
Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase

 , , , , , and
, , , , , and
Abstract
:1. Introduction
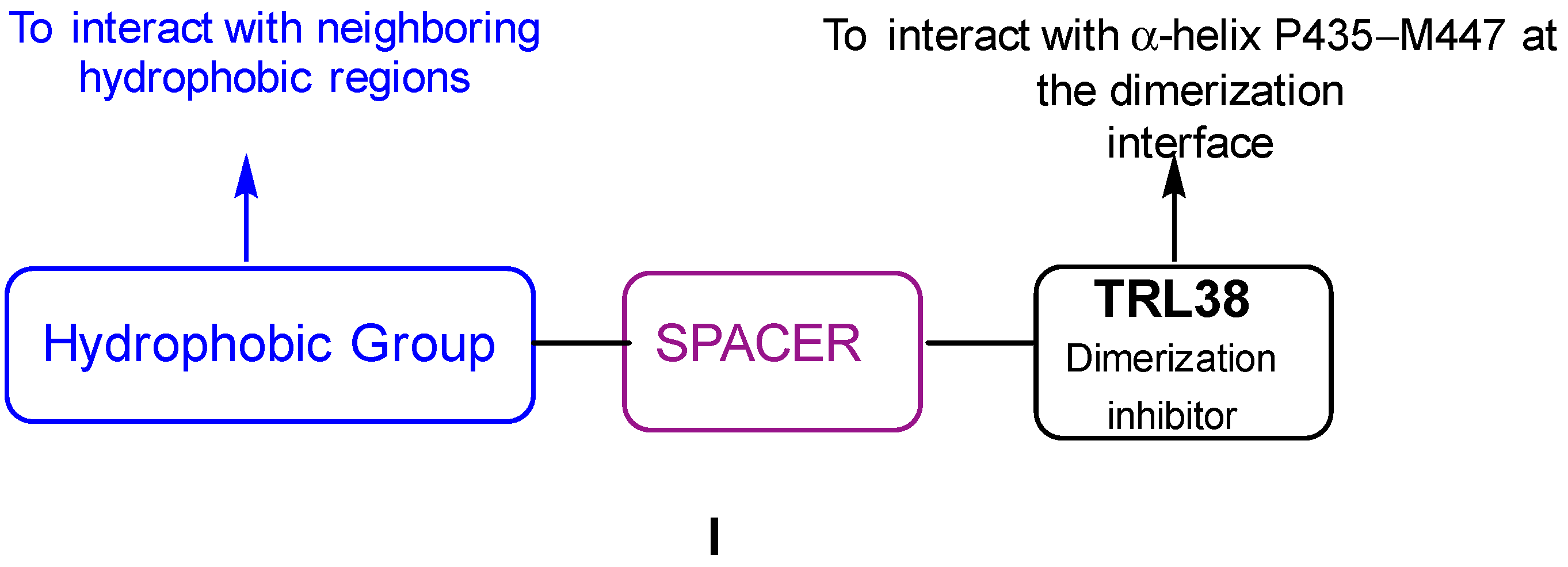
