
Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4


 ,
,  , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
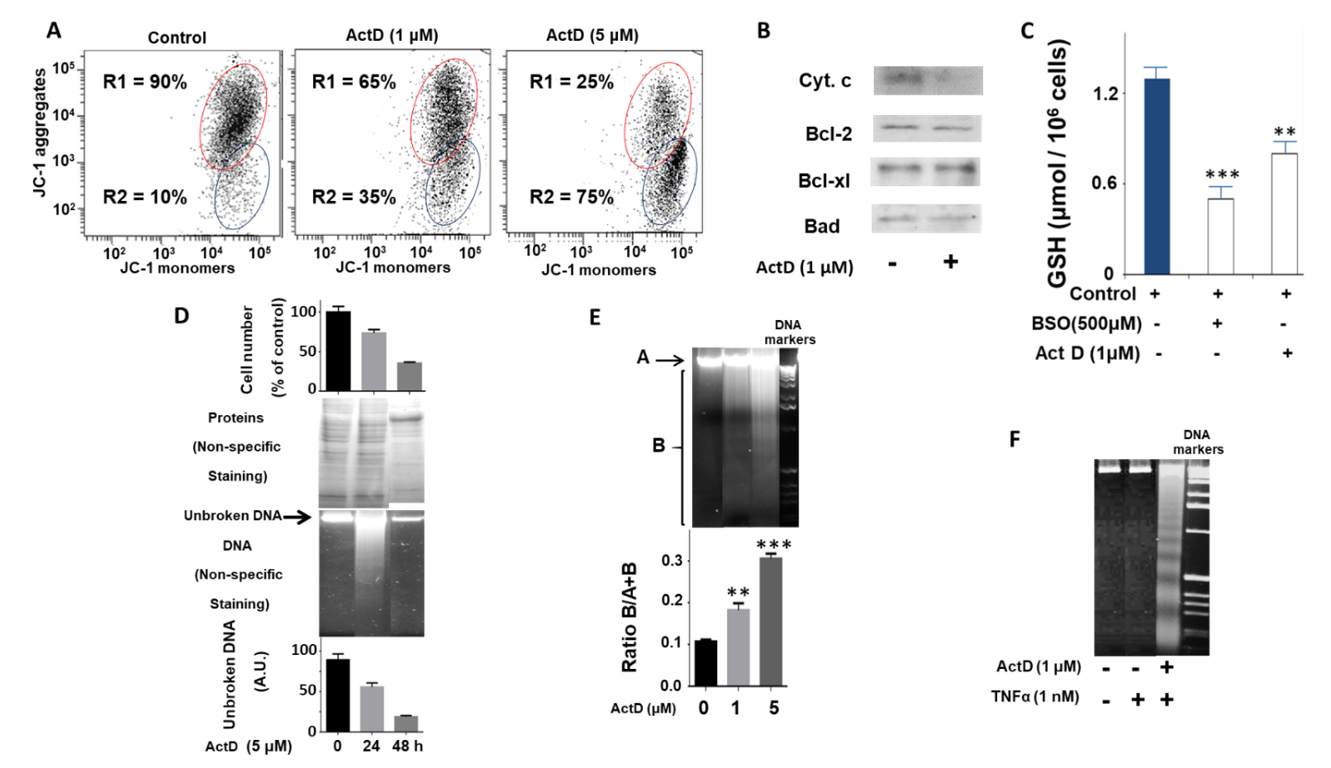
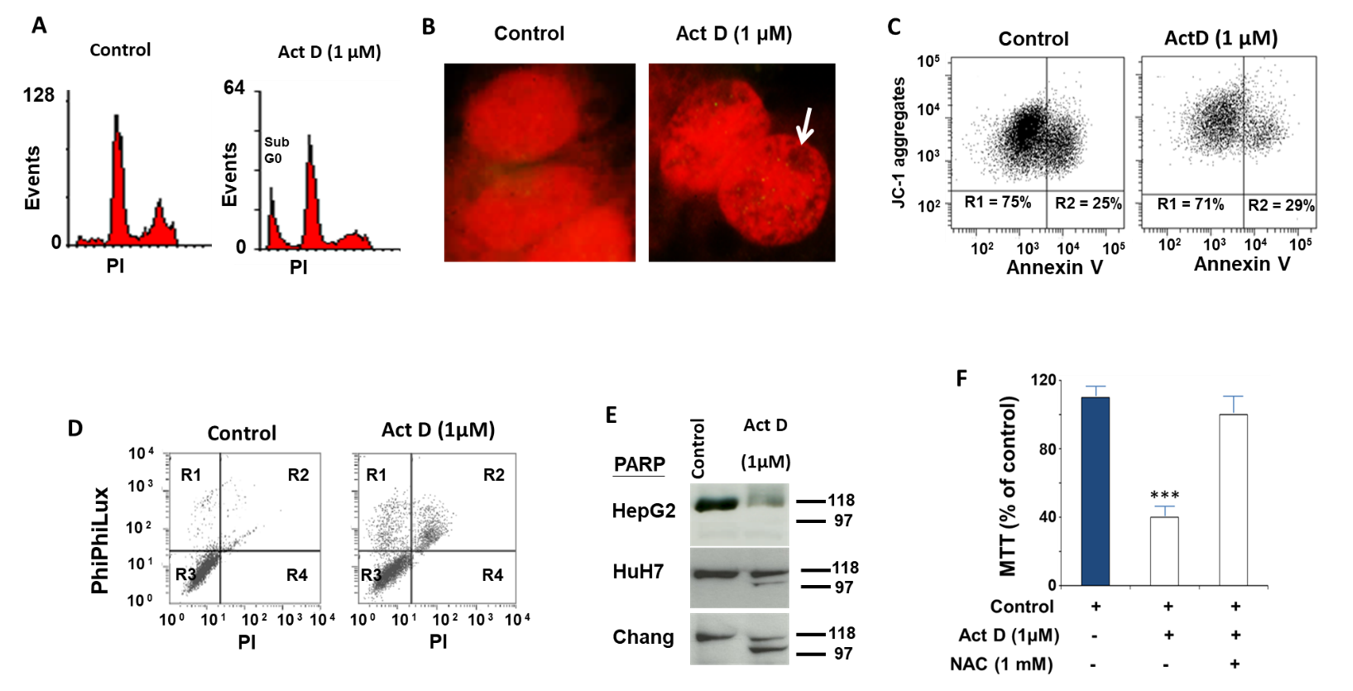
2.1. Study of the Effect of ActD on the Biochemical and Cellular Parameters of the HepG2 Cell Line
2.2. Effect of ActD on the MAP Kinases and AKT Cascades in HepG2 Cells
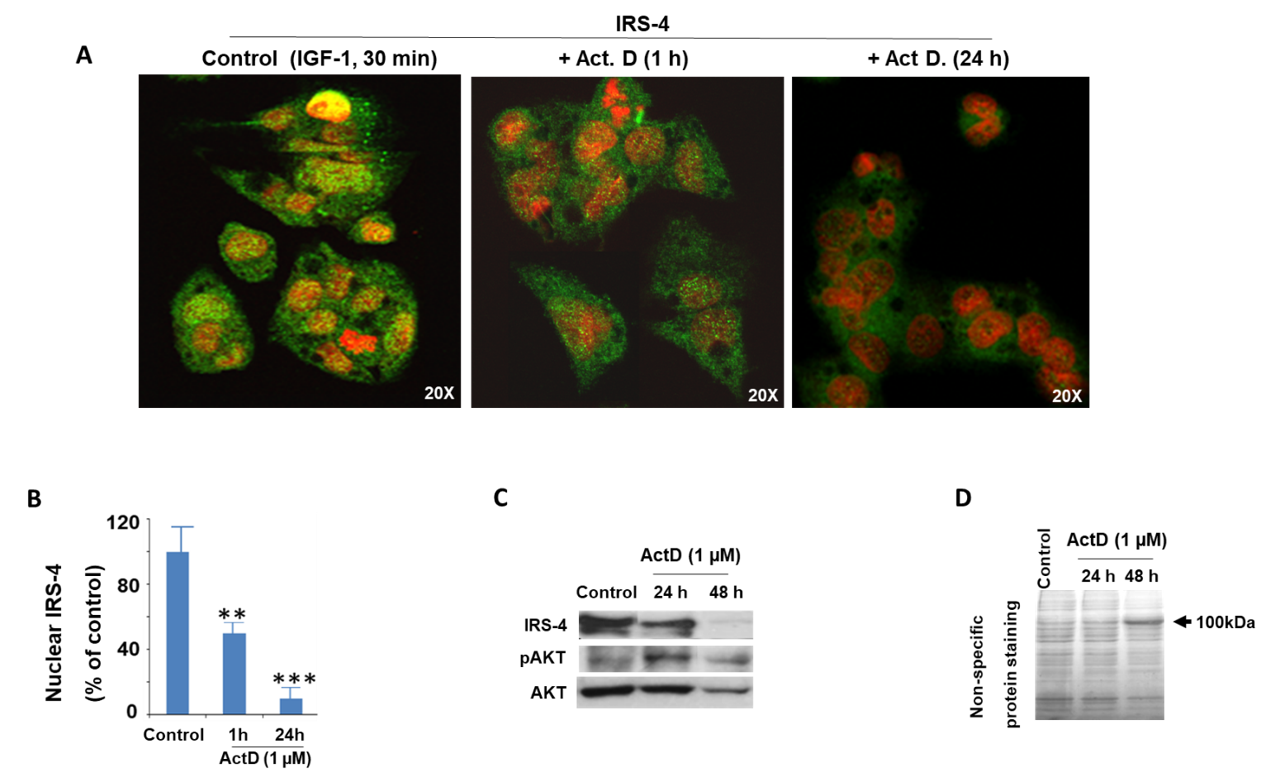
2.3. Modulation of IRS-4 Expression by ActD in the HepG2 Cell Line
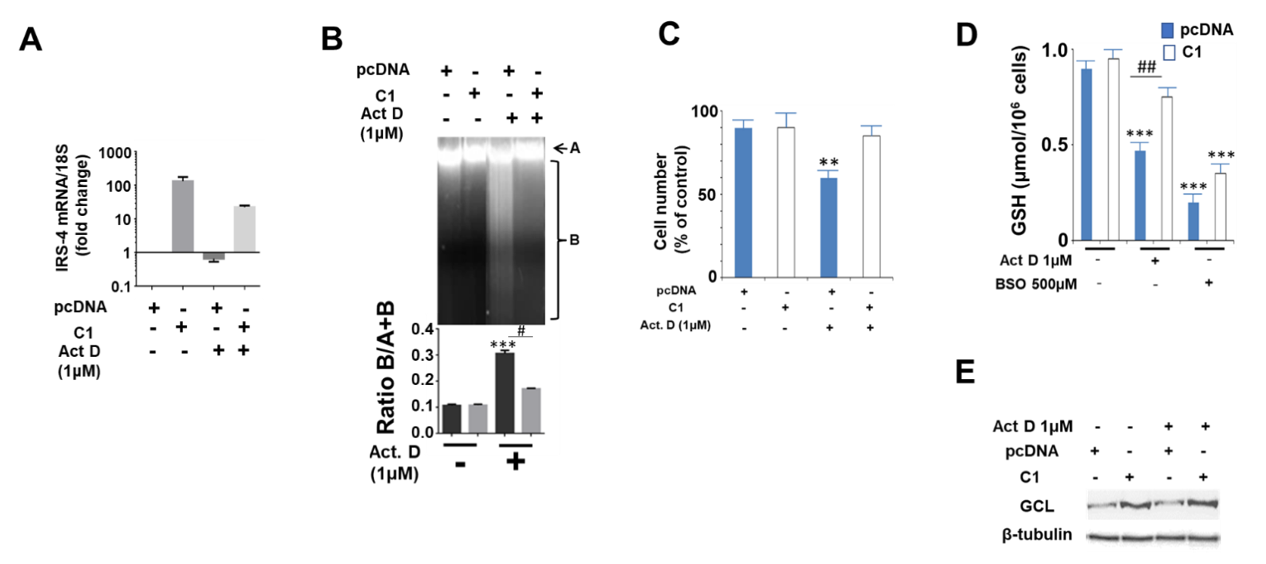
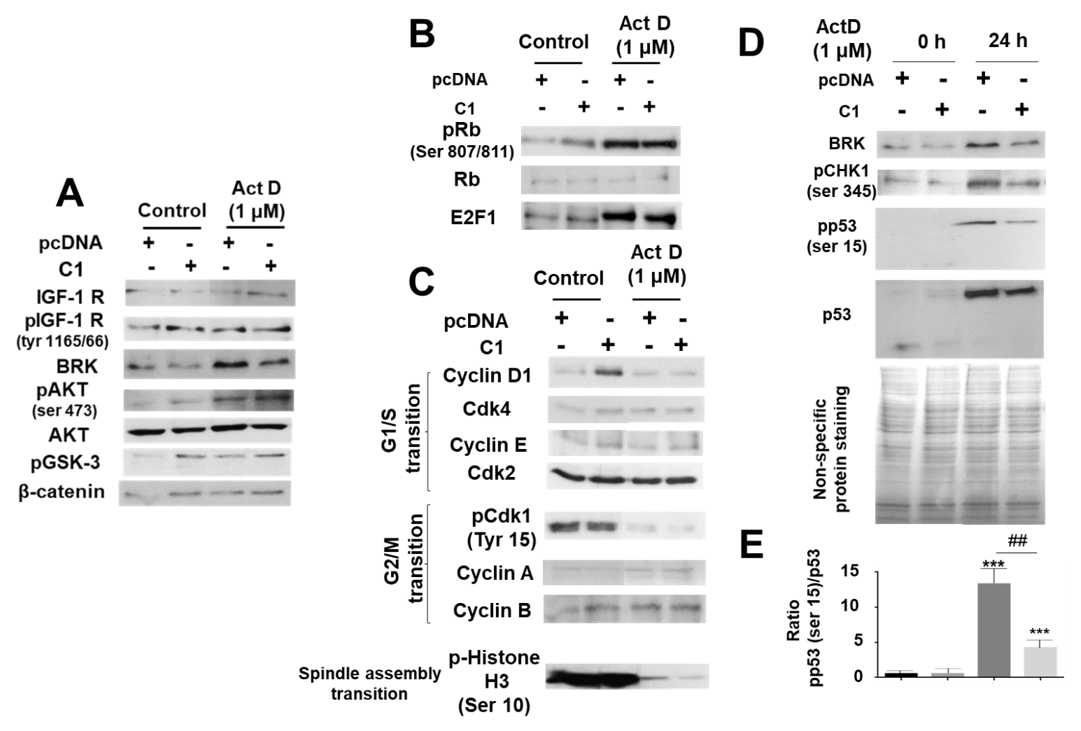
2.4. Study of the Protective Role of IRS-4 against ActD in HepG2 Cells
2.5. Study of the Mechanisms Involved in the Protective Role of IRS-4 against ActD in HepG2 Cells
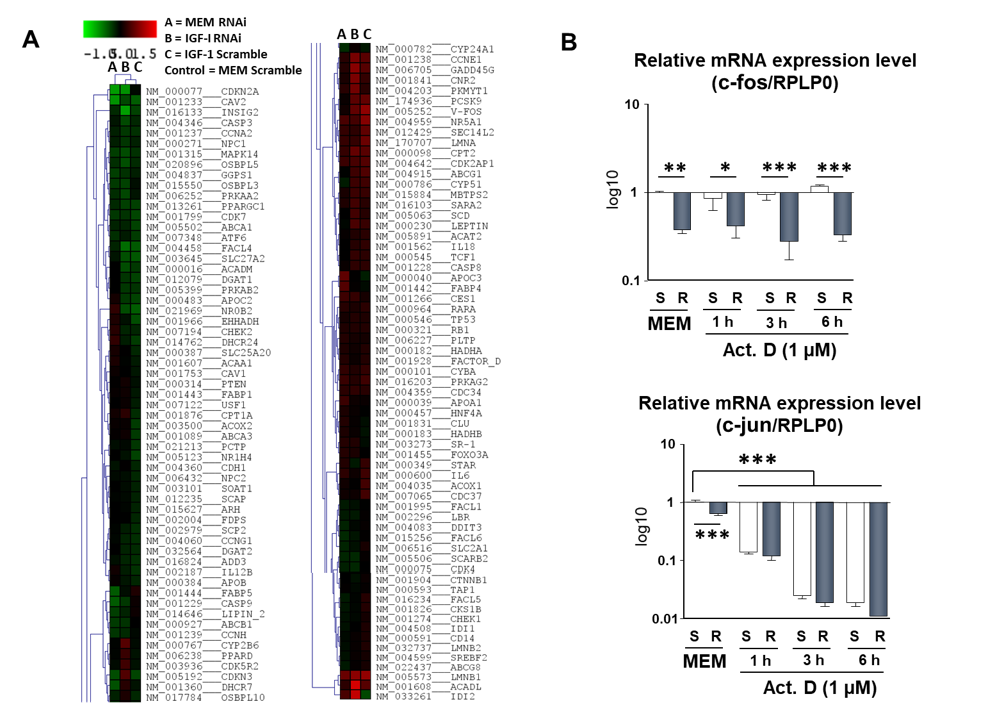
2.6. Study of the Effect of IRS-4 Downregulation and ActD Treatment on Gene Expression in HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Incubation Conditions
4.2. Transfection Assays
4.3. Immunocytochemistry
4.4. Biochemical and Molecular Biology Methods
4.5. Cytometry: Mitochondrial Activity, Apoptosis, and Cell Cycle
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.L.; Maibach, R.; Hiyama, E.; Häberle, B.; Krailo, M.; Rangaswami, A.; Aronson, D.C.; Malogolowkin, M.H.; Perilongo, G.; von Schweinitz, D.; et al. Risk-stratified staging in paediatric hepatoblastoma: A unified analysis from the Children’s Hepatic tumors International Collaboration. Lancet Oncol. 2016, 18, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.J.G.; Cives-Losada, C.; Asensio, M.; Lozano, E.; Briz, O.; Macias, R.I.R. Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers 2019, 11, 407. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.J.; Macias, R.I.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Yeh, C.-T.; Lin, K.-H. Cancer Stem Cell Functions in Hepatocellular Carcinoma and Comprehensive Therapeutic Strategies. Cells 2020, 9, 1331. [Google Scholar] [CrossRef]
