
Metabolic Soft Spot and Pharmacokinetics: Functionalization of C-3 Position of an Eph–Ephrin Antagonist Featuring a Bile Acid Core as an Effective Strategy to Obtain Oral Bioavailability in Mice

 ,
,  , ,
, ,  ,
,  ,
, 
 and
and
Abstract
:1. Introduction
2. Results
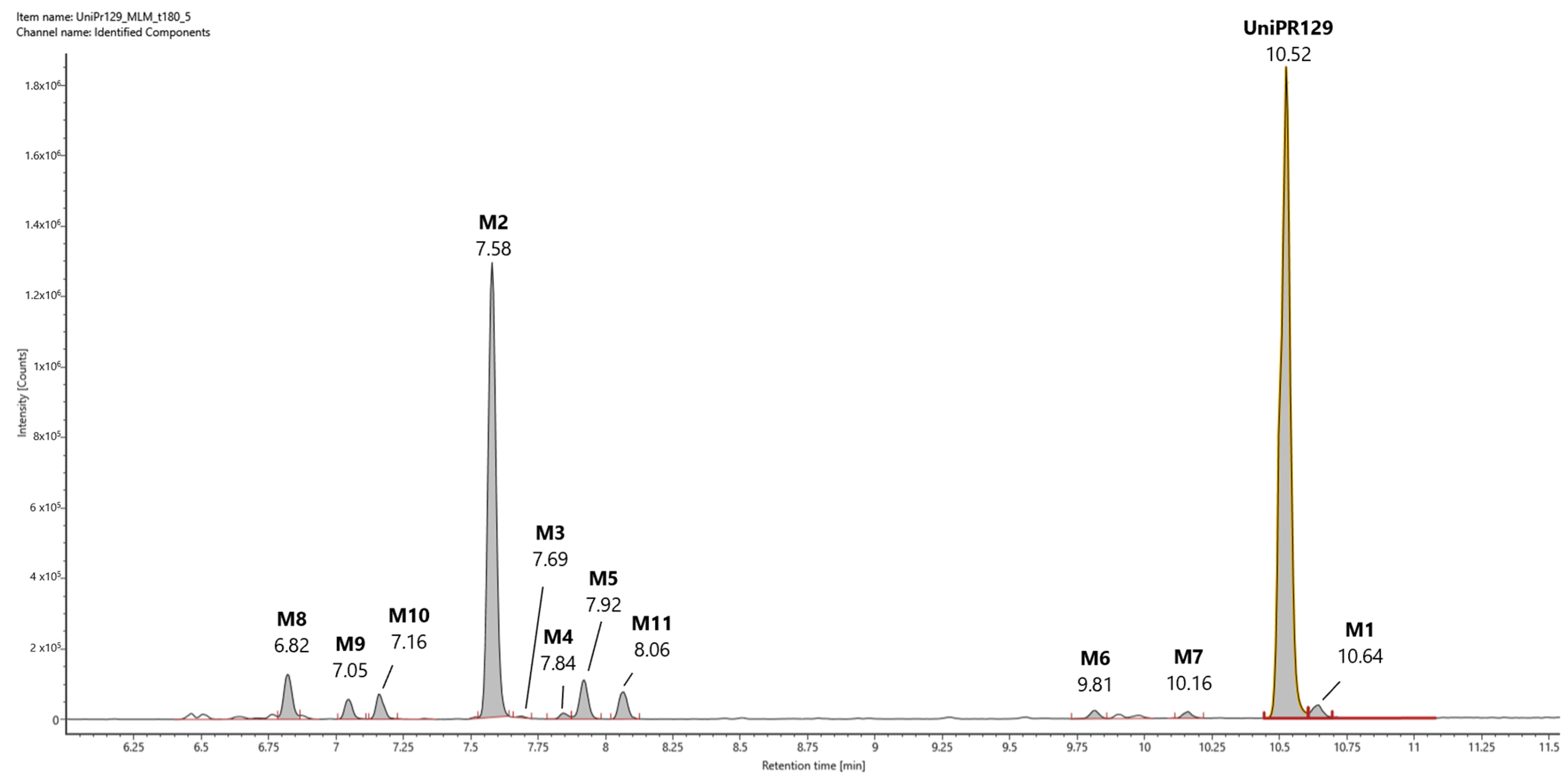
2.1. In Vitro Metabolism of UniPR129
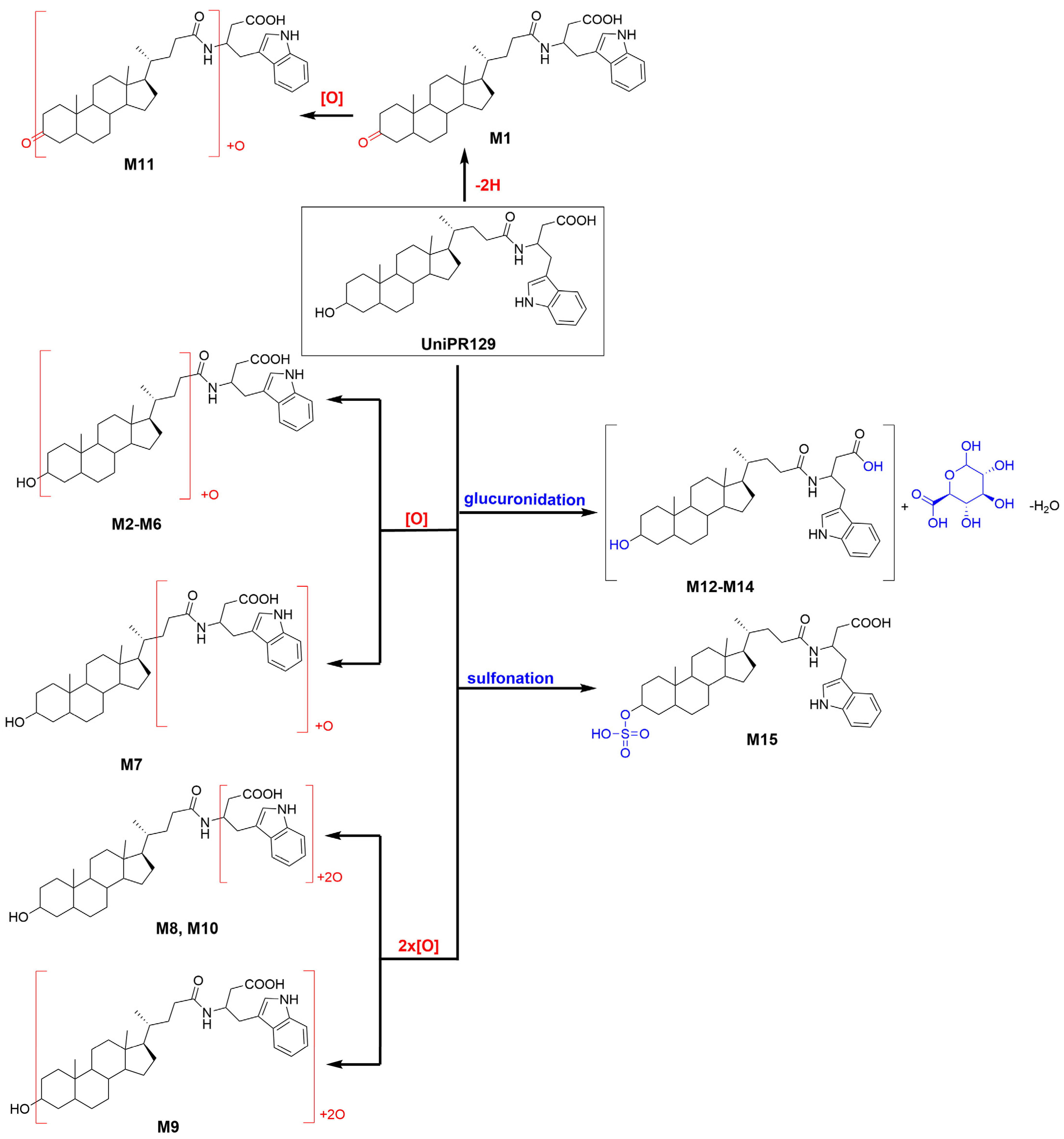
2.2. Profiling of UniPR129 in Vitro Phase I Metabolites
2.2.1. Metabolite M1
2.2.2. Metabolite M2
2.2.3. Metabolites M3-M7
2.2.4. Metabolites M8−M10
2.2.5. Metabolite M11
2.3. Profiling of UniPR129 in Vitro Metabolites: Phase II Metabolism
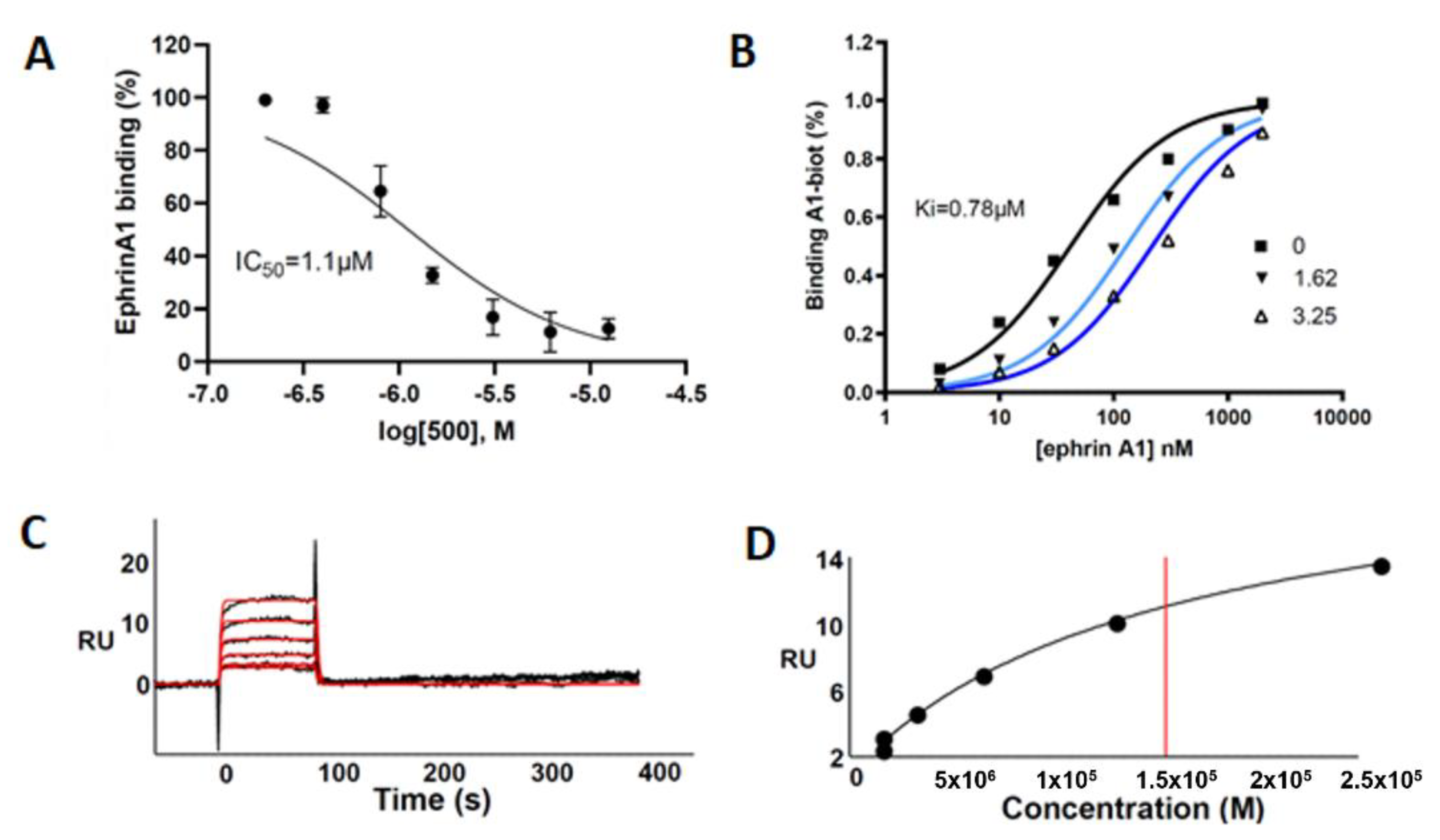
2.4. Design and in Vitro Characterization of UniPR500
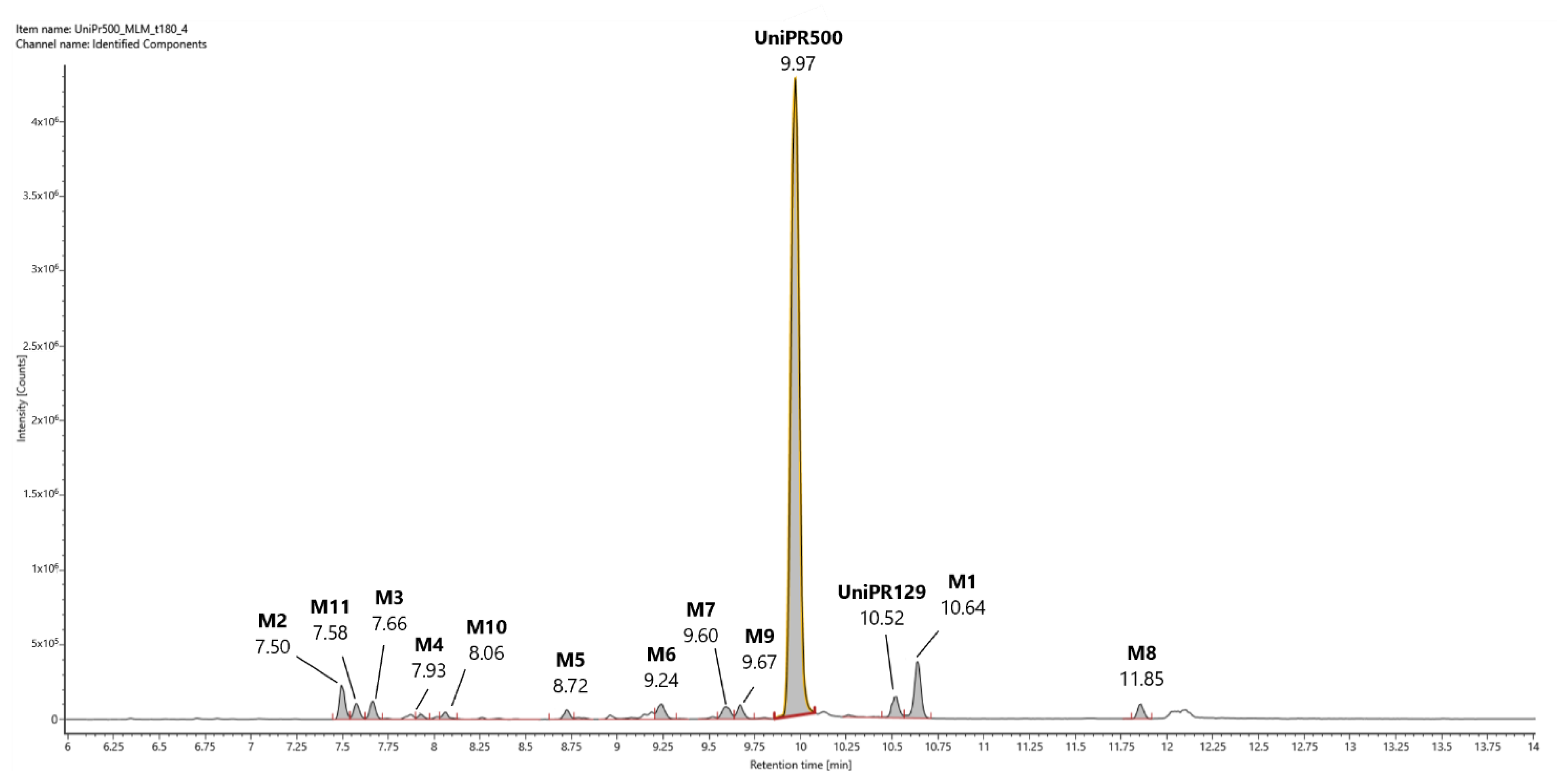
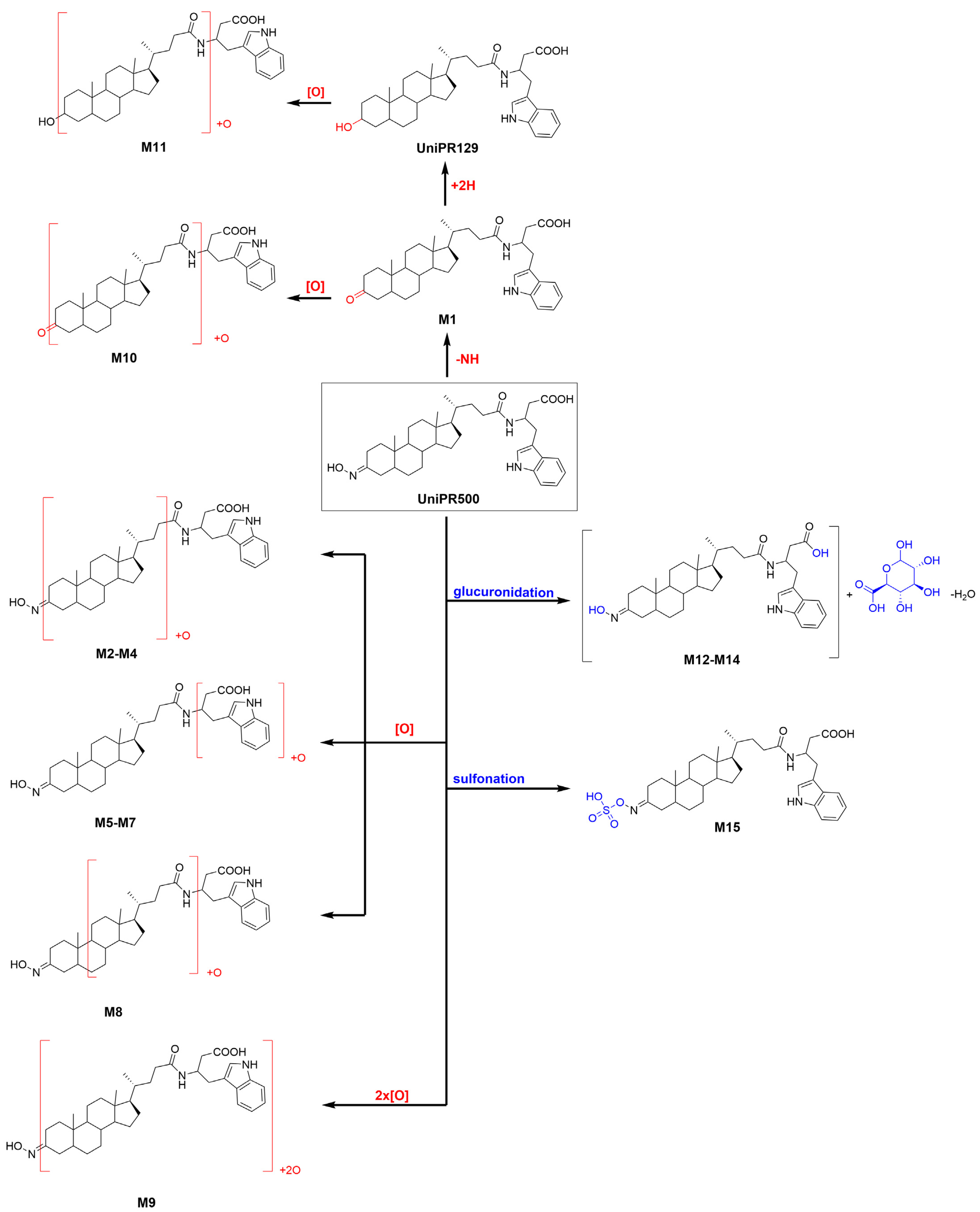
2.5. Profiling of UniPR500 in Vitro Phase I Metabolites in Mouse Liver Microsomes
2.5.1. Metabolites M1 and UniPR129
2.5.2. Mono-Hydroxylated Metabolites M2–M8
2.5.3. Di-Hydroxylated Metabolite M9
2.5.4. Second-Generation Metabolites Derived from M1 and UniPR129
2.6. Profiling of UniPR500 in Vitro Phase II Metabolites in MLM and HLS9 Fraction
2.7. In Vitro Metabolic stability of UniPR500
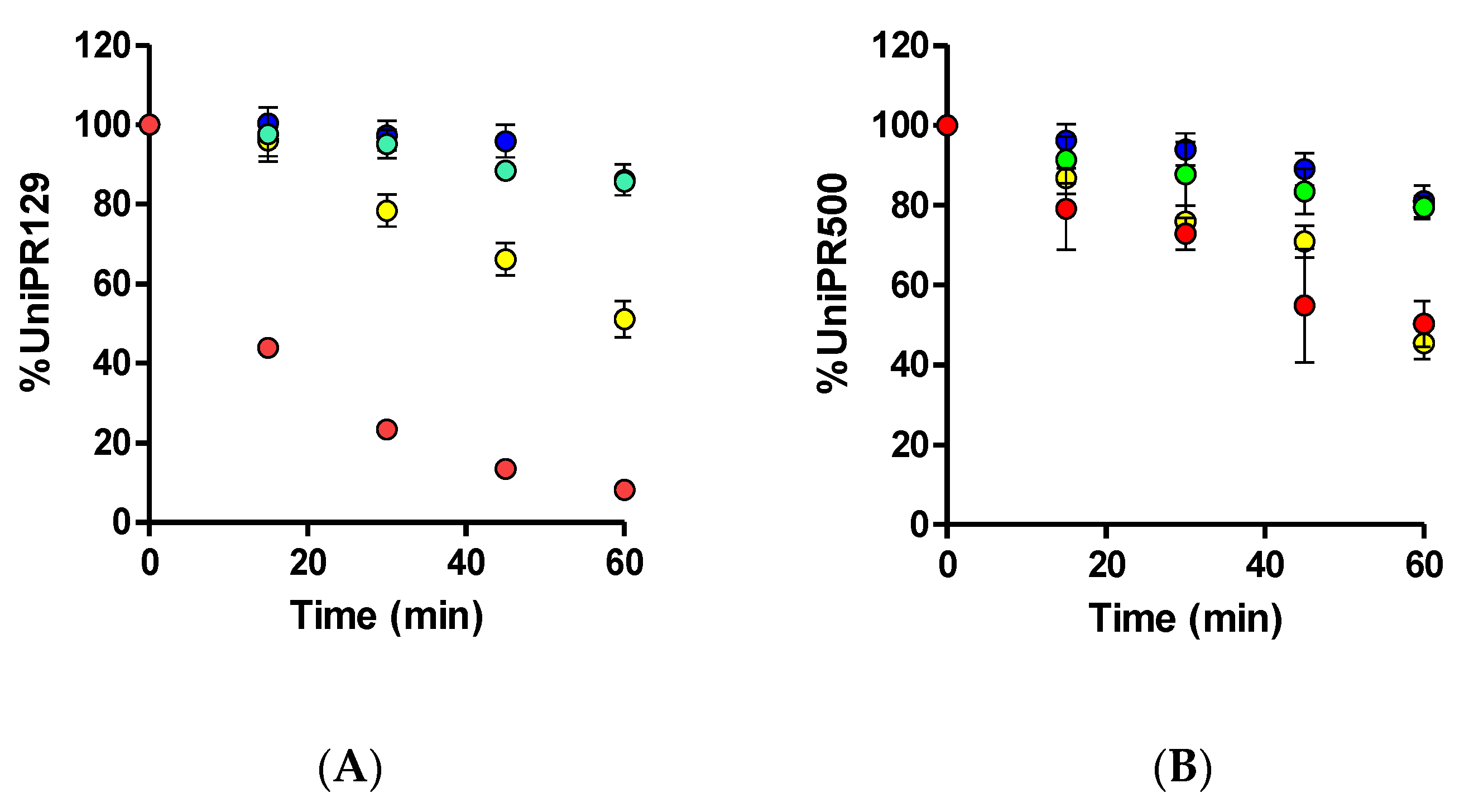
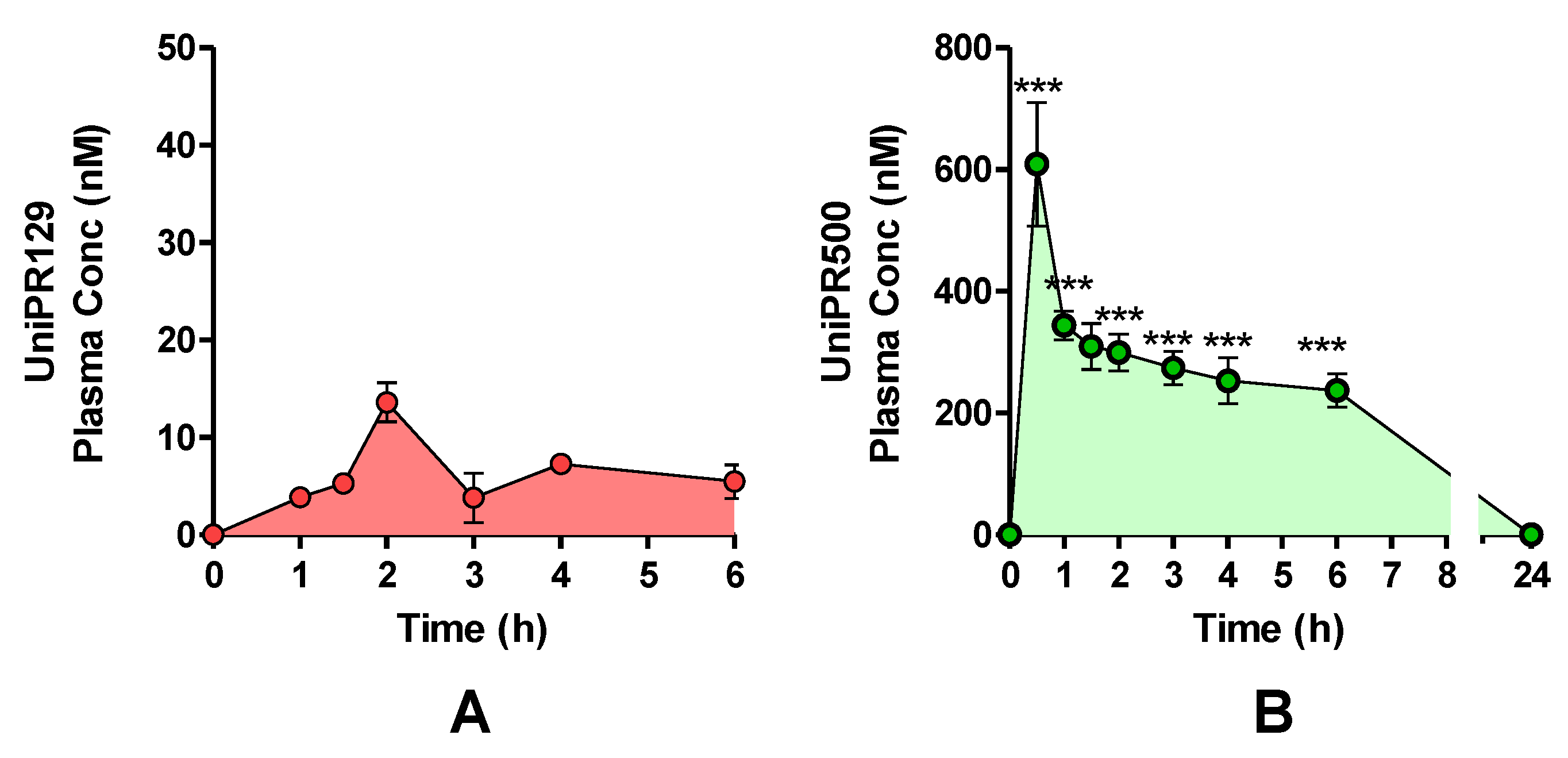
2.8. In Vivo Pharmacokinetics of UniPR129 and UniPR500 in Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
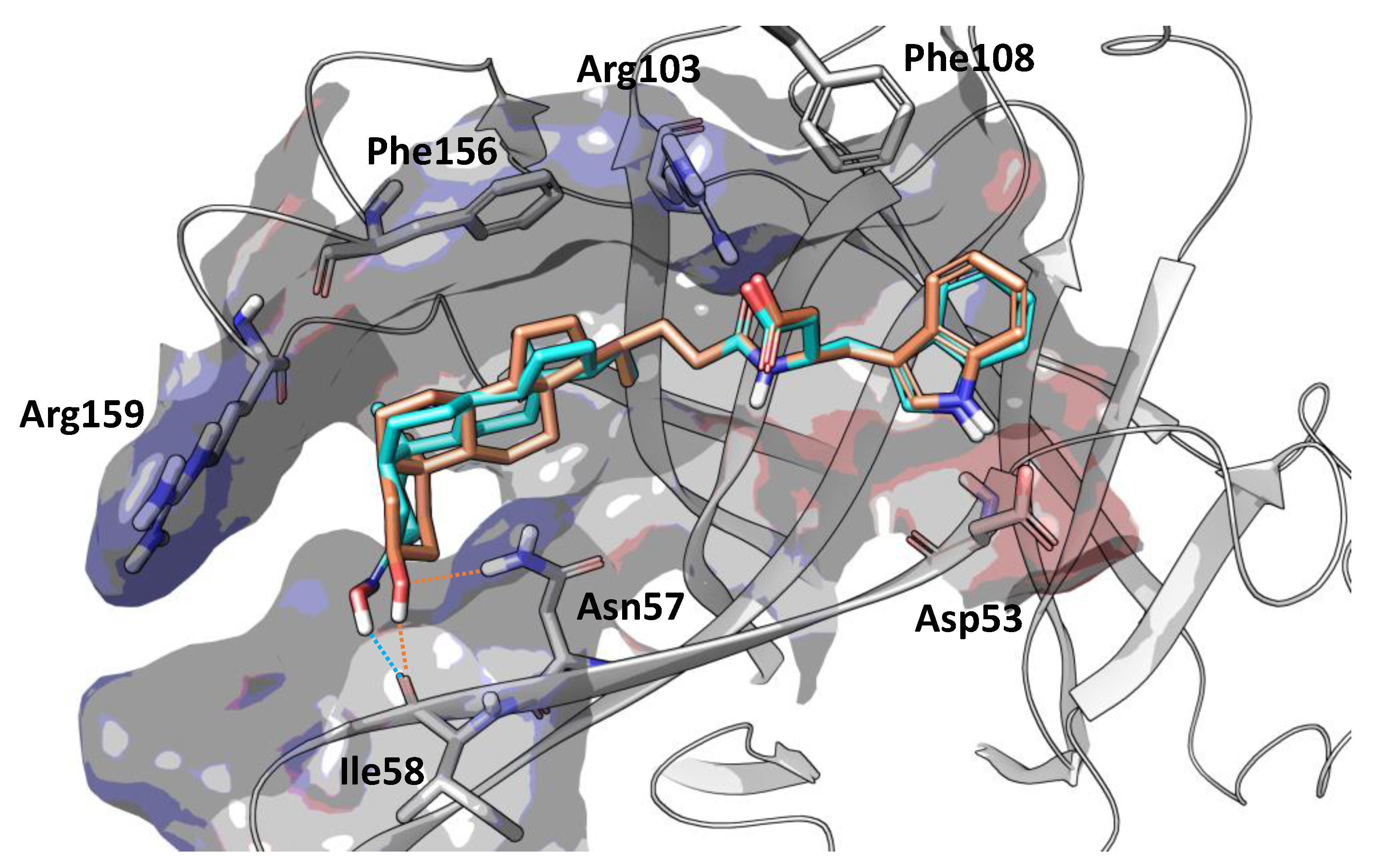
4.2. Docking Simulations
4.3. ELISA Binding Assay
4.4. Surface Plasmon Resonance (SPR) Assay
4.5. In Vitro Phase I and II Liver Metabolism: Clearance and Metabolite ID
4.6. In Vivo Dosing of UniPR129 and UniPR500 in Mouse Plasma
4.7. IM-Enabled Data Acquisition for Phase I Metabolite ID
4.8. HPLC-ESI-HR-MS Analytical Conditions for Phase II Metabolite ID
