
Facilitation of TRKB Activation by the Angiotensin II Receptor Type-2 (AT2R) Agonist C21


 ,
,  and
and
Abstract
:1. Introduction
2. Results
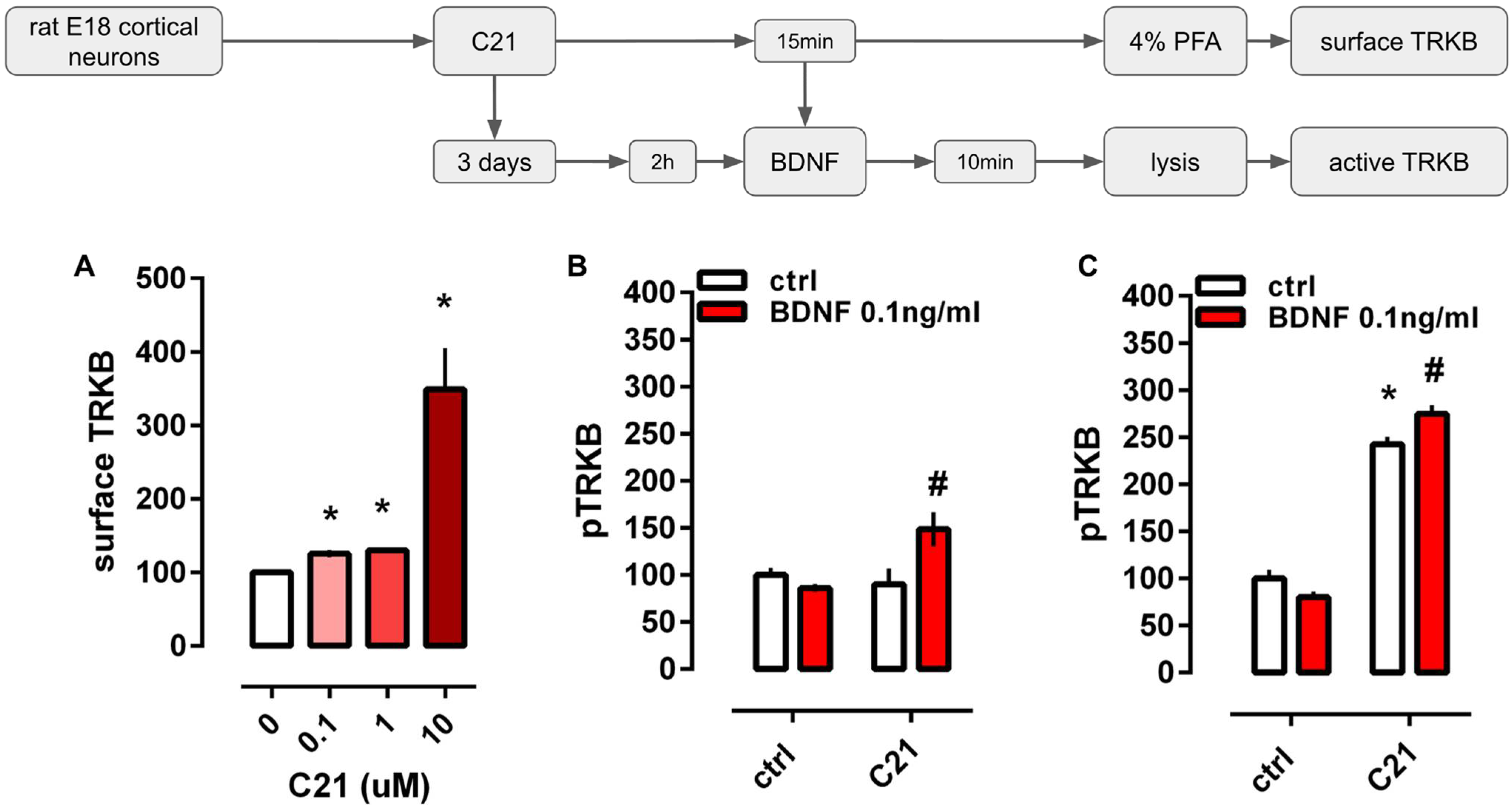
2.1. C21 Exposes TRKB on the Cell Surface and Facilitates BDNF Effect on TRKB
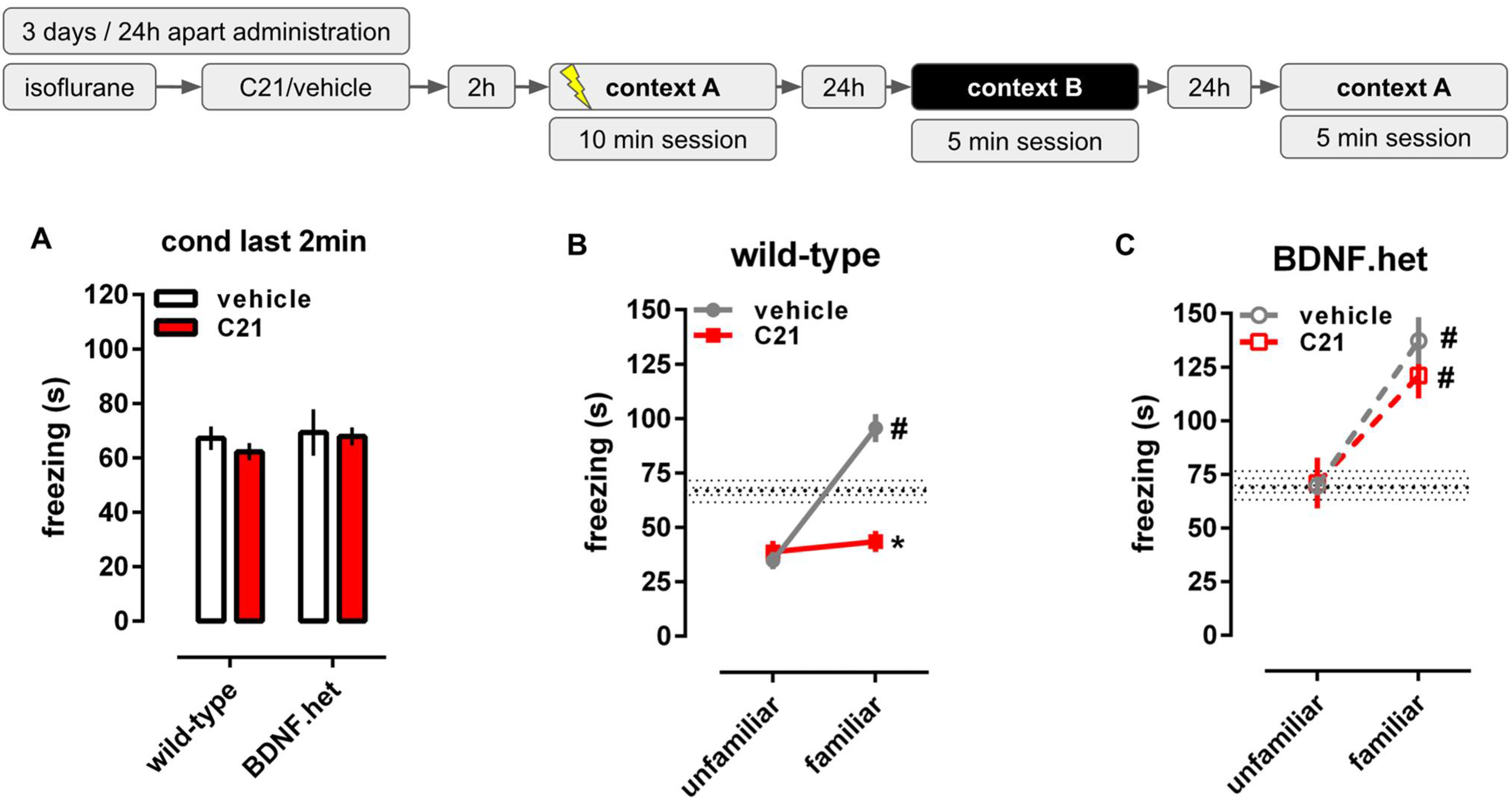
2.2. C21 Reduces Behavioral Consequences of Stress
2.3. C21 Lacks Anxiolytic-like Effect
3. Discussion
4. Methods
4.1. Animals
4.2. Drugs
4.3. Primary Cultures of Cortical Cells
4.4. Surface TRKB
4.5. Phospho-TRKB Interaction ELISA
4.6. Behavioral Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, J.W.; Harding, J.W. Brain Renin-Angiotensin—A New Look at an Old System. Prog. Neurobiol. 2011, 95, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Brain Angiotensin II: New Developments, Unanswered Questions and Therapeutic Opportunities. Cell. Mol. Neurobiol. 2005, 25, 485–512. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Saavedra, J.M. Characterization and Development of Angiotensin II Receptor Subtypes (AT1 and AT2) in Rat Brain. Am. J. Physiol. 1991, 261, R209–R216. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, A.D.; Wang, L.; Ludin, J.A.; Smith, J.A.; Pioquinto, D.J.; Hiller, H.; Steckelings, U.M.; Scheuer, D.A.; Sumners, C.; Krause, E.G. Reporter Mouse Strain Provides a Novel Look at Angiotensin Type-2 Receptor Distribution in the Central Nervous System. Brain Struct. Funct. 2016, 221, 891–912. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef] [Green Version]
- Pryce, C.R.; Azzinnari, D.; Spinelli, S.; Seifritz, E.; Tegethoff, M.; Meinlschmidt, G. Helplessness: A Systematic Translational Review of Theory and Evidence for Its Relevance to Understanding and Treating Depression. Pharmacol. Ther. 2011, 132, 242–267. [Google Scholar] [CrossRef]
- Castren, E.; Saavedra, J.M. Repeated Stress Increases the Density of Angiotensin II Binding Sites in Rat Paraventricular Nucleus and Subfornical Organ. Endocrinology 1988, 122, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Gard, P.R.; Mandy, A.; Sutcliffe, M.A. Evidence of a Possible Role of Altered Angiotensin Function in the Treatment, but Not Etiology, of Depression. Biol. Psychiatry 1999, 45, 1030–1034. [Google Scholar] [CrossRef]