- Wang, H.; Yang, M.; Lin, L.; Ren, H.; Lin, C.; Lin, S.; Shen, G.; Ji, B.; Meng, C. HepG2 cells acquire stem cell-like characteristics after immune cell stimulation. Cell. Oncol. 2015, 39, 35–45. [Google Scholar] [CrossRef]
- Cheng, S.-W.; Tsai, H.-W.; Lin, Y.-J.; Cheng, P.-N.; Chang, Y.-C.; Yen, C.-J.; Huang, H.-P.; Chuang, Y.-P.; Chang, T.-T.; Lee, C.-T.; et al. Lin28B Is an Oncofetal Circulating Cancer Stem Cell-Like Marker Associated with Recurrence of Hepatocellular Carcinoma. PLoS ONE 2013, 8, e80053. [Google Scholar] [CrossRef]
- Das, T.; Nair, R.R.; Green, R.; Padhee, S.; Howell, M.; Banerjee, J.; Mohapatra, S.S. Actinomycin D Down-regulates SOX2 Expression and Induces Death in Breast Cancer Stem Cells. Anticancer. Res. 2017, 37, 1655–1663. [Google Scholar] [CrossRef]
- Taylor, J.T.; Ellison, S.; Pandele, A.; Wood, S.; Nathan, E.; Forte, G.; Parker, H.; Zindy, E.; Elvin, M.; Dickson, A.; et al. Actinomycin D downregulates Sox2 and improves survival in preclinical models of recurrent glioblastoma. Neuro-Oncology 2020, 22, 1289–1301. [Google Scholar] [CrossRef]
- Zsíros, J.; Maibach, R.; Shafford, E.; Brugieres, L.; Brock, P.; Czauderna, P.; Roebuck, D.; Childs, M.; Zimmermann, A.; Laithier, V.; et al. Successful Treatment of Childhood High-Risk Hepatoblastoma With Dose-Intensive Multiagent Chemotherapy and Surgery: Final Results of the SIOPEL-3HR Study. J. Clin. Oncol. 2010, 28, 2584–2590. [Google Scholar] [CrossRef]