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Himanen, J.P.; Saha, N.; Nikolov, D.B. Cell-cell signaling via Eph receptors and ephrins. Curr. Opin. Cell Biol. 2007, 19, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ieguchi, K.; Tomita, T.; Takao, T.; Omori, T.; Mishima, T.; Shimizu, I.; Tognolini, M.; Lodola, A.; Tsunoda, T.; Kobayashi, S.; et al. Analysis of ADAM12-Mediated Ephrin-A1 Cleavage and Its Biological Functions. Int. J. Mol. Sci. 2021, 22, 2480. [Google Scholar] [CrossRef]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5, e14845. [Google Scholar] [CrossRef] [Green Version]
- Konstantinova, I.; Nikolova, G.; Ohara-Imaizumi, M.; Meda, P.; Kucera, T.; Zarbalis, K.; Wurst, W.; Nagamatsu, S.; Lammert, E. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 2007, 129, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, C.; Zanotti, I.; Lodola, A.; Tognolini, M. Ephrin or not? Six tough questions on Eph targeting. Expert Opin. Ther. Targets 2020, 24, 403–415. [Google Scholar] [CrossRef]
- Castelli, R.; Tognolini, M.; Vacondio, F.; Incerti, M.; Pala, D.; Callegari, D.; Bertoni, S.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; et al. Delta(5)-Cholenoyl-amino acids as selective and orally available antagonists of the Eph-ephrin system. Eur. J. Med. Chem. 2015, 103, 312–324. [Google Scholar] [CrossRef]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino acid conjugates of lithocholic acid as antagonists of the EphA2 receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef]