- Giardina, W.J.; Ebert, D.M. Positive Effects of Captopril in the Behavioral Despair Swim Test. Biol. Psychiatry 1989, 25, 697–702. [Google Scholar] [CrossRef]
- Martin, P.; Massol, J.; Puech, A.J. Captopril as an Antidepressant? Effects on the Learned Helplessness Paradigm in Rats. Biol. Psychiatry 1990, 27, 968–974. [Google Scholar] [CrossRef]
- Okuyama, S.; Sakagawa, T.; Sugiyama, F.; Fukamizu, A.; Murakami, K. Reduction of Depressive-like Behavior in Mice Lacking Angiotensinogen. Neurosci. Lett. 1999, 261, 167–170. [Google Scholar] [CrossRef]
- Ola, M.S.; Ahmed, M.M.; Abuohashish, H.M.; Al-Rejaie, S.S.; Alhomida, A.S. Telmisartan Ameliorates Neurotrophic Support and Oxidative Stress in the Retina of Streptozotocin-Induced Diabetic Rats. Neurochem. Res. 2013, 38, 1572–1579. [Google Scholar] [CrossRef]
- Krikov, M.; Thone-Reineke, C.; Müller, S.; Villringer, A.; Unger, T. Candesartan but Not Ramipril Pretreatment Improves Outcome after Stroke and Stimulates Neurotrophin BNDF/TrkB System in Rats. J. Hypertens. 2008, 26, 544–552. [Google Scholar] [CrossRef]
- Ping, G.; Qian, W.; Song, G.; Zhaochun, S. Valsartan Reverses Depressive/anxiety-like Behavior and Induces Hippocampal Neurogenesis and Expression of BDNF Protein in Unpredictable Chronic Mild Stress Mice. Pharmacol. Biochem. Behav. 2014, 124, 5–12. [Google Scholar] [CrossRef]
- Saarelainen, T.; Hendolin, P.; Lucas, G.; Koponen, E.; Sairanen, M.; MacDonald, E.; Agerman, K.; Haapasalo, A.; Nawa, H.; Aloyz, R.; et al. Activation of the TrkB Neurotrophin Receptor Is Induced by Antidepressant Drugs and Is Required for Antidepressant-Induced Behavioral Effects. J. Neurosci. 2003, 23, 349–357. [Google Scholar] [CrossRef]
- Rantamäki, T.; Hendolin, P.; Kankaanpää, A.; Mijatovic, J.; Piepponen, P.; Domenici, E.; Chao, M.V.; Männistö, P.T.; Castrén, E. Pharmacologically Diverse Antidepressants Rapidly Activate Brain-Derived Neurotrophic Factor Receptor TrkB and Induce Phospholipase-Cgamma Signaling Pathways in Mouse Brain. Neuropsychopharmacology 2007, 32, 2152–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpova, N.N.; Pickenhagen, A.; Lindholm, J.; Tiraboschi, E.; Kulesskaya, N.; Agústsdóttir, A.; Antila, H.; Popova, D.; Akamine, Y.; Bahi, A.; et al. Fear Erasure in Mice Requires Synergy between Antidepressant Drugs and Extinction Training. Science 2011, 334, 1731–1734. [Google Scholar] [CrossRef] [Green Version]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant Drugs Act by Directly Binding to TRKB Neurotrophin Receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef] [PubMed]
