
Repurposing FDA Drug Compounds against Breast Cancer by Targeting EGFR/HER2

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Docking Studies
2.2. Convergence and Equilibrium
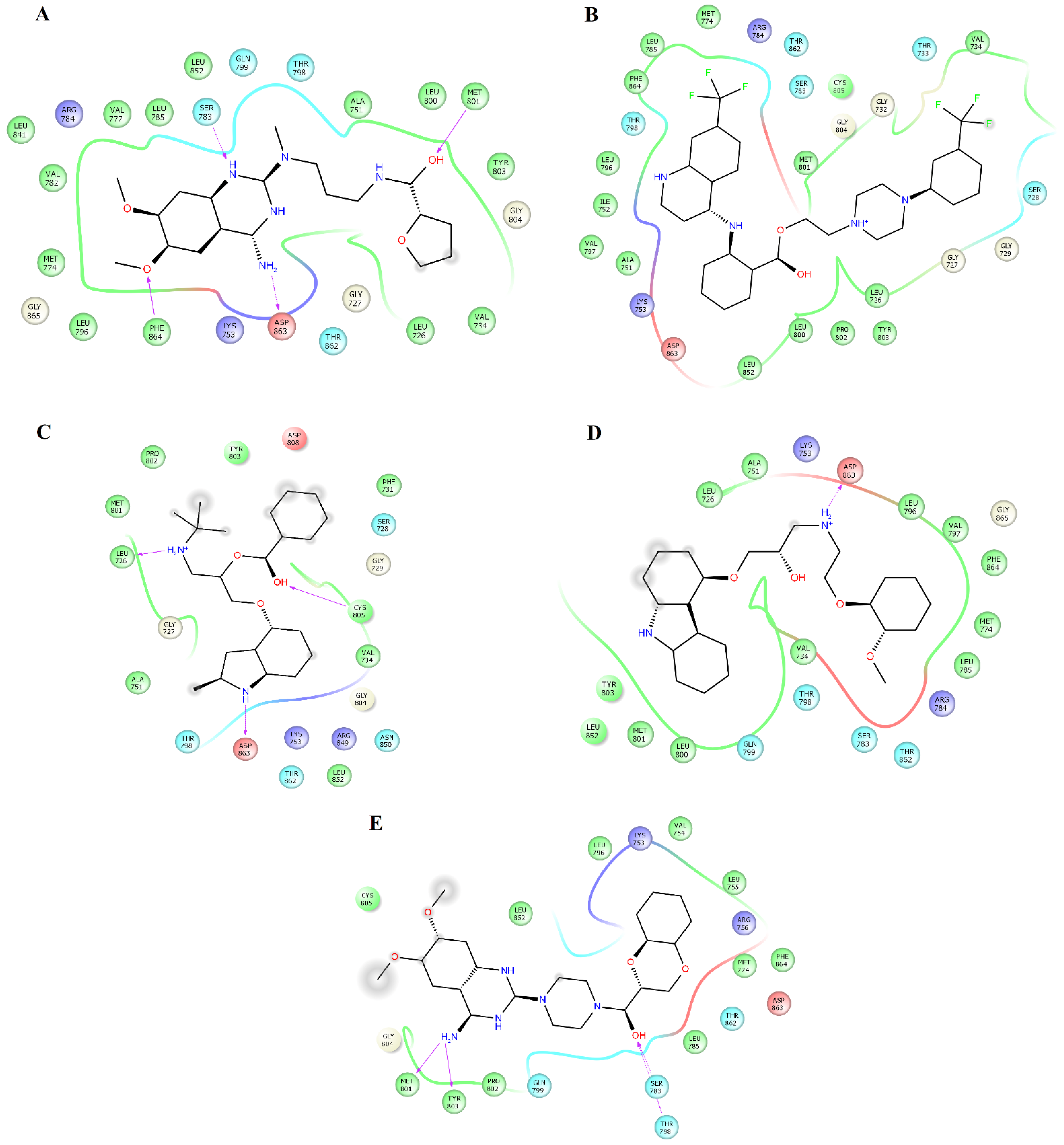
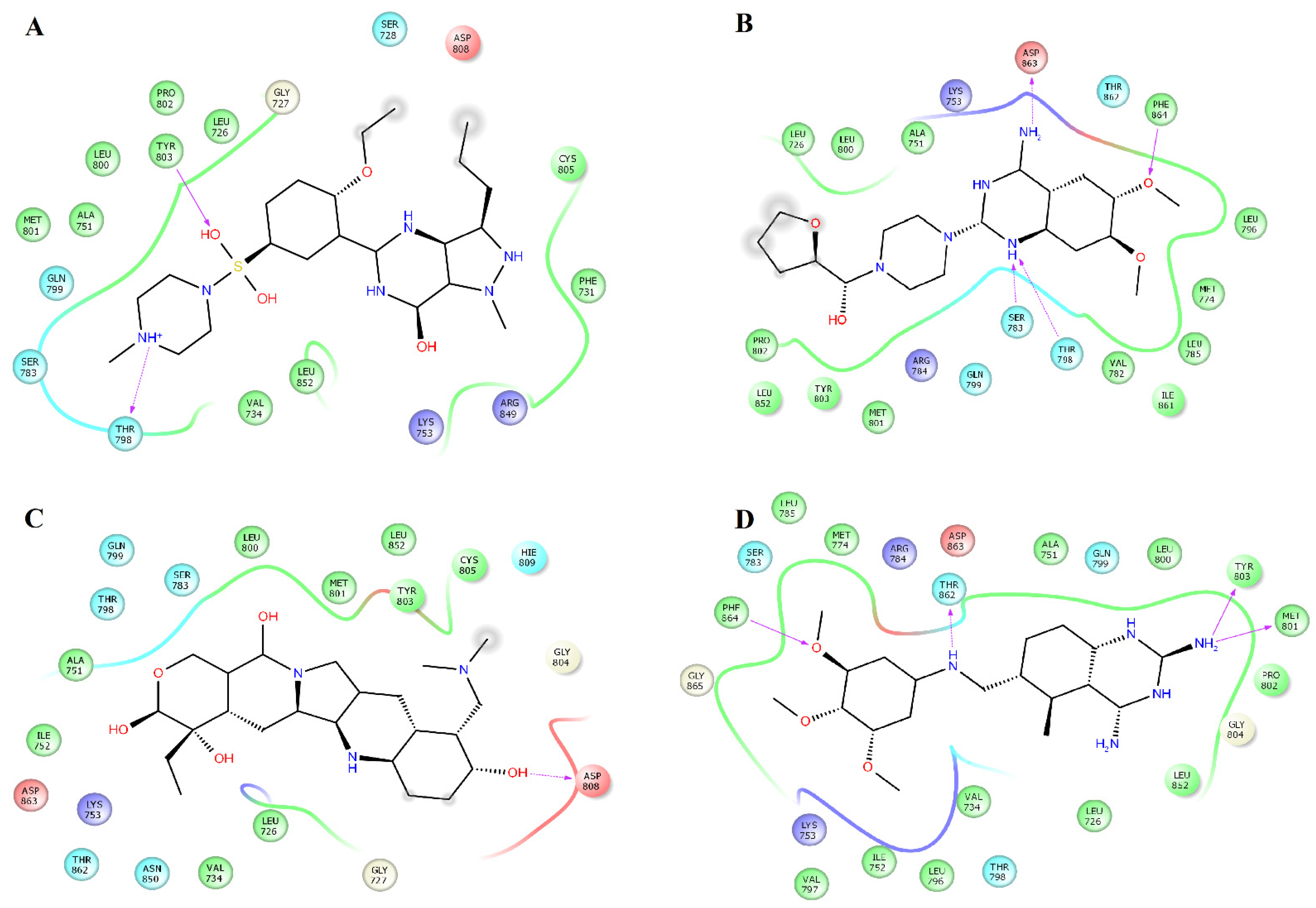
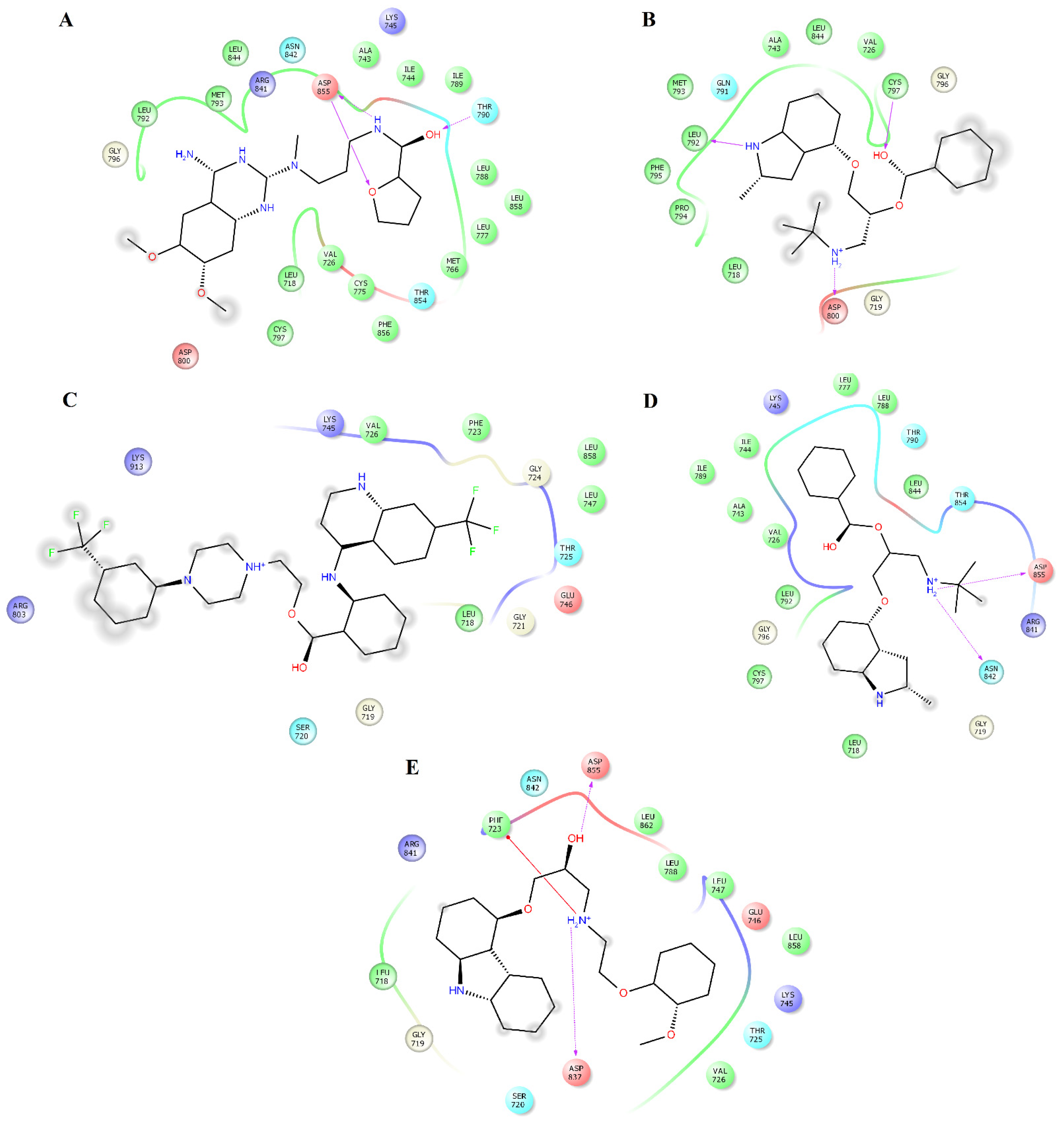
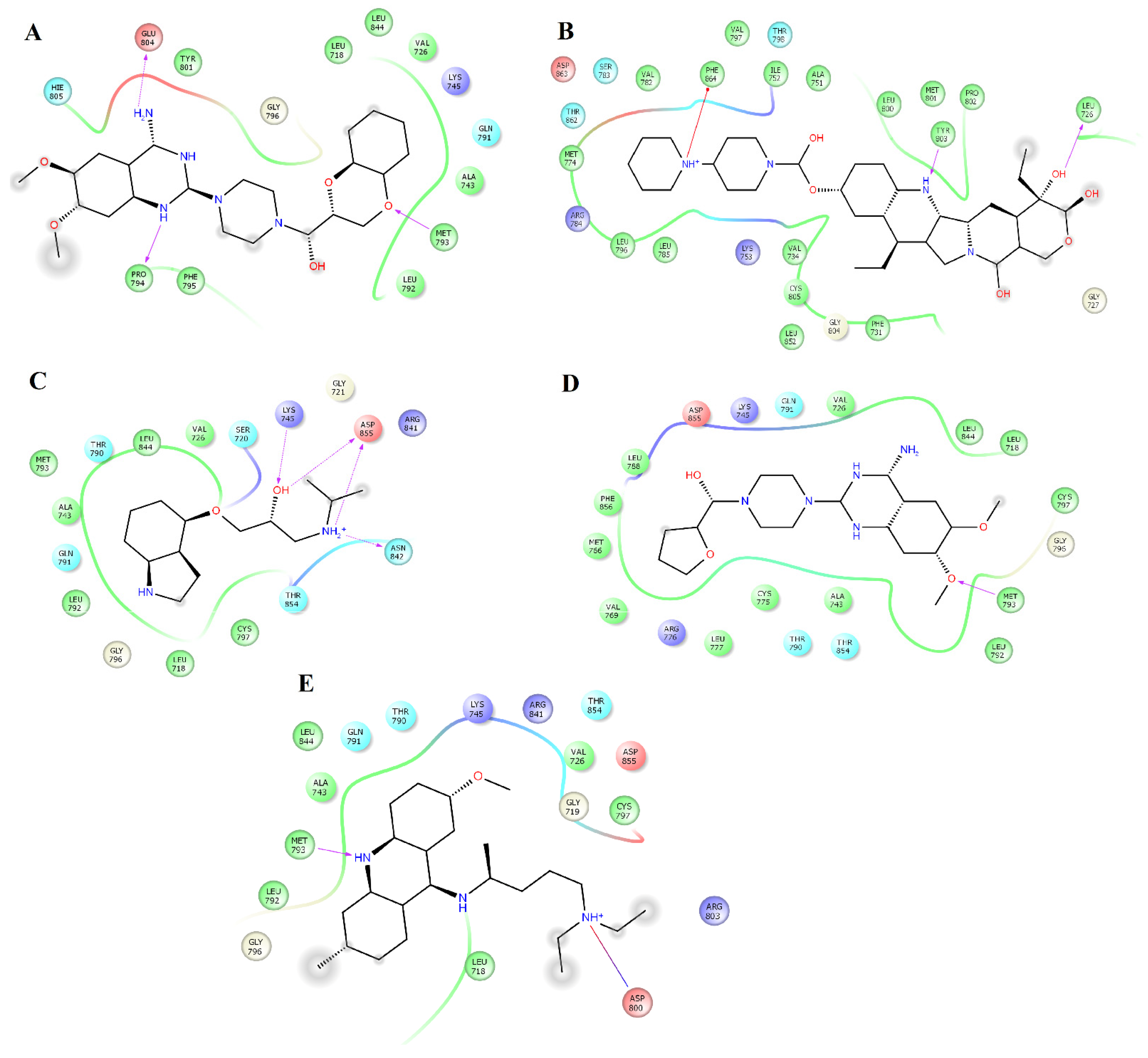
2.3. Structural Analysis of HER2-Ligand Complexes
2.4. Structural Analysis of EGFR-Ligand Complexes
2.5. Affinity of Compounds
2.6. Per-Residue Decomposition Free Energy
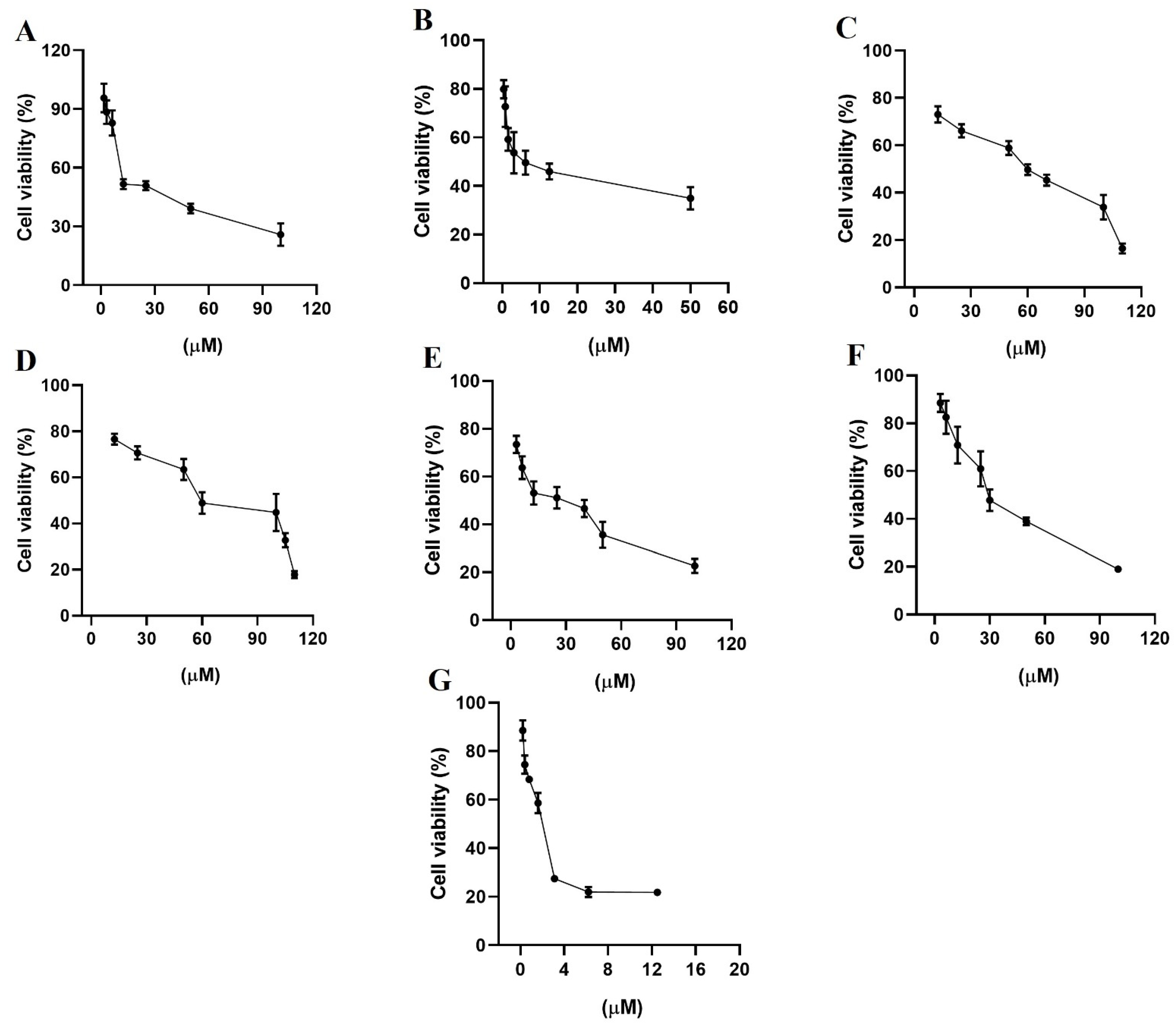
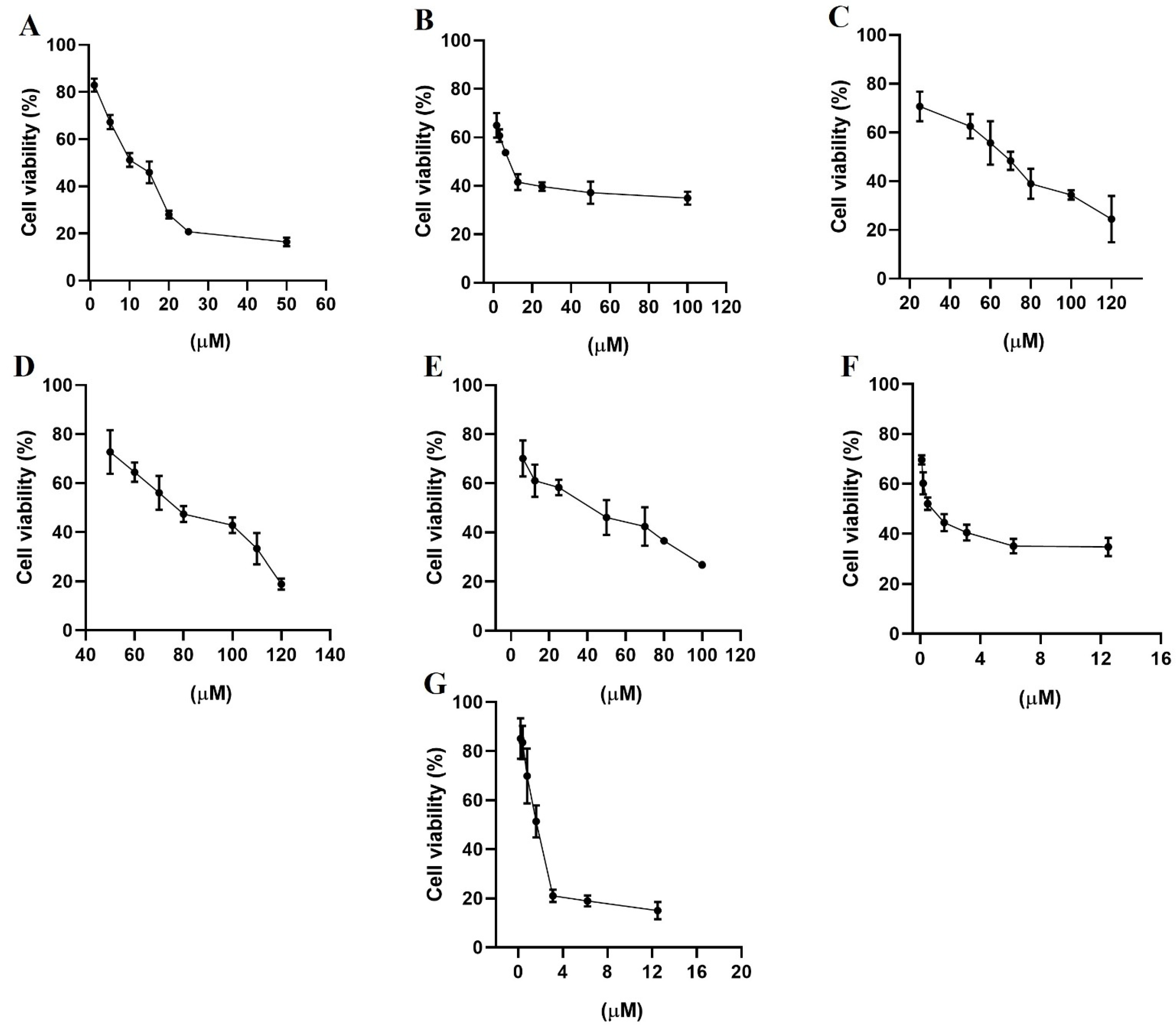
2.7. Antiproliferative Assays
3. Methods
3.1. Preparation of Systems
3.2. Docking Studies
3.3. Molecular Dynamics Simulations
3.4. Affinity Prediction and Per-Residue Decomposition
3.5. Biological Assays on Cell Lines
3.6. Antiproliferative Assays on Cell Cultures
3.7. Cell Viability Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Bono, J.S.; Rowinsky, E.K. The ErbB receptor family: A therapeutic target for cancer. Trends Mol. Med. 2002, 8, S19–S26. [Google Scholar] [CrossRef]
- Lurje, G.; Lenz, H.-J. EGFR signaling and drug discovery. Oncology 2009, 77, 400–410. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; LeVea, C.M.; Freeman, J.K.; Dougall, W.C.; Greene, M.I. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: A mechanism of interreceptor kinase activation and transphosphorylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1500–1504. [Google Scholar] [CrossRef] [Green Version]
- Riese, D.J., II; Gallo, R.M.; Settleman, J. Mutational activation of ErbB family receptor tyrosine kinases: Insights into mechanisms of signal transduction and tumorigenesis. BioEssays 2007, 29, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 2007, 19, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Balius, T.E.; Rizzo, R.C. Quantitative prediction of fold resistance for inhibitors of EGFR. Biochemistry 2009, 48, 435–8448. [Google Scholar] [CrossRef] [Green Version]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [Green Version]
- Bello, M.; Guadarrama-García, C.; Rodriguez-Fonseca, R.A. Dissecting the Molecular Recognition of Dual Lapatinib Derivatives for EGFR/HER2. J. Comput. Aided. Mol. Des. 2020, 34, 293–303. [Google Scholar] [CrossRef]