- Incerti, M.; Russo, S.; Callegari, D.; Pala, D.; Giorgio, C.; Zanotti, I.; Barocelli, E.; Vicini, P.; Vacondio, F.; Rivara, S.; et al. Metadynamics for Perspective Drug Design: Computationally Driven Synthesis of New Protein-Protein Interaction Inhibitors Targeting the EphA2 Receptor. J. Med. Chem. 2017, 60, 787–796. [Google Scholar] [CrossRef]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 is a competitive small molecule Eph-ephrin antagonist blocking in vitro angiogenesis at low micromolar concentrations. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, C.; Incerti, M.; Corrado, M.; Rusnati, M.; Chiodelli, P.; Russo, S.; Callegari, D.; Ferlenghi, F.; Ballabeni, V.; Barocelli, E.; et al. Pharmacological evaluation of new bioavailable small molecules targeting Eph/ephrin interaction. Biochem. Pharmacol. 2018, 147, 21–29. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Castelli, R.; Scalvini, L.; Giorgio, C.; Corrado, M.; Tognolini, M.; Mor, M.; Lodola, A.; Vacondio, F. Drug-gut microbiota metabolic interactions: The case of UniPR1331, selective antagonist of the Eph-ephrin system, in mice. J. Pharm. Biomed. Anal. 2020, 180, 113067. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Maccioni, P.; Mugnaini, C.; Brizzi, A.; Fara, F.; Mostallino, R.; Castelli, M.P.; Colombo, G.; Mor, M.; Vacondio, F.; et al. The GABAB receptor positive allosteric modulator COR659: In vitro metabolism, in vivo pharmacokinetics in rats, synthesis and pharmacological characterization of metabolically protected derivatives. Eur. J. Pharm. Sci. 2020, 155, 105544. [Google Scholar] [CrossRef]
- Huang, J.; Bathena, S.P.; Csanaky, I.L.; Alnouti, Y. Simultaneous characterization of bile acids and their sulfate metabolites in mouse liver, plasma, bile, and urine using LC-MS/MS. J. Pharm. Biomed. Anal. 2011, 55, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. Detoxification of lithocholic acid, a toxic bile acid: Relevance to drug hepatotoxicity. Drug Metab. Rev. 2004, 36, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. Drug-Like