- Rana, A.N.; Qidwai, A.; Pritchard, J.; Ashraf, M.S. Successful treatment of multifocal unresectable hepatoblastoma with chemotherapy only. Pediatr. Hematol. Oncol. 2006, 23, 153–158. [Google Scholar] [CrossRef]
- Noguchi, K.; Yokoo, H.; Nakanishi, K.; Kakisaka, T.; Tsuruga, Y.; Kamachi, H.; Matsushita, M.; Kamiyama, T. A long-term survival case of adult undifferentiated embryonal sarcoma of liver. World J. Surg. Oncol. 2012, 10, 65. [Google Scholar] [CrossRef]
- Estlin, E.; Veal, G. Clinical and cellular pharmacology in relation to solid tumours of childhood. Cancer Treat. Rev. 2003, 29, 253–273. [Google Scholar] [CrossRef]
- Sato, S.; Yamamoto, E.; Niimi, K.; Ino, K.; Nishino, K.; Suzuki, S.; Kotani, T.; Kajiyama, H.; Kikkawa, F. The efficacy and toxicity of 4-day chemotherapy with methotrexate, etoposide and actinomycin D in patients with choriocarcinoma and high-risk gestational trophoblastic neoplasia. Int. J. Clin. Oncol. 2019, 25, 203–209. [Google Scholar] [CrossRef]
- Song, Y.; Park, I.-S.; Kim, J.; Seo, H.R. Actinomycin D inhibits the expression of the cystine/glutamate transporter xCT via attenuation of CD133 synthesis in CD133+ HCC. Chem. Interact. 2019, 309, 108713. [Google Scholar] [CrossRef]
- Chen, C.-S.; Ho, D.-R.; Chen, F.-Y.; Chen, C.-R.; Ke, Y.-D.; Su, J.-G.J. AKT mediates actinomycin D-induced p53 expression. Oncotarget 2014, 5, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Escribano, O.; Fernández-Moreno, M.D.; Zueco, J.A.; Menor, C.; Fueyo, J.; Ropero, R.M.; Díaz-Laviada, I.; Román, I.D.; Guijarro, L.G. Insulin receptor substrate-4 signaling in quiescent rat hepatocytes and in regenerating rat liver. Hepatology 2003, 37, 1461–1469. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Dissanayake, K.; Mackintosh, C. Effect of IRS4 Levels on PI 3-Kinase Signalling. PLoS ONE 2013, 8, e73327. [Google Scholar] [CrossRef]