2. Results
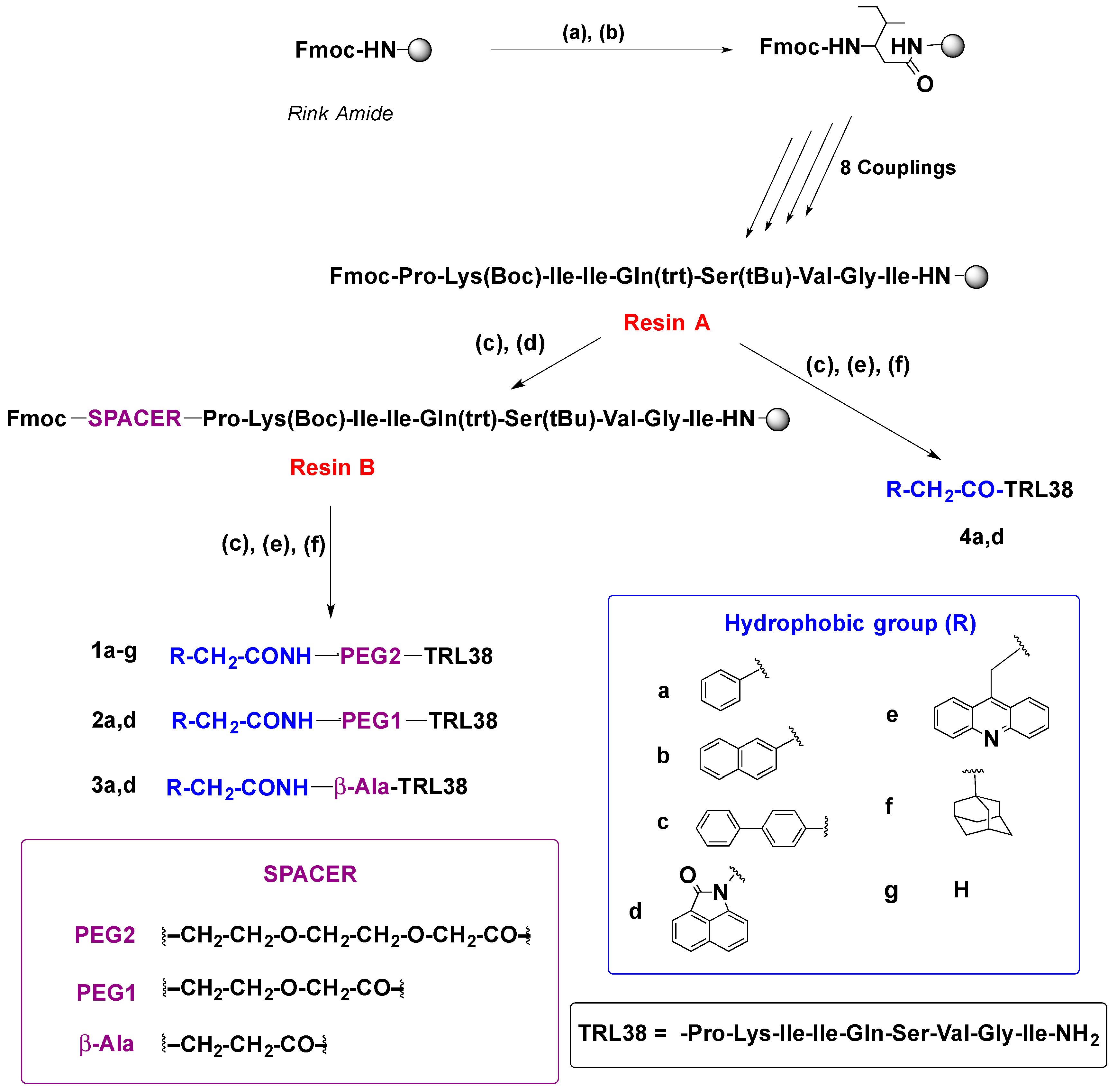