properties: Concepts, structure design and methods from ADME to Toxicity Optimization, 1st ed.; Elsevier: Oxford, UK, 2008; ISBN 9780123695208. [Google Scholar]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An ex-amination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994, 47, 1469–1479. [Google Scholar] [CrossRef]
- Incerti, M.; Russo, S.; Corrado, M.; Giorgio, C.; Ballabeni, V.; Chiodelli, P.; Rusnati, M.; Scalvini, L.; Callegari, D.; Castelli, R.; et al. Optimization of EphA2 antagonists based on a lithocholic acid core led to the identification of UniPR505, a new 3α-carbamoyloxy derivative with antiangiogenetic properties. Eur. J. Med. Chem. 2020, 189, 112083. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Deo, A.K.; Bandiera, S.M. Biotransformation of lithocholic acid by rat hepatic microsomes: Metabolite analysis by liquid chromatography/mass spectrometry. Drug Metab. Dispos. 2008, 36, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodin, K.; Lindbom, U.; Diczfalusy, U. Novel pathways of bile acid metabolism involving CYP3A4. Biochim. Biophys. Acta 2005, 1687, 84–93. [Google Scholar] [CrossRef]
- Penning, T.M.; Smithgall, T.E.; Askonas, L.J.; Sharp, R.B. Rat liver 3 alpha-hydroxysteroid dehydrogenase. Steroids 1986, 47, 221–247. [Google Scholar] [CrossRef]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. PNAS 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [Green Version]
- Zimniak, P.; Holsztynska, E.J.; Lester, R.; Waxman, D.J.; Radominska, A. Detoxification of lithocholic acid. Elucidation of the pathways of oxidative metabolism in rat liver microsomes. J. Lipid Res. 1989, 30, 907–918. [Google Scholar] [CrossRef]