- Diniz, C.R.A.F.; Casarotto, P.C.; Fred, S.M.; Biojone, C.; Castrén, E.; Joca, S.R.L. Antidepressant-like Effect of Losartan Involves TRKB Transactivation from Angiotensin Receptor Type 2 (AGTR2) and Recruitment of FYN. Neuropharmacology 2018, 135, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Maya Vetencourt, J.F.; Sale, A.; Viegi, A.; Baroncelli, L.; De Pasquale, R.; O’Leary, O.F.; Castrén, E.; Maffei, L. The Antidepressant Fluoxetine Restores Plasticity in the Adult Visual Cortex. Science 2008, 320, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Steinzeig, A.; Cannarozzo, C.; Castrén, E. Fluoxetine-Induced Plasticity in the Visual Cortex Outlasts the Duration of the Naturally Occurring Critical Period. Eur. J. Neurosci. 2019, 50, 3663–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namsolleck, P.; Boato, F.; Schwengel, K.; Paulis, L.; Matho, K.S.; Geurts, N.; Thöne-Reineke, C.; Lucht, K.; Seidel, K.; Hallberg, A.; et al. AT2-Receptor Stimulation Enhances Axonal Plasticity after Spinal Cord Injury by Upregulating BDNF Expression. Neurobiol. Dis. 2013, 51, 177–191. [Google Scholar] [CrossRef]
- Fred, S.M.; Laukkanen, L.; Brunello, C.A.; Vesa, L.; Göös, H.; Cardon, I.; Moliner, R.; Maritzen, T.; Varjosalo, M.; Casarotto, P.C.; et al. Pharmacologically Diverse Antidepressants Facilitate TRKB Receptor Activation by Disrupting Its Interaction with the Endocytic Adaptor Complex AP-2. J. Biol. Chem. 2019, 294, 18150–18161. [Google Scholar] [CrossRef] [Green Version]
- Speth, R.C. [125I] CGP 42112 Binding Reveals Differences between Rat Brain and Adrenal AT2 Receptor Binding Sites. Regul. Pept. 1993, 44, 189–197. [Google Scholar] [CrossRef]
- Heemskerk, F.M.; Saavedra, J.M. Quantitative Autoradiography of Angiotensin II AT2 Receptors with [125I] CGP 42112. Brain Res. 1995, 677, 29–38. [Google Scholar] [CrossRef]
- Wan, Y.; Wallinder, C.; Plouffe, B.; Beaudry, H.; Mahalingam, A.K.; Wu, X.; Johansson, B.; Holm, M.; Botoros, M.; Karlén, A.; et al. Design, Synthesis, and Biological Evaluation of the First Selective Nonpeptide AT2 Receptor Agonist. J. Med. Chem. 2004, 47, 5995–6008. [Google Scholar] [CrossRef] [PubMed]