- Bello, M.; Saldaña-Rivero, L.; Correa-Basurto, J.; García, B.; Sánchez-Espinosa, V.A. Structural and energetic basis for the molecular recognition of dual synthetic vs. natural inhibitors of EGFR/HER2. Int. J. Biol. Macromol. 2018, 111, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Protein kinase inhibitors: Contributions from structure to clinical compounds. Q. Rev. Biophys. 2009, 42, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, J.R.C.; Farndon, J.R.; Needham, G.K.; Malcolm, A.J.; Harris, A.L. Epidermalgrowth-factor receptor status as predictor of early recurrence of and death from breast-cancer. Lancet 1987, 1, 1398–1402. [Google Scholar]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Rexer, B.N.; Ghosh, R.; Narasanna, A. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin. Cancer Res. 2013, 19, 5390–5401. [Google Scholar] [CrossRef] [Green Version]
- Gonzaga, I.M.; Soares-Lima, S.C.; de Santos, P.T. Alterations in epidermal growth factor receptors 1 and 2 in esophageal squamous cell carcinomas. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 2009, 69, 2384–2392. [Google Scholar]
- Bello, M. Binding mechanism of kinase inhibitors to EGFR and T790M, L858R and L858R/T790M mutants through structural and energetic analysis. Int. J. Biol. Macromol. 2018, 118, 1948–1962. [Google Scholar] [CrossRef]
- Saldaña-Rivera, L.; Bello, M.; Méndez-Luna, D. Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. J. Biomol. Struct. Dyn. 2019, 37, 4671–4684. [Google Scholar] [CrossRef]
- Bello, M.; Mejía, M.V. Structural Insight of the Anticancer Properties of Doxazosin on Overexpressing EGFR/HER2 Cell Lines. In Breast Cancer; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [Green Version]
- Rusnak, D.W.; Lackey, K.; Affleck, K. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–89. [Google Scholar] [PubMed]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2- overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.R.D.; Leary, A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today 2006, 42, 441–453. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Hu, Y.P.; Wang, J.; Hauser, J. Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res. 2006, 66, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Medina, P.J.; Goodin, S. Lapatinib: A dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin. Ther. 2008, 30, 1426–1447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Camp, E.R.; Summy, J.; Bauer, T.W.; Liu, W.; Gallick, G.E.; Ellis, L.M. Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor. Clin. Cancer Res. 2005, 11, 397–405. [Google Scholar] [PubMed]
- Liao, Q.H.; Gao, Q.Z.; Wei, J.; Chou, K.C. Docking and molecular dynamics study on the inhibitory activity of novel inhibitors on epidermal growth factor receptor (EGFR). Med. Chem. 2011, 7, 24–31. [Google Scholar] [CrossRef]
- Waterson, A.G.; Petrov, K.G.; Hornberger, K.R.; Hubbard, R.D.; Sammond, D.M.; Smith, S.C.; Dickson, H.D.; Caferro, T.R.; Hinkle, K.W.; Stevens, K.L.; et al. Synthesis and evaluation of aniline headgroups for alkynyl thienopyrimidine dual EGFR/ErbB-2kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1332–1336. [Google Scholar] [CrossRef]
- Rheault, T.R.; Caferro, T.R.; Dickerson, S.H.; Donaldson, K.H.; Gaul, M.D.; Goetz, A.S.; Mullin, R.J.; McDonald, O.B.; Petrov, K.G.; Rusnak, D.W.; et al. Thienopyrimidine-based dual EGFR/ErbB-2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Johnson, S.G.; Connolly, P.J.; Wetter, S.K.; Binnun, E.; Hughes, T.V.; Murray, W.V.; Pandey, N.B.; Moreno-Mazza, S.J.; Adams, M.; et al. Synthesis and evaluation of 2,7-diamino-thiazolo [4,5-d] pyrimidine analogues as anti-tumor epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Abad, M.C.; Connolly, P.J.; Neeper, M.P.; Struble, G.T.; Springer, B.A.; Emanuel, S.L.; Pandey, N.; Gruninger, R.H.; Adams, M.; et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4615–4619. [Google Scholar] [CrossRef]
- Ducray, R.; Ballard, P.; Barlaam, B.C.; Hickinson, M.D.; Kettle, J.G.; Ogilvie, D.J.; Trigwell, C.B. Novel 3-alkoxy-1H-pyrazolo [3,4-d]pyrimidines as EGFR and erbB2 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Sadek, M.; Abouzid, K.A.; Wang, F. In silico design: Extended molecular dynamic simulations of a new series of dually acting inhibitors against EGFR and HER2. J. Mol. Graph. Model. 2013, 44, 220–231. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Katiyar, S.; Kannan, N. Computational and experimental characterization of patient derived mutations reveal an unusual mode of regulatory spine assembly and drug sensitivity in EGFR kinase. Biochemistry 2017, 56, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef]