- Voigt, W.; Thomas, P.J.; Hsia, S.L. Enzymic studies of bile acid metabolism. I. 6-beta-Hydroxylation of chenodeoxycholic and taurochenodeoxycholic acids by microsomal preparations of rat liver. J. Biol. Chem. 1968, 243, 3493–3499. [Google Scholar] [CrossRef]
- Araya, Z.; Wikvall, K. 6-alpha-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes. Biochim. Biophys. Acta 1999, 1438, 47–54. [Google Scholar] [CrossRef]
- Dear, G.J.; Munoz-Muriedas, J.; Beaumont, C.; Roberts, A.; Kirk, J.; Williams, J.P.; Campuzano, I. Sites of metabolic substitution: Investigating metabolite structures utilising ion mobility and molecular modelling. Rapid Commun. Mass Spec. 2010, 24, 3157–3162. [Google Scholar] [CrossRef]
- Shimizu, A.; Chiba, M. Ion mobility spectrometry-mass spectrometry analysis for the site of aromatic hydroxylation. Drug Metab. Dispos. 2013, 41, 1295–1299. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.J.; Hsia, S.L.; Matschiner, J.T.; Doisy, E.A., Jr.; Elliott, W.H.; Thayer, S.A.; Doisy, E.A. Bile Acids. XIX. Metabolism of lithocholic acid-24-14c in the rat. J. Biol. Chem. 1964, 239, 102–105. [Google Scholar] [CrossRef]
- Kitada, H.; Miyata, M.; Nakamura, T.; Tozawa, A.; Honma, W.; Shimada, M.; Nagata, K.; Sinal, C.J.; Guo, G.L.; Gonzalez, F.J.; et al. Protective role of hydroxysteroid sulfotransferase in lithocholic acid-induced liver toxicity. J. Biol. Chem. 2003, 278, 17838–17844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, M.; Gauthier-Landry, L.; Trottier, J.; Verreault, M.; Caron, P.; Finel, M.; Barbier, O. The Human UDP-glucuronosyltransferase UGT2A1 and UGT2A2 enzymes are highly active in bile acid glucuronidation. Drug Metab. Dispos. 