- Bosnyak, S.; Jones, E.S.; Christopoulos, A.; Aguilar, M.-I.; Thomas, W.G.; Widdop, R.E. Relative Affinity of Angiotensin Peptides and Novel Ligands at AT1 and AT2 Receptors. Clin. Sci. 2011, 121, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Guimond, M.-O.; Wallinder, C.; Alterman, M.; Hallberg, A.; Gallo-Payet, N. Comparative Functional Properties of Two Structurally Similar Selective Nonpeptide Drug-like Ligands for the Angiotensin II Type-2 (AT2) Receptor. Effects on Neurite Outgrowth in NG108-15 Cells. Eur. J. Pharmacol. 2013, 699, 160–171. [Google Scholar] [CrossRef]
- Pereira, D.B.; Chao, M.V. The Tyrosine Kinase Fyn Determines the Localization of TrkB Receptors in Lipid Rafts. J. Neurosci. 2007, 27, 4859–4869. [Google Scholar] [CrossRef]
- Rajagopal, R.; Chao, M.V. A Role for Fyn in Trk Receptor Transactivation by G-Protein-Coupled Receptor Signaling. Mol. Cell. Neurosci. 2006, 33, 36–46. [Google Scholar] [CrossRef]
- Haapasalo, A.; Sipola, I.; Larsson, K.; Akerman, K.E.O.; Stoilov, P.; Stamm, S.; Wong, G.; Castren, E. Regulation of TRKB Surface Expression by Brain-Derived Neurotrophic Factor and Truncated TRKB Isoforms. J. Biol. Chem. 2002, 277, 43160–43167. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; et al. Endocannabinoids Regulate Interneuron Migration and Morphogenesis by Transactivating the TrkB Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 19115–19120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, C.R.A.F.; Biojone, C.; Joca, S.R.L.; Rantamäki, T.; Castrén, E.; Guimarães, F.S.; Casarotto, P.C. Dual Mechanism of TRKB Activation by Anandamide through CB1 and TRPV1 Receptors. PeerJ 2019, 7, e6493. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Swiercz, A.P.; Moshfegh, C.M.; Hopkins, L.; Wiaderkiewicz, J.; Speth, R.C.; Park, J.; Marvar, P.J. Angiotensin II Type 2 Receptor-Expressing Neurons in the Central Amygdala Influence Fear-Related Behavior. Biol. Psychiatry 2019, 86, 899–909. [Google Scholar] [CrossRef]