- Mehine, M.; Mäkinen, N.; Heinonen, H.-R.; Aaltonen, L.A.; Vahteristo, P. Genomics of uterine leiomyomas: Insights from high-throughput sequencing. Fertil. Steril. 2014, 102, 621–629. [Google Scholar] [CrossRef]
- Mertens, F.; Möller, E.; Mandahl, N.; Picci, P.; Samson, I.; Sciot, R.; Perez-Atayde, A.R.; Debiec-Rychter, M. The t(X;6) in subungual exostosis results in transcriptional deregulation of the gene for insulin receptor substrate 4. Int. J. Cancer 2010, 128, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Ikink, G.J.; Boer, M.; Bakker, E.R.M.; Hilkens, J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat. Commun. 2016, 7, 13567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrman, K.; Isaksson, M.; Paulsson, K.; Johansson, B. The insulin receptor substrate 4 gene (IRS4) is mutated in paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2011, 155, 516–519. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2016, 49, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmartín-Salinas, P.; Guijarro, L.G. Overexpression of IRS-4 Correlates with Procaspase 3 Levels in Tumoural Tissue of Patients with Colorectal Cancer. J. Oncol. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Sanmartín-Salinas, P.; Lobo, M.D.V.T.; Noguerales-Fraguas, F.; Londoño, M.T.; Jiménez-Ruiz, A.; Guijarro, L.G. Insulin receptor substrate-4 is overexpressed in colorectal cancer and promotes retinoblastoma–cyclin-dependent kinase activation. J. Gastroenterol. 2018, 53, 932–944. [Google Scholar] [CrossRef]