2013, 41, 1616–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousserandot, A.; Boucher, J.L.; Henry, Y.; Niklaus, B.; Clement, B.; Mansuy, D. Microsomal cytochrome P450 dependent oxidation of N-hydroxyguanidines, amidoximes, and ketoximes: Mechanism of the oxidative cleavage of their C=N(OH) bond with formation of nitrogen oxides. Biochemistry 1998, 37, 17179–17191. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, H.; Mähönen, N.; Laitinen, M.L.; Jaurakkajärvi, M.; Raunio, H.; Juvonen, R.O.; Vepsäläinen, J.; Järvinen, T.; Rautio, J. Evaluation of hydroxyimine as cytochrome P450-selective prodrug structure. J. Med. Chem. 2006, 49, 1207–1211. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ion | RT (min) | Calculated Mass | Experimental Mass | Δm (ppm) | Observed CCS (Ų) | Chemical Formula | Mass Shift |
|---|---|---|---|---|---|---|---|---|
| UniPR129 | [M + H]+ | 10.52 | 577.4000 | 577.4000 | 0.1 | 228.89 | C36H52N2O4 | -- |
| M1 | [M + H]+ | 10.64 | 575.3843 | 575.3841 | −0.4 | 228.93 | C36H50N2O4 | −2H |
| M2 | [M + H]+ | 7.58 | 593.3949 | 593.3944 | −0.9 | 231.09 | C36H52N2O5 | +O |
| M3 | [M + H]+ | 7.69 | 593.3949 | 593.3942 | −1.1 | 250.25 | C36H52N2O5 | +O |
| M4 | [M + H]+ | 7.84 | 593.3949 | 593.3942 | −1.1 | 230.52 | C36H52N2O5 | +O |
| M5 | [M + H]+ | 7.92 | 593.3949 | 593.3946 | −0.4 | 231.13 | C36H52N2O5 | +O |
| M6 | [M + H]+ | 9.81 | 593.3949 | 593.3934 | −2.6 | 234.45 | C36H52N2O5 | +O |
| M7 | [M + H]+ | 10.16 | 593.3949 | 593.3934 | −2.5 | 243.15 | C36H52N2O5 | +O |
| M8 | [M + H]+ | 6.82 | 609.3898 | 609.3893 | −0.8 | 237.80 | C36H52N2O6 | +2O |
| M9 | [M + H]+ | 7.05 | 609.3898 | 609.3895 | −0.5 | 233.19 | C36H52N2O6 | +2O |
| M10 | [M + H]+ | 7.16 | 609.3898 | 609.3893 | −0.7 | 248.46 | C36H52N2O6 | +2O |
| Compound | Ion | RT (min) | Calculated Mass | Experimental Mass | Δm (ppm) | Chemical Formula | Mass Shift |
|---|---|---|---|---|---|---|---|
| M12 | [M − H]− | 25.09 | 751.4175 | 751.4178 | 0.44 | C42H60N2O10 | +176 |