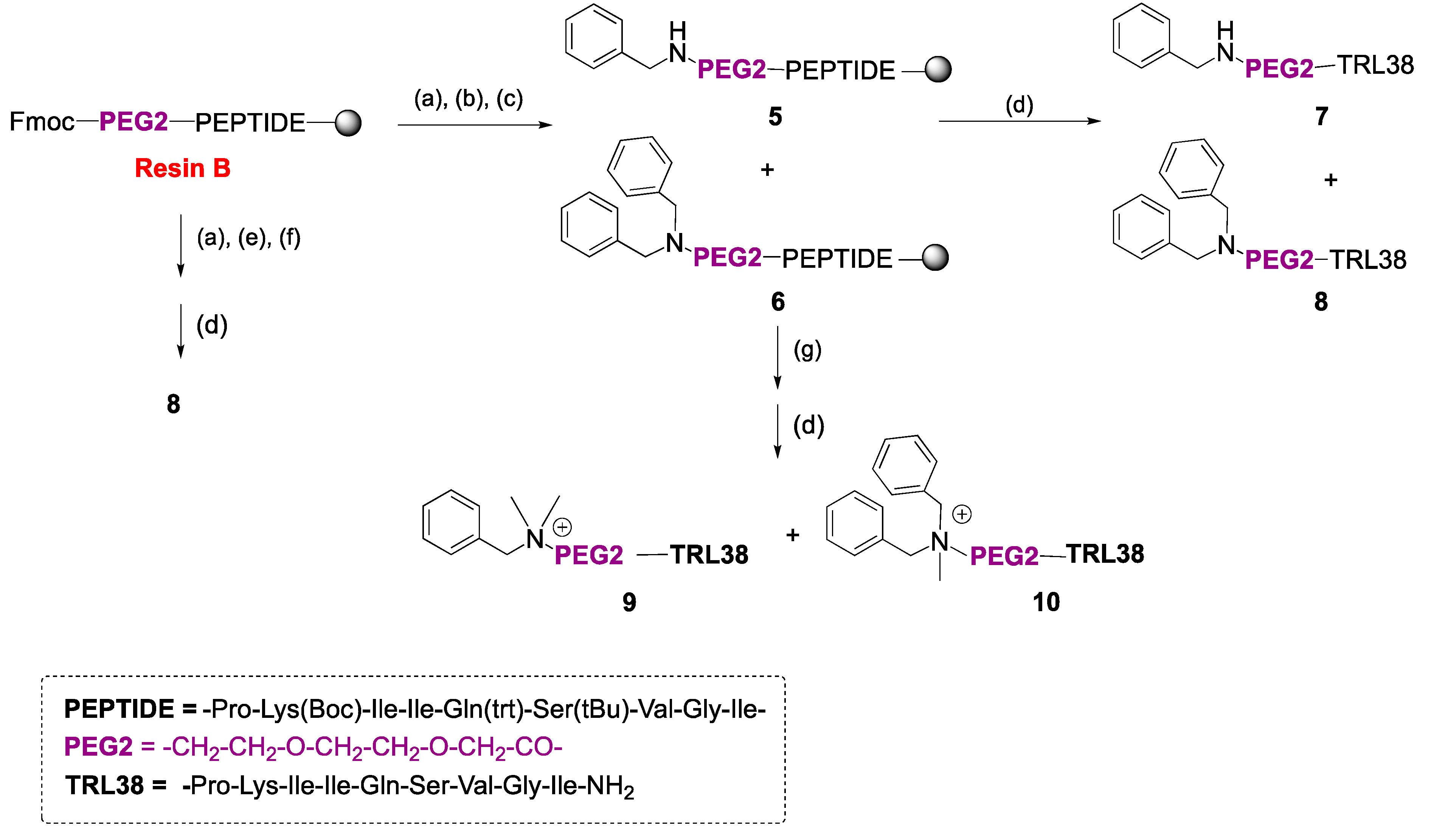
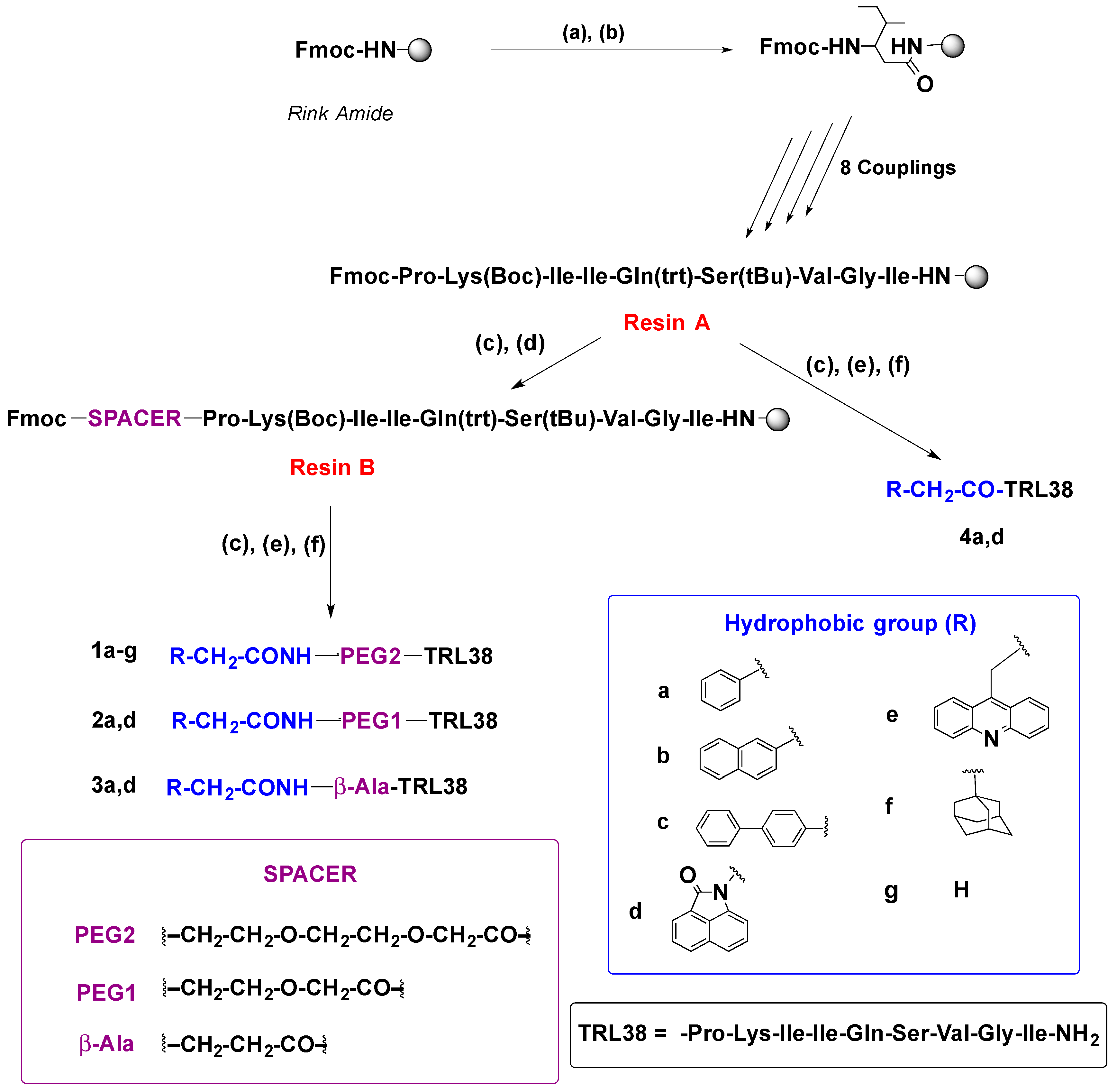