- Ford, C.H.; Al-Bader, M.; Al-Ayadhi, B.; Francis, I. Reassessment of estrogen receptor expression in human breast cancer cell lines. Anticancer Res. 2011, 2, 521–527. [Google Scholar]
- Stanley, A.; Ashrafi, G.H.; Seddon, A.M.; Modjtahedi, H. Synergistic effects of various Her inhibitors in combination with IGF-1R, C-MET and Src targeting agents in breast cancer cell lines. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, C.; et al. DrugBank 5.0, a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Frisch, M.J.T.; Schlegel, H.B.G.W.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4, automated docking with selective receptor fexibility. J. Comput Chem 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. Apoint-charge force field for molecular mechanics simulations of proteinsbased on condensedphase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development andtesting of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- van Gunsteren, W.F.; Berendsen, H.J.C. Algorithms for macromoleculardynamics and constraint dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald-an N. log(N) method forewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Moleculardynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Francisco, CA, USA; Palo Alto: Santa Clara, CA, USA, 2002. [Google Scholar]
- Maestro, Version 10.1; Schrödinger, LLC: New York, NY, USA, 2015; Volume 1.
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A.J. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Hou, T.J.; Xu, X.J. Recent advances in free energy calculations with a combination of molecular mechanics and continuum model. Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.; Bashford, V.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Residues |
|---|---|
| Alfuzosin | L726, F731, V734, A751, I752, K753, M774, S783, R784, L785, L796, V797, T798, Q799, L800, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Antrafenine | L726, F731, V734, A751, I752, K753, M774, S783, L785, L796, V797, T798, L800, M801, Y803, G804, C805, D808, H809, L852, V853, T862, D863, F864. |