| M13 | [M − H]− | 23.33 | 751.4175 | 751.4174 | −0.21 | C42H60N2O10 | +176 |
| M14 | [M − H]− | 34.88 | 655.3421 | 655.3411 | −0.22 | C36H52N2O7S | +79 |
| Compound | Ion | RT (min) | Calculated Mass | Experimental Mass | Δm (ppm) | Observed CCS (Ų) | Chemical Formula | Mass Shift |
|---|---|---|---|---|---|---|---|---|
| UniPR500 | [M + H]+ | 9.97 | 590.3952 | 590.3950 | −0.5 | 235.54 | C36H51N3O4 | -- |
| M1 | [M + H]+ | 10.64 | 575.3843 | 575.3838 | −0.9 | 230.52 | C36H50N2O4 | −NH |
| UniPR129 | [M + H]+ | 10.52 | 577.4000 | 577.3988 | −2.1 | 228.92 | C36H52N2O4 | −N + H |
| M2 | [M + H]+ | 7.50 | 606.3901 | 606.3890 | −1.9 | 239.09 | C36H51N3O5 | +O |
| M3 | [M + H]+ | 7.66 | 606.3901 | 606.3892 | −1.6 | 237.17 | C36H51N3O5 | +O |
| M4 | [M + H]+ | 7.93 | 606.3901 | 606.3886 | −2.5 | 235.81 | C36H51N3O5 | +O |
| M5 | [M + H]+ | 8.72 | 606.3901 | 606.3889 | −2.1 | 239.62 | C36H51N3O5 | +O |
| M6 | [M + H]+ | 9.24 | 606.3901 | 606.3886 | −2.5 | 235.55 | C36H51N3O5 | +O |
| M7 | [M + H]+ | 9.59 | 606.3901 | 606.3891 | −1.7 | 239.24 | C36H51N3O5 | +O |
| M8 | [M + H]+ | 11.85 | 606.3901 | 606.3894 | −1.2 | 235.89 | C36H51N3O5 | +O |
| M9 | [M + H]+ | 9.68 | 622.3851 | 622.3848 | −0.4 | 237.29 | C36H51N3O6 | +2O |
| M10 | [M + H]+ | 8.06 | 591.3792 | 591.3785 | −1.4 | 233.68 | C36H50N2O5 | −NH + O |
| M11 | [M + H]+ | 7.58 | 593.3949 | 593.3942 | −1.2 | 232.71 | C36H52N2O5 | − N + OH |
| Compound | Ion | RT (min) | Calculated Mass | Experimental Mass | Δm (ppm) | Chemical Formula | Mass Shift |
|---|---|---|---|---|---|---|---|
| M12 | [M − H] | 22.77 | 764.4128 | 764.4132 | 0.58 | C42H59N3O10 | +176 |
| M13 | [M − H] | 22.41 | 764.4128 | 764.4128 | 0.01 | C42H59N3O10 | +176 |
| M14 | [M − H] | 24.10 | 764.4128 | 764.4130 | 0.33 | C42H59N3O10 | +176 |