- Guijarro, L.; Sanmartin-Salinas, P.; Pérez-Cuevas, E.; Toledo-Lobo, M.; Monserrat, J.; Zoullas, S.; Sáez, M.; Álvarez-Mon, M.; Bujan, J.; Noguerales-Fraguas, F.; et al. Possible Role of IRS-4 in the Origin of Multifocal Hepatocellular Carcinoma. Cancers 2021, 13, 2560. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, F.; Fonseca, N.A.; He, Y.; Fujita, M.; Nakagawa, H.; Zhang, Z.; PCAWG Transcriptome Working Group; PCAWG Structural Variation Working Group; Creighton, C.J.; et al. High-coverage whole-genome analysis of 1220 cancers reveals hundreds of genes deregulated by rearrangement-mediated cis-regulatory alterations. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cui, A.; Jin, Z.; Gao, Z.; Jin, M.; Zhu, L.; Li, L.; Jin, C.; An, Y. Downregulation of miR-493 promoted melanoma proliferation by suppressing IRS4 expression. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Lei, P.; Zeng, W.; Gao, J.; Wu, N. Long Noncoding RNA LINC00173 Promotes the Malignancy of Melanoma by Promoting the Expression of IRS4 Through Competitive Binding to microRNA-493. Cancer Manag. Res. 2020, 12, 3131–3144. [Google Scholar] [CrossRef]
- Hao, P.; Huang, Y.; Peng, J.; Yu, J.; Guo, X.; Bao, F.; Dian, Z.; An, S.; Xu, T.-R. IRS4 promotes the progression of non-small cell lung cancer and confers resistance to EGFR-TKI through the activation of PI3K/Akt and Ras-MAPK pathways. Exp. Cell Res. 2021, 403, 112615. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, E.P.; Escribano, O.; Monserrat, J.; Martinez-Botas, J.; Sã¡nchez, M.G.; Chiloeches, A.; Hernã¡ndez-Breijo, B.; Sã¡nchez-Alonso, V.; Romã¡n, I.D.; Fernã¡ndez-Moreno, M.D.; et al. RNAi-mediated silencing of insulin receptor substrate-4 enhances actinomycin D- and tumor necrosis factor-α-induced cell death in hepatocarcinoma cancer cell lines. J. Cell. Biochem. 2009, 108, 1292–1301. [Google Scholar] [CrossRef]