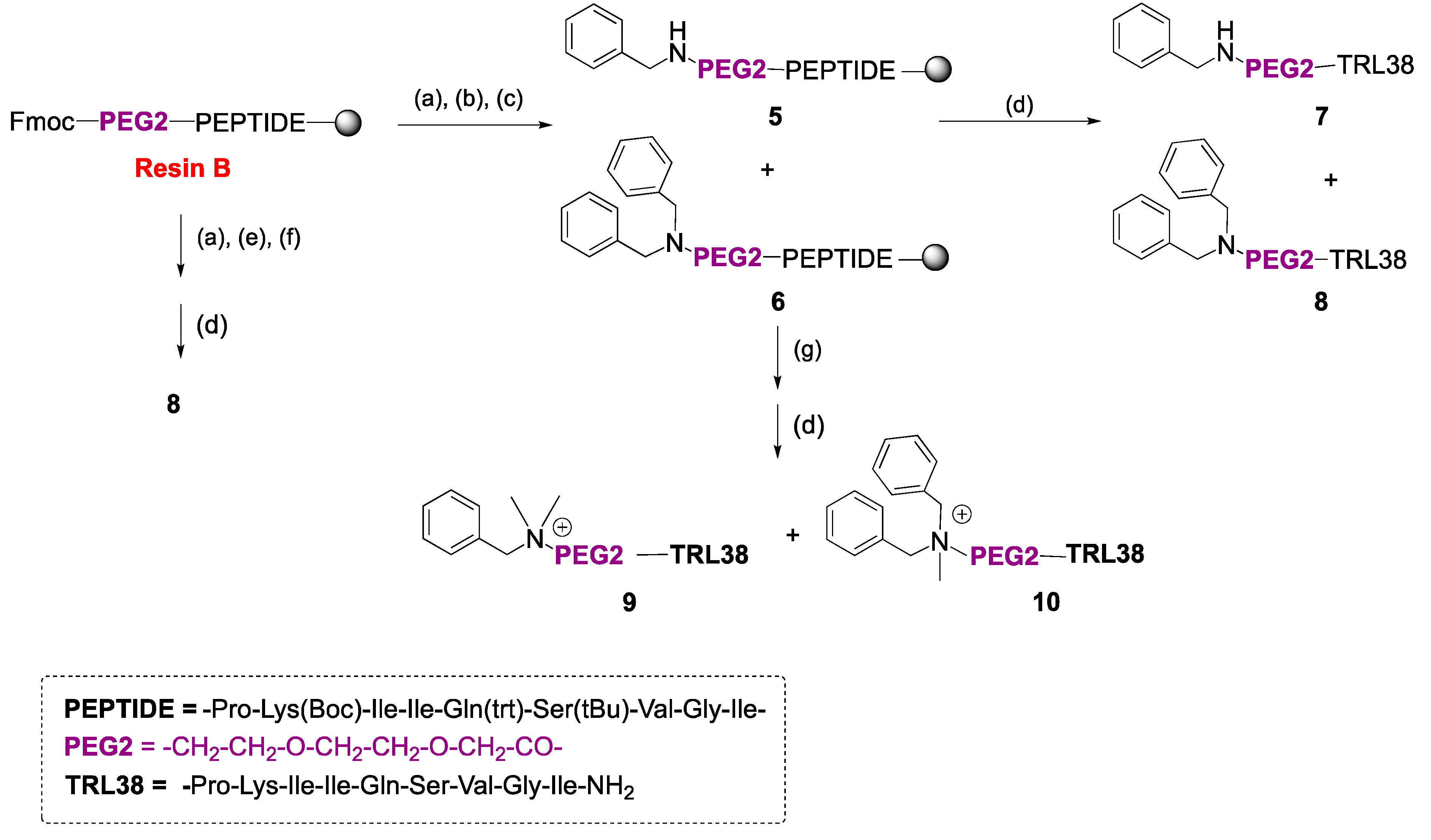
2.1. Synthesis
2.2. Biological Evaluation
