E.; Rivas, L.; Gil, C. Towards discovery of new leishmanicidal scaffolds able to inhibit Leishmania GSK-3. J. Enzyme Inhib. Med. Chem. 2020, 35, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salas-Sarduy, E.; Landaburu, L.U.; Karpiak, J.; Madauss, K.P.; Cazzulo, J.J.; Agüero, F.; Alvarez, V.E. Novel scaffolds for inhibition of Cruzipain identified from high-throughput screening of anti-kinetoplastid chemical boxes. Sci. Rep. 2017, 7, 12073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, G.F.; Pritchard, K. In Vitro Screening for Drug Repositioning. J. Biomol. Screen. 2015, 20, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Elisi, G.; Santucci, M.; D’Arca, D.; Lauriola, A.; Marverti, G.; Losi, L.; Scalvini, L.; Bolognesi, M.L.; Mor, M.; Costi, M.P. Repurposing of Drugs Targeting YAP-TEAD Functions. Cancers 2018, 10, 329. [Google Scholar] [CrossRef] [Green Version]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Dallanoce, C.; Bazza, P.; Grazioso, G.; De Amici, M.; Gotti, C.; Riganti, L.; Clementi, F.; de Micheli, C. Synthesis of Epibatidine-Related Δ2-Isoxazoline Derivatives and Evaluation of Their Binding Affinity at Neuronal Nicotinic Acetylcholine Receptors. Eur. J. Org. Chem. 2006, 2006, 3746–3754. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, Synthesis, and Biological Evaluation of Conformationally Restricted Rivastigmine Analogues. J. Med. Chem. 2004, 47, 5945–5952. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-Based Design of Pteridine Reductase Inhibitors Targeting African Sleeping Sickness and the Leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, Y.; Albay, C.; Arslan, T.; Ece, A.; Turkoglu, E.A.; Efe, A.; Senturk, M.; Supuran, C.T.; Ekinci, D. Design, synthesis and molecular modelling studies of some pyrazole derivatives as carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Voicu, A.; Duteanu, N.; Voicu, M.; Vlad, D.; Dumitrascu, V. The rcdk and cluster R packages applied to drug candidate selection. J. Cheminform. 2020, 12, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Luciani, R.; Pozzi, C.; Poehner, I.; Henrich, S.; Trande, M.; Cordeiro-Da-Silva, A.; Santarem, N.; Baptista, C.; Tait, A.; et al. Profiling of Flavonol Derivatives for the Development of Antitrypanosomatidic Drugs. J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Shanks, E.J.; Ong, H.B.; Robinson, D.A.; Thompson, S.; Sienkiewicz, N.; Fairlamb, A.H.; Frearson, J.A. Development and validation of a cytochrome c-coupled assay for pteridine reductase 1 and dihydrofolate reductase. Anal. Biochem. 2010, 396, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Dawson, A.; Gibellini, F.; Sienkiewicz, N.; Tulloch, L.B.; Fyfe, P.K.; McLuskey, K.; Fairlamb, A.H.; Hunter, W.N. Structure and reactivity of Trypanosoma brucei pteridine reductase: Inhibition by the archetypal antifolate methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, M.; Naesens, L.; Gazzarrini, S.; Santucci, M.; Cichero, E.; Tasso, B.; Moroni, A.; Costi, M.P.; Loddo, R. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur. J. Med. Chem. 2017, 135, 467–478. [Google Scholar] [CrossRef]