- Hussein, K.H.; Park, K.M.; Ghim, J.H.; Yang, S.R.; Woo, H.M. Three dimensional culture of HepG2 liver cells on a rat decellularized liver matrix for pharmacological studies. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 104, 263–273. [Google Scholar] [CrossRef]
- Alessi, D.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-activated Protein Kinase Kinase in Vitro and in Vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Hernández, M.A.; Chapresto-Garzón, R.; Cadenas, M.; Navarro-Villarán, E.; Negrete, M.; Gómez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntané, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339–410. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; Dijke, P.T.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and p38 Signaling Pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Möhle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef]
- Smith, M.J.; Prochownik, E.V. Inhibition of c-jun causes reversible proliferative arrest and withdrawal from the cell cycle. Blood 1992, 79, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Experientia 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Lai, W.S.; Arvola, R.; Goldstrohm, A.C.; Blackshear, P.J. Inhibiting transcription in cultured metazoan cells with actinomycin D to monitor mRNA turnover. Methods 2019, 155, 77–87. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Lu, B.; Dong, L.; Wang, H.; Bi, C.; Wu, G.; Guo, H.; Wu, M.; Guo, Y. TIP30 Induces Apoptosis under Oxidative Stress through Stabilization of p53 Messenger RNA in Human Hepatocellular Carcinoma. Cancer Res. 2008, 68, 4133–4141. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.-J.; Park, H.-J. Novel Molecular Mechanism for Actinomycin D Activity as an Oncogenic Promoter G-Quadruplex Binder. Biochemistry 2009, 48, 7392–7398. [Google Scholar] [CrossRef]