| Bopindolol | L726, G727, F731, V734, A751, K753, S783, T798, Q799, L800, M801, P802, Y803, G804, C805, L852, T862, D863. |
| Carvedilol | L726, F731, V734, A751, K753, M774, S783, L785, L796, T798, Q799, L800, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Doxasozin | V725, L726, S728, G727, K736, V734, K753, G804, C805, L807, D808, R849, N850, L852, T862, D863, L866 |
| Irinotecan | L726, F731, V734, A751, K753, V754, L755, R756, I767, M774, S783, L785, L796, V797, T798, Q799, L800, M801, P802, Y803, G804, C805, L852, T862, D863, F864, G865. |
| Pindolol | L726, V734, A751, I752, K753, M774, S783, L785, L796, V797, T798, Q799, L800, M801, Y803, G804, L852, T862, D863, F864. |
| Prazosin | L726, F731, V734, A751, I752, K753, R756, M774, S783, R784, L785, L796, V797, T798, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Quinacrine | L726, F731, V734, A751, I752, K753, M774, S783, L785, L796, V797, T798, Q799, L800, M801, Y803, G804, C805, R849, N850, L852, T862, D863, F864. |
| Saprisartan | L726, F731, V734, A751, I752, K753, V754, L755, R756, M774, S783, L785, L796, V797, T798, Q799, L800, M801, P802, Y803, G804, C805, L852, T862, D863, F864. |
| Sildenafil | L726, F731, V734, A751, K753, S783, L785, L796, V797, T798, Q799, L800, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Terazosin | L726, F731, V734, A751, I752, K753, M774, S783, R784, L785, L796, V797, T798, Q799, L800, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Topotecan | L726, F731, V734, A751, K753, S783, T798, Q799, L800, M801, Y803, G804, C805, D808, L852, T862, D863. |
| Trimetrexate | F731, V734, A751, I752, K753, M774, S783, R784, L785, L796, V797, T798, M801, Y803, G804, C805, L852, T862, D863, F864. |
| Drug | Residues |
|---|---|
| Alfuzosin | L718, V726, A743, I744, K745, M766, C775, L777, L788, I789, T790, L792, M793, G796, C797, D800, R841, L844, N842, T854, D855, F856, L858. |
| Amodiaquine | L718, G719, V726, A743, Q791, L792, M793, P794, F795, G796, C797, D800, L844. |
| Antrafenine | L718, G719, S720, G721, G724, F723, T725, V726, K745, E746, L747, R803, L858, K913. |
| Bopindolol | L718, G719, V726, A743, I744, K745, L777, L788, I789, T790, L792, G796, C797, R841, N842, L844, T854, D855. |