- Francesconi, V.; Giovannini, L.; Santucci, M.; Cichero, E.; Costi, M.P.; Naesens, L.; Giordanetto, F.; Tonelli, M. Synthesis, biological evaluation and molecular modeling of novel azaspiro dihydrotriazines as influenza virus inhibitors targeting the host factor dihydrofolate reductase (DHFR). Eur. J. Med. Chem. 2018, 155, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.W.; Dewar, S.; Ong, H.B.; Sienkiewicz, N.; Fairlamb, A.H. Trypanosoma brucei DHFR-TS Revisited: Characterisation of a Bifunctional and Highly Unstable Recombinant Dihydrofolate Reductase-Thymidylate Synthase. PLoS Negl. Trop. Dis. 2016, 10, e0004714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.; Morandi, F.; Motiejunas, D.; Nerini, E.; Henrich, S.; Luciani, R.; Venturelli, A.; Lazzari, S.; Calo, S.; Gupta, S.; et al. Virtual Screening Identification of Nonfolate Compounds, Including a CNS Drug, as Antiparasitic Agents Inhibiting Pteridine Reductase. J. Med. Chem. 2011, 54, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Spyrakis, F.; Cross, S.; Quotadamo, A.; Farina, D.; Tondi, D.; de Luca, F.; Docquier, J.-D.; Prieto, A.I.; Ibacache, C.; et al. Computational and biological profile of boronic acids for the detection of bacterial serine- and metallo-β-lactamases. Sci. Rep. 2017, 7, 17716. [Google Scholar] [CrossRef] [Green Version]
- Czerwonka, A.; Lemieszek, M.K.; Karpińska, M.; Matysiak, J.; Niewiadomy, A.; Rzeski, W. Evaluation of the effect of 2-(2,4-dihydroxyphenyl)-4H-benzofuro[3,2-d][1,3]thiazin-4-one on colon cells and its anticancer potential. Med. Chem. Res. 2018, 27, 2150–2159. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Milletti, F.; Storchi, L.; Sforna, G.; Cross, S.; Cruciani, G. Tautomer Enumeration and Stability Prediction for Virtual Screening on Large Chemical Databases. J. Chem. Inf. Model. 2009, 49, 68–75. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Hashimoto, Y. Improvement in Aqueous Solubility in Small Molecule Drug Discovery Programs by Disruption of Molecular Planarity and Symmetry. J. Med. Chem. 2011, 54, 1539–1554. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.I.; Huggan, J.K.; Suckling, C.J.; Gibson, C.L.; Stewart, K.; Giordani, F.; Barrett, M.P.; Wong, P.E.; Barrack, K.L.; Hunter, W.N. Structure-Based Design and Synthesis of Antiparasitic Pyrrolopyrimidines Targeting Pteridine Reductase 1. J. Med. Chem. 2014, 57, 6479–6494. [Google Scholar] [CrossRef] [Green Version]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major Pteridine Reductase Complexes Reveal the Active Site Features Important for Ligand Binding and to Guide Inhibitor Design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Gibellini, F.; Carvalho, P.; Avery, M.A.; Hunter, W.N. Inhibition of Leishmania major pteridine reductase by 2,4,6-triaminoquinazoline: structure of the NADPH ternary complex. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1780–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal Dihydrofolate Reductase Reveals Natural Antifolate Resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Property | Minimum Value | Maximum Value | Average | Drug-Likeness Criteria | % Compounds According to RO5 * |
|---|---|---|---|---|---|
| MW (g/mol) | 210 | 547 | 375 | ≤500 | 99.2% |
| AlogP | −2.7 | 5.8 | 2.8 | ≤5 | 98.7% |
| HBA | 1 | 8 | 3.8 | ≤10 | 100% |
| HBD | 0 | 5 | 1.3 | ≤5 | 100% |
| Total Polar Surface Area (Å2) | 19 | 184 | 89.5 | ≤140 | 94.3% |
| N° of Rotatable Bonds | 1 | 11 | 5.1 | ≤10 | 99.8% |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Substituents | IC50 (µM) | EC50 (µM) | |||||||