- Kerr, D.S.; Bevilaqua, L.R.M.; Bonini, J.S.; Rossato, J.I.; Köhler, C.A.; Medina, J.H.; Izquierdo, I.; Cammarota, M. Angiotensin II Blocks Memory Consolidation through an AT2 Receptor-Dependent Mechanism. Psychopharmacology 2005, 179, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.V.; de Souza, A.M.A.; Ji, H.; West, C.A.; Wu, X.; Lee, D.L.; Aguilar, B.L.; Forcelli, P.A.; de Menezes, R.C.; Sandberg, K. The Angiotensin Type 1 Receptor Antagonist Losartan Prevents Ovariectomy-Induced Cognitive Dysfunction and Anxiety-Like Behavior in Long Evans Rats. Cell. Mol. Neurobiol. 2020, 40, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Wincewicz, D.; Braszko, J.J. Telmisartan Attenuates Cognitive Impairment Caused by Chronic Stress in Rats. Pharmacol. Rep. 2014, 66, 436–441. [Google Scholar] [CrossRef]
- Wincewicz, D.; Juchniewicz, A.; Waszkiewicz, N.; Braszko, J.J. Angiotensin II Type 1 Receptor Blockade by Telmisartan Prevents Stress-Induced Impairment of Memory via HPA Axis Deactivation and up-Regulation of Brain-Derived Neurotrophic Factor Gene Expression. Pharmacol. Biochem. Behav. 2016, 148, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Suresh, B.; Ramanathan, M. Differential Anxiolytic Effect of Enalapril and Losartan in Normotensive and Renal Hypertensive Rats. Physiol. Behav. 2003, 78, 585–591. [Google Scholar] [CrossRef]
- Nade, V.S.; Kawale, L.A.; Valte, K.D.; Shendye, N.V. Cognitive Enhancing Effect of Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers on Learning and Memory. Indian J. Pharmacol. 2015, 47, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Pavel, J.; Benicky, J.; Murakami, Y.; Sanchez-Lemus, E.; Saavedra, J.M. Peripherally Administered Angiotensin II AT1 Receptor Antagonists Are Anti-Stress Compounds in Vivo. Ann. N. Y. Acad. Sci. 2008, 1148, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, M.P.; Nikkilä, O.; Lågas, S.; Kolehmainen, S.; Castrén, E. Culturing Primary Neurons from Rat Hippocampus and Cortex. Neuronal Signal. 2019, 3, 567. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Shen, W.-H.; Lu, T.-J.; Zhou, Y.; Chen, Q.; Wang, Z.; Xiang, T.; Zhu, Y.-C.; Zhang, C.; Duan, S.; et al. Clathrin-Dependent Endocytosis Is Required for TrkB-Dependent Akt-Mediated Neuronal Protection and Dendritic Growth. J. Biol. Chem. 2008, 283, 13280–13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantamäki, T.; Vesa, L.; Antila, H.; Di Lieto, A.; Tammela, P.; Schmitt, A.; Lesch, K.