- Hudson, J.S.; Brooks, S.C.; Graves, D.E. Interactions of Actinomycin D with Human Telomeric G-Quadruplex DNA. Biochemistry 2009, 48, 4440–4447. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.R.; Jamieson, D.; Thomas, H.D.; Brown, C.D.; Boddy, A.V.; Veal, G.J. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem. Pharmacol. 2013, 85, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Breijo, B.; Monserrat, J.; Ramírez-Rubio, S.; Cuevas, E.P.; Vara, D.; Díaz-Laviada, I.; Fernández-Moreno, M.D.; Román, I.D.; Gisbert, J.P.; Guijarro, L.G. Preclinical evaluation of azathioprine plus buthionine sulfoximine in the treatment of human hepatocarcinoma and colon carcinoma. World J. Gastroenterol. 2011, 17, 3899–3911. [Google Scholar] [CrossRef]
- Salinas, P.S.; Toledo-Lobo, M.V.; Noguerales-Fraguas, F.; Fernández-Contreras, M.-E.; Guijarro, L.G. Overexpression of insulin receptor substrate-4 is correlated with clinical staging in colorectal cancer patients. J. Mol. Histol. 2017, 49, 39–49. [Google Scholar] [CrossRef]
- Cuevas, E.P.; Escribano, O.; Chiloeches, A.; Rubio, S.R.; Román, I.D.; Fernández-Moreno, M.D.; Guijarro, L.G. Role of insulin receptor substrate-4 in IGF-I-stimulated HEPG2 proliferation. J. Hepatol. 2007, 46, 1089–1098. [Google Scholar] [CrossRef]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- A Scudiero, D.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
- Rodríguez-Acebes, S.; Palacios, N.; Botella-Carretero, J.I.; Olea, N.; Crespo, L.; Peromingo, R.; Gómez-Coronado, D.; A Lasunción, M.; Vázquez, C.; Martínez-Botas, J. Gene expression profiling of subcutaneous adipose tissue in morbid obesity using a focused microarray: Distinct expression of cell-cycle- and differentiation-related genes. BMC Med. Genom. 