| Carvedilol | L718, G719, S720, F723, T725, V726, K745, D746, L747, L788, D837, R841, N842, D855, L858, L862. |
| Doxasozin | L718, V726, A743, K745, Q791, L792, M793, P794, F795, G796, Y801, E804, H805, L844. |
| Pindolol | L718, S720, G721, V726, A743, K745, T790, Q791, L792, M793, G796, C797, R841, N842, L844, T854, D855. |
| Prazosin | L718, V726, A743, K745, M766, V769, C775, R776, L777, L788, T790, Q791, L792, M793, G796, C797, L844, T854, D855, F856. |
| Quinacrine | L718, G719, V726, A743, K745, T790, Q791, L792, M793, G796, C797, D800, R803, R841, L844, T854, D855. |
| Saprisartan | L718, G719, S720, G721, F723, G724, T725, V726, A743, K745, L747, T790, Q791, L792, M793, G796, C797, R841, L844, T854, L858, L862, K875, P877. |
| Terazosin | L718, G719, V726, A743, K745, C775, R776, M766, L777, L788, T790, M793, G796, C797, D800, L844, T854, D855, F856. |
| Topotecan | L718, G719, S720, G721, V726, A743, L792, M793, P794, G796, C797, D800, R841, N842, L844, D855. |
| Trimetrexate | L718, G721, A722, F723, V726, A743, K745, T790, Q791, L792, M793, G796, C797, R841, N842, L844, T854, D855, L858. |
| Udenafil | V717, L718, A743, Q791, L792, M793, P794, F795, G796, C797, L799, D800, R803, R841, L844. |
| Vardenafil | L718, G719, S720, G721, F723, G724, T725, V726, K745, E746, L747, L788, L792, G796, C797, D800, R841, N842, D855, L858. |
| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | ΔGmmgbsa |
|---|---|---|---|---|---|
| HER2alfuzosin | −57.0 ± 4 | −33.2 ± 6 | 45.4 ± 4 | −7.1 ± 0.3 | −51.9 ± 6 |
| HER2antrafenine | −66.3 ± 3 | 24.4 ± 8 | −0.9 ± 0.1 | −8.3 ± 0.2 | −51.1 ± 3 |
| HER2bopindolol | −40.0 ± 4 | 18.7 ± 10 | −7.8 ± 1 | −5.5 ± 0.5 | −34.6 ± 5 |
| HER2carvedilol | −51.1 ± 6 | 16.0 ± 4 | 4.6 ± 1 | −6.8 ± 0.6 | −37.3 ± 5 |
| HER2doxasozin | −49.0 ± 4 | 24.0 ± 9 | −7.4 ± 1 | −6.0 ± 0.3 | −38.4 ± 4 |
| HER2irinotecan | −68.0 ± 4 | 40.2 ± 12 | −21.0 ± 8 | −7.6 ± 0.3 | −56.4 ± 5 |
| HER2pindolol | −26.0 ± 3 | 4.0 ± 0.5 | 3.4 ± 0.6 | −3.8 ± 0.4 | −22.4 ± 4 |
| HER2prazosin | −56.0 ± 3 | −24.5 ± 7 | 42.7 ± 5 | −6.9 ± 0.2 | −44.7 ± 4 |
| HER2quinacrine | −57.5 ± 3 | 34.0 ± 8 | −24.0 ± 7 | −7.4 ± 0.2 | −54.9 ± 3 |