| TCMDC ID | R1 | R2 | HTS_BOX | TbPTR1 | LmPTR1 | TbDHFR | LmDHFR | T. brucei | L. donovani |
| 143232 | H |  | CHAGAS | 10.9 | 5.9 | - | 24.4 | 28.4 | 29.5 |
| 143295 | CH3 | 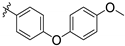 | LEISH | 7.9 | 6.7 | - | 16.6 | N.D. | 0.06 |
| 143296 | CH3 | 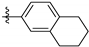 | LEISH | 8.6 | 6.5 | 9.7 | 6.9 | 14.2 | 1.8 |
| 143297 | CH3 | 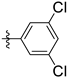 | LEISH | 9.8 | 5.7 | 11.6 | 20.1 | 35.7 | 0.6 |
| 143298 | CH3 | 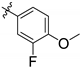 | CHAGAS | 8.1 | 6.0 | - | - | 8.8 | 10.8 |
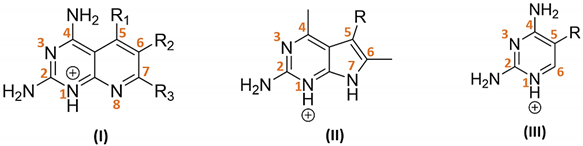 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | ||||||||||
| Substituents | IC50 (µM) | EC50 (µM) | ||||||||
| TCMDC ID | R1 | R2 | R3 | HTS_BOX | TbPTR1 | LmPTR1 | TbDHFR | LmDHFR | T. brucei | L. donovani |
| 143606 (VI) | H | CH2CH3 | (CH2)2CH3 | CHAGAS | 6.4 | 6.8 | - | 19.4 | 22.5 | 27.2 |
| 143607 (VI) | CH3 | H | 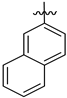 | LEISH | 7.3 | 8.1 | - | - | 25.8 | 2.7 |
| II | ||||||||||
| Substituents | IC50 (µM) | EC50 (µM) | ||||||||
| TCMDC ID | R | HTS_BOX | TbPTR1 | LmPTR1 | TbDHFR | LmDHFR | T. brucei | L. donovani | ||
| 143610 | 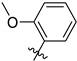 | CHAGAS | 7.3 | 9.3 | 38.2 | 40.0 | 32.5 | 17.9 | ||
| III | ||||||||||
| Substituents | IC50 (µM) | EC50 (µM) | ||||||||
| TCMDC ID | R | HTS_BOX | TbPTR1 | LmPTR1 | TbDHFR | LmDHFR | T. brucei | L. donovani | ||
| 143611 (XI) | 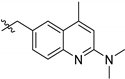 | CHAGAS | 8.9 | 9.8 | - | - | 5.0 | - | ||
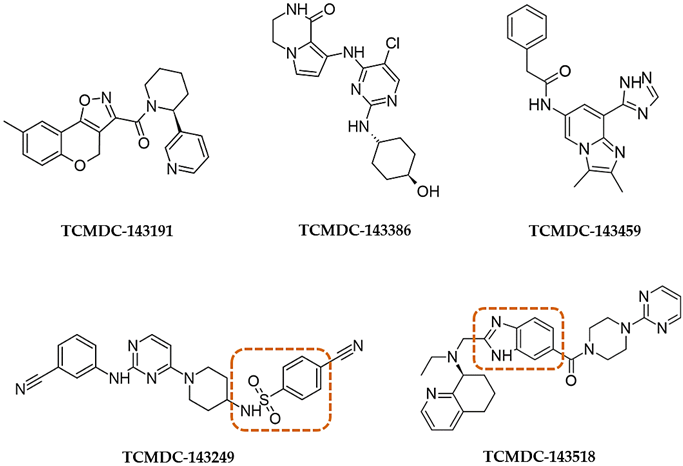 | |||||||
|---|---|---|---|---|---|---|---|
| IC50 (µM) | EC50 (µM) | ||||||
| TCMDC ID | HTS_BOX | TbPTR1 | LmPTR1 | TbDHFR | LmDHFR | T. brucei | L. donovani |
| 143191 | CHAGAS | 9.8 | 38.5 | - | - | 39.8 | - |
| 143249 (XVI) | LEISH | 13.5 | 6.0 | - | - | 6.3 | 5.6 |
| 143518 (X) | LEISH | 33.3 | 8.5 | - | 25 | 3.8 | 3.5 |
| 143386 | HAT | 35.0 | 6.7 | - | 25.8 | 0.6 | 1.4 |
| 143459 | LEISH | 9.8 | - | - | - | 6.6 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santucci, M.; Luciani, R.; Gianquinto, E.; Pozzi, C.; Pisa, F.d.; dello Iacono, L.; Landi, G.; Tagliazucchi, L.; Mangani, S.; Spyrakis, F.; et al. Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases. Pharmaceuticals 2021, 14, 1246. https://doi.org/10.3390/ph14121246
Santucci M, Luciani R, Gianquinto E, Pozzi C, Pisa Fd, dello Iacono L, Landi G, Tagliazucchi L, Mangani S, Spyrakis F, et al. Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases. Pharmaceuticals. 2021; 14(12):1246. https://doi.org/10.3390/ph14121246
Chicago/Turabian StyleSantucci, Matteo, Rosaria Luciani, Eleonora Gianquinto, Cecilia Pozzi, Flavio di Pisa, Lucia dello Iacono, Giacomo Landi, Lorenzo Tagliazucchi, Stefano Mangani, Francesca Spyrakis, and et al. 2021. "Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases" Pharmaceuticals 14, no. 12: 1246. https://doi.org/10.3390/ph14121246
APA StyleSantucci, M., Luciani, R., Gianquinto, E., Pozzi, C., Pisa, F. d., dello Iacono, L., Landi, G., Tagliazucchi, L., Mangani, S., Spyrakis, F., & Costi, M. P. (2021). Repurposing the Trypanosomatidic GSK Kinetobox for the Inhibition of Parasitic Pteridine and Dihydrofolate Reductases. Pharmaceuticals, 14(12), 1246. https://doi.org/10.3390/ph14121246