2010, 3, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazet, U.; Schröder, A.; Grässel, S.; Muschter, D.; Proff, P.; Kirschneck, C. Housekeeping gene validation for RT-qPCR studies on synovial fibroblasts derived from healthy and osteoarthritic patients with focus on mechanical loading. PLoS ONE 2019, 14, e0225790. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guijarro, L.G.; Sanmartin-Salinas, P.; Pérez-Cuevas, E.; Toledo-Lobo, M.V.; Monserrat, J.; Zoullas, S.; Sáez, M.A.; Álvarez-Mon, M.A.; Bujan, J.; Noguerales-Fraguas, F.; et al. Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4. Pharmaceuticals 2021, 14, 845. https://doi.org/10.3390/ph14090845
Guijarro LG, Sanmartin-Salinas P, Pérez-Cuevas E, Toledo-Lobo MV, Monserrat J, Zoullas S, Sáez MA, Álvarez-Mon MA, Bujan J, Noguerales-Fraguas F, et al. Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4. Pharmaceuticals. 2021; 14(9):845. https://doi.org/10.3390/ph14090845
Chicago/Turabian StyleGuijarro, Luis G., Patricia Sanmartin-Salinas, Eva Pérez-Cuevas, M. Val Toledo-Lobo, Jorge Monserrat, Sofia Zoullas, Miguel A. Sáez, Miguel Angel Álvarez-Mon, Julia Bujan, Fernando Noguerales-Fraguas, and et al. 2021. "Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4" Pharmaceuticals 14, no. 9: 845. https://doi.org/10.3390/ph14090845
APA StyleGuijarro, L. G., Sanmartin-Salinas, P., Pérez-Cuevas, E., Toledo-Lobo, M. V., Monserrat, J., Zoullas, S., Sáez, M. A., Álvarez-Mon, M. A., Bujan, J., Noguerales-Fraguas, F., Arilla-Ferreiro, E., Álvarez-Mon, M., & Ortega, M. A. (2021). Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4. Pharmaceuticals, 14(9), 845. https://doi.org/10.3390/ph14090845