| HER2saprisartan | −57.0 ± 4 | −17.8 ± 5 | 46.3 ± 4 | −7.2 ± 0.4 | −35.7 ± 4 |
| HER2sildenafil | −49.1 ± 3 | 34.7 ± 9 | −22.7 ± 9 | −6.2 ± 0.3 | −43.3 ± 3 |
| HER2terazosin | −46.0 ± 4 | −38.3 ± 11 | 50.0 ± 7 | −5.7 ± 0.4 | −40.0 ± 7 |
| HER2topotecan | −45.1 ± 3 | −26.3 ± 4 | 43.0 ± 12 | −5.3 ± 0.4 | −33.7 ± 5 |
| HER2trimetrexate | −51.6 ± 3 | −23.6 ± 4 | 33.9 ± 3 | −6.5 ± 0.3 | −47.8 ± 4 |
| EGFRalfuzosin | −55.5 ± 4 | −30.3 ± 6 | 47.4 ± 5 | −7.0 ± 0.3 | −45.4 ± 4 |
| EGFRamodiaquine | −30.8 ± 4 | 43.3 ± 20 | −35.2 ± 15 | −4.1 ± 0.6 | −26.8 ± 8 |
| EGFRantrafenine | −32.6 ± 4 | 65.4 ± 10 | −48.5 ± 10 | −4.0 ± 0.6 | −19.7 ± 3 |
| EGFRbopindolol | −40.6 ± 4 | 17.7 ± 9 | −8.4 ± 1 | −5.8 ± 0.4 | −37.1 ± 3 |
| EGFRcarvedilol | −35.8 ± 5 | −21.4 ± 11 | 25.8 ± 7 | −5.4 ± 0.5 | −36.8 ± 7 |
| EGFRdoxasozin | −38.8 ± 4 | 3.2 ± 0.9 | 9.7 ± 3 | −4.6 ± 0.4 | −30.5 ± 4 |
| EGFRpindolol | −25.4 ± 5 | 4.7 ± 0.5 | −4.1 ± 1 | −4.4 ± 0.7 | −29.2 ± 5 |
| EGFRprazosin | −52.8 ± 3 | −19.0 ± 6 | 38.2 ± 4 | −6.6 ± 0.2 | −40.2 ± 4 |
| EGFRquinacrine | −40.6 ± 4 | 12.9 ± 3 | −4.7 ± 0.5 | −5.4 ± 0.5 | −37.8 ± 4 |
| EGFRsaprisartan | −46.4 ± 3 | −15.3 ± 2 | 46.2 ± 10 | −6.4 ± 0.5 | −21.9 ± 5 |
| EGFRterazosin | −55.8 ± 4 | −22.7 ± 7 | 40.4 ± 6 | −6.6 ± 0.3 | −44.7 ± 6 |
| EGFRtopotecan | −35.2 ± 7 | −6.3 ± 1 | 22.7 ± 9 | −3.9 ± 0.8 | −22.7 ± 7 |
| EGFRtrimetrexate | −42.6 ± 3 | −29.8 ± 7 | 48.5 ± 6 | −5.6 ± 0.4 | −29.5 ± 4 |
| EGFRudenafil | −40.3 ± 3 | 10.0 ± 2 | 5.6 ± 0.5 | −5.0 ± 0.3 | −29.7 ± 3 |
| EGFRvardenafil | −53.6 ± 5 | −35.0 ± 12 | 46.8 ± 11 | −6.4 ± 0.7 | −48.2 ± 7 |
| Residue | HER2alfuzosin | HER2antrafenine | HER2irinotecan | HER2quinacrine |
|---|---|---|---|---|
| L726 | −1.0 | −1.1 | −2.6 | −1.8 |
| G727 | −1.0 | −0.9 | ||
| F731 | −1.1 | −1.9 | −1.6 | |
| T733 | −0.7 | |||
| V734 | −1.6 | −3.0 | −2.6 | −2.1 |
| A751 | −0.9 | −1.0 | −1.6 | −1.1 |
| K753 | −1.3 | −1.2 | −1.7 | −1.5 |
| M774 | −1.1 | |||
| S783 | −0.6 | −0.5 | −0.7 | |
| L785 | −1.8 | −0.9 | −1.0 | −0.9 |
| L796 | −1.4 | −1.9 | −0.5 | |
| V797 | −0.5 | −0.9 | −1.0 | |
| T798 | −1.8 | −1.8 | −1.8 | −5.7 |
| L800 | −0.7 | |||
| M801 | −0.9 | −1.8 | ||
| P802 | −1.7 | −1.0 | ||
| Y803 | −1.7 | −2.0 | ||
| G804 | −0.6 | |||
| C805 | −0.6 | −0.6 | −0.5 | −0.6 |
| L852 | −1.8 | −1.4 | −1.3 | −1.9 |