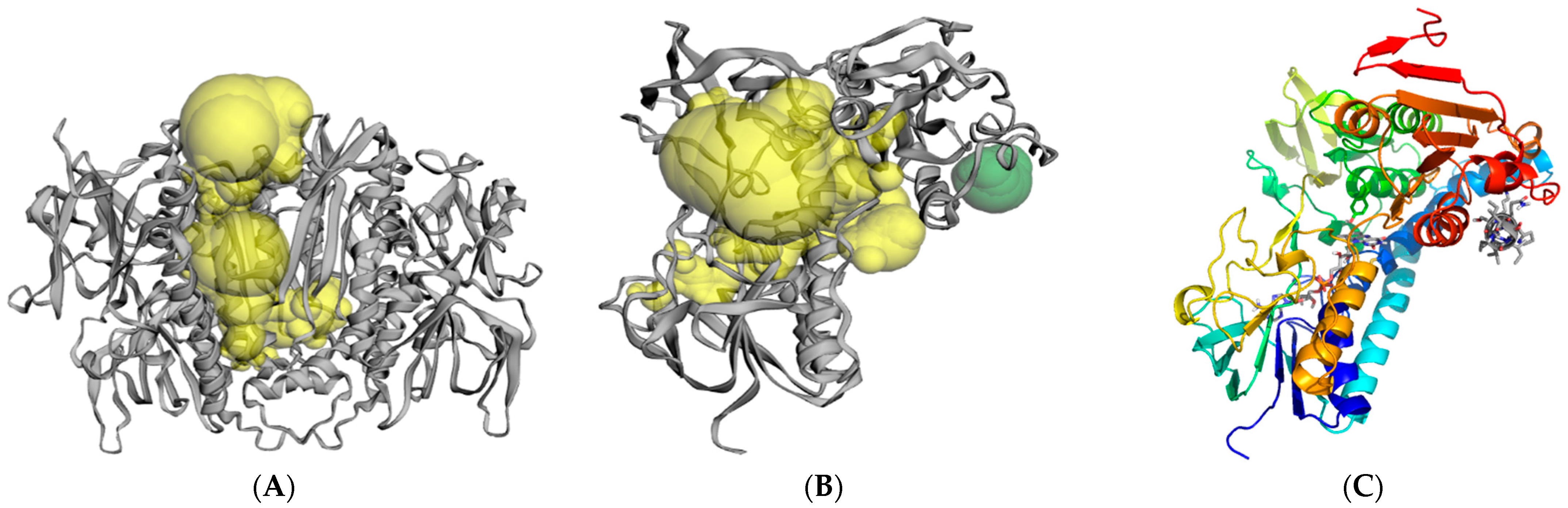
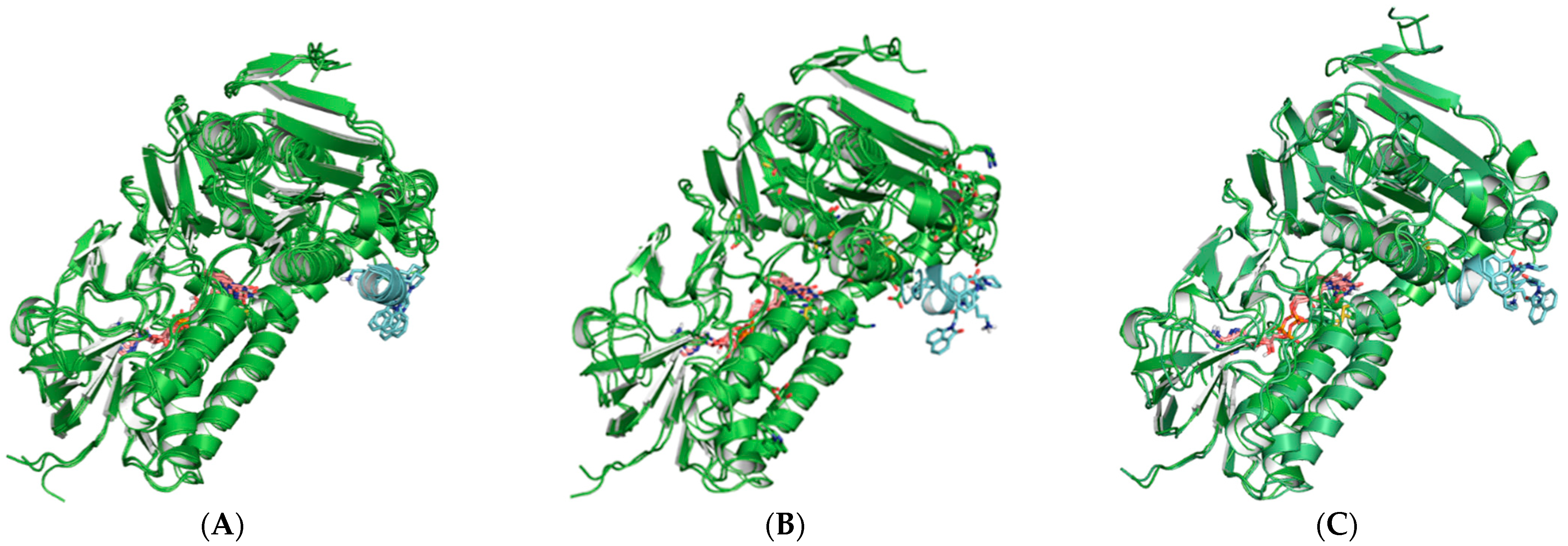