| M15 | [M − H] | 32.88 | 668.3375 | 668.3372 | 0.21 | C36H51N3O7S | +78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferlenghi, F.; Giorgio, C.; Incerti, M.; Guidetti, L.; Chiodelli, P.; Rusnati, M.; Tognolini, M.; Vacondio, F.; Mor, M.; Lodola, A. Metabolic Soft Spot and Pharmacokinetics: Functionalization of C-3 Position of an Eph–Ephrin Antagonist Featuring a Bile Acid Core as an Effective Strategy to Obtain Oral Bioavailability in Mice. Pharmaceuticals 2022, 15, 41. https://doi.org/10.3390/ph15010041
Ferlenghi F, Giorgio C, Incerti M, Guidetti L, Chiodelli P, Rusnati M, Tognolini M, Vacondio F, Mor M, Lodola A. Metabolic Soft Spot and Pharmacokinetics: Functionalization of C-3 Position of an Eph–Ephrin Antagonist Featuring a Bile Acid Core as an Effective Strategy to Obtain Oral Bioavailability in Mice. Pharmaceuticals. 2022; 15(1):41. https://doi.org/10.3390/ph15010041
Chicago/Turabian StyleFerlenghi, Francesca, Carmine Giorgio, Matteo Incerti, Lorenzo Guidetti, Paola Chiodelli, Marco Rusnati, Massimiliano Tognolini, Federica Vacondio, Marco Mor, and Alessio Lodola. 2022. "Metabolic Soft Spot and Pharmacokinetics: Functionalization of C-3 Position of an Eph–Ephrin Antagonist Featuring a Bile Acid Core as an Effective Strategy to Obtain Oral Bioavailability in Mice" Pharmaceuticals 15, no. 1: 41. https://doi.org/10.3390/ph15010041
APA StyleFerlenghi, F., Giorgio, C., Incerti, M., Guidetti, L., Chiodelli, P., Rusnati, M., Tognolini, M., Vacondio, F., Mor, M., & Lodola, A. (2022). Metabolic Soft Spot and Pharmacokinetics: Functionalization of C-3 Position of an Eph–Ephrin Antagonist Featuring a Bile Acid Core as an Effective Strategy to Obtain Oral Bioavailability in Mice. Pharmaceuticals, 15(1), 41. https://doi.org/10.3390/ph15010041