| T862 | −1.1 | −1.2 | −1.1 | −2.0 |
| D863 | −1.9 | −0.7 | ||
| F864 | −1.6 | −0.5 |
| Residue | EGFRalfuzosin | EGFRprazosin | EGFRterazosin | EGFRvardenafil |
|---|---|---|---|---|
| L718 | −2.2 | −1.2 | −2.2 | −2.4 |
| G719 | −0.9 | |||
| G721 | −1.2 | |||
| F723 | −0.6 | |||
| T725 | −0.7 | |||
| V726 | −1.9 | −2.0 | −2.1 | −3.0 |
| A743 | −1.0 | −0.9 | −1.0 | −0.7 |
| I744 | −0.7 | |||
| K745 | −1.4 | −1.2 | −2.0 | −3.2 |
| M766 | −0.6 | |||
| L777 | −1.9 | −1.1 | ||
| L788 | −1.0 | −1.1 | −1.6 | |
| I789 | −1.3 | −0.5 | ||
| T790 | −0.5 | −1.2 | −1.1 | |
| L792 | −0.9 | −0.8 | −0.9 | |
| M793 | −1.0 | −1.1 | −0.7 | −0.5 |
| G796 | −1.0 | −0.5 | −1.0 | −0.7 |
| C797 | −1.0 | −0.4 | −0.9 | −1.3 |
| R841 | −2.0 | |||
| L844 | −2.0 | −2.2 | −2.2 | −0.8 |
| T854 | −1.3 | −1.7 | −1.7 | |
| F856 | −1.0 | −0.6 | −0.5 | |
| L858 | −0.6 | −0.6 |
| Compounds | MCF-7 | MDA-MB-231 |
|---|---|---|
| Gefitinib | 10 ± 1 | 13 ± 1 |
| Lapatinib | 7 ± 1 | 10 ± 1 |
| Terazosin hydrochloride | 70 ± 1 | 68 ± 2 |
| Alfuzosin hydrochloride | 85 ± 2 | 69 ± 1 |
| Prazosin hydrochloride | 41 ± 2 | 25 ± 1 |
| Irinotecan hydrochloride | 0.37 ± 0.8 | 27 ± 1 |
| Quinacrine dihydrochloride | 1.4 ± 1 | 1.3 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balbuena-Rebolledo, I.; Padilla-Martínez, I.I.; Rosales-Hernández, M.C.; Bello, M. Repurposing FDA Drug Compounds against Breast Cancer by Targeting EGFR/HER2. Pharmaceuticals 2021, 14, 791. https://doi.org/10.3390/ph14080791
Balbuena-Rebolledo I, Padilla-Martínez II, Rosales-Hernández MC, Bello M. Repurposing FDA Drug Compounds against Breast Cancer by Targeting EGFR/HER2. Pharmaceuticals. 2021; 14(8):791. https://doi.org/10.3390/ph14080791
Chicago/Turabian StyleBalbuena-Rebolledo, Irving, Itzia Irene Padilla-Martínez, Martha Cecilia Rosales-Hernández, and Martiniano Bello. 2021. "Repurposing FDA Drug Compounds against Breast Cancer by Targeting EGFR/HER2" Pharmaceuticals 14, no. 8: 791. https://doi.org/10.3390/ph14080791
APA StyleBalbuena-Rebolledo, I., Padilla-Martínez, I. I., Rosales-Hernández, M. C., & Bello, M. (2021). Repurposing FDA Drug Compounds against Breast Cancer by Targeting EGFR/HER2. Pharmaceuticals, 14(8), 791. https://doi.org/10.3390/ph14080791