-P.; Rios, M.; Castrén, E. Antidepressant Drugs Transactivate TrkB Neurotrophin Receptors in the Adult Rodent Brain Independently of BDNF and Monoamine Transporter Blockade. PLoS ONE 2011, 6, e20567. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal Administration of CNS Therapeutics to Awake Mice. J. Vis. Exp. 2013, 74, 4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, B.; Zwaka, H.; Bartels, R.; Lushchak, O.; Franke, K.; Endres, T.; Fendt, M.; Song, I.; Bakr, M.; Budragchaa, T.; et al. Memory Enhancement by Ferulic Acid Ester across Species. Sci. Adv. 2018, 4, eaat6994. [Google Scholar] [CrossRef] [Green Version]
- Hedges, L.V.; Olkin, I. Statistical Methods for Meta-Analysis; Academic Press: Cambridge, MA, USA, 2014; ISBN 9780080570655. [Google Scholar]
- Reinecke, A.; Browning, M.; Klein Breteler, J.; Kappelmann, N.; Ressler, K.J.; Harmer, C.J.; Craske, M.G. Angiotensin Regulation of Amygdala Response to Threat in High-Trait-Anxiety Individuals. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 826–835. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Vehicle | C21 | |
|---|---|---|
| %OAT | 45.08 ± 7.30 | 41.25 ± 7.22 |
| %OAE | 49.20 ± 5.39 | 46.01 ± 4.66 |
| EAE (number) | 54.50 ± 9.75 | 60.67 ± 5.47 |
| dist trav (cm) | 1838.28 ± 333.71 | 1833.49 ± 154.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laukkanen, L.; Diniz, C.R.A.F.; Foulquier, S.; Prickaerts, J.; Castrén, E.; Casarotto, P.C. Facilitation of TRKB Activation by the Angiotensin II Receptor Type-2 (AT2R) Agonist C21. Pharmaceuticals 2021, 14, 773. https://doi.org/10.3390/ph14080773
Laukkanen L, Diniz CRAF, Foulquier S, Prickaerts J, Castrén E, Casarotto PC. Facilitation of TRKB Activation by the Angiotensin II Receptor Type-2 (AT2R) Agonist C21. Pharmaceuticals. 2021; 14(8):773. https://doi.org/10.3390/ph14080773
Chicago/Turabian StyleLaukkanen, Liina, Cassiano R. A. F. Diniz, Sebastien Foulquier, Jos Prickaerts, Eero Castrén, and Plinio C. Casarotto. 2021. "Facilitation of TRKB Activation by the Angiotensin II Receptor Type-2 (AT2R) Agonist C21" Pharmaceuticals 14, no. 8: 773. https://doi.org/10.3390/ph14080773
APA StyleLaukkanen, L., Diniz, C. R. A. F., Foulquier, S., Prickaerts, J., Castrén, E., & Casarotto, P. C. (2021). Facilitation of TRKB Activation by the Angiotensin II Receptor Type-2 (AT2R) Agonist C21. Pharmaceuticals, 14(8), 773. https://doi.org/10.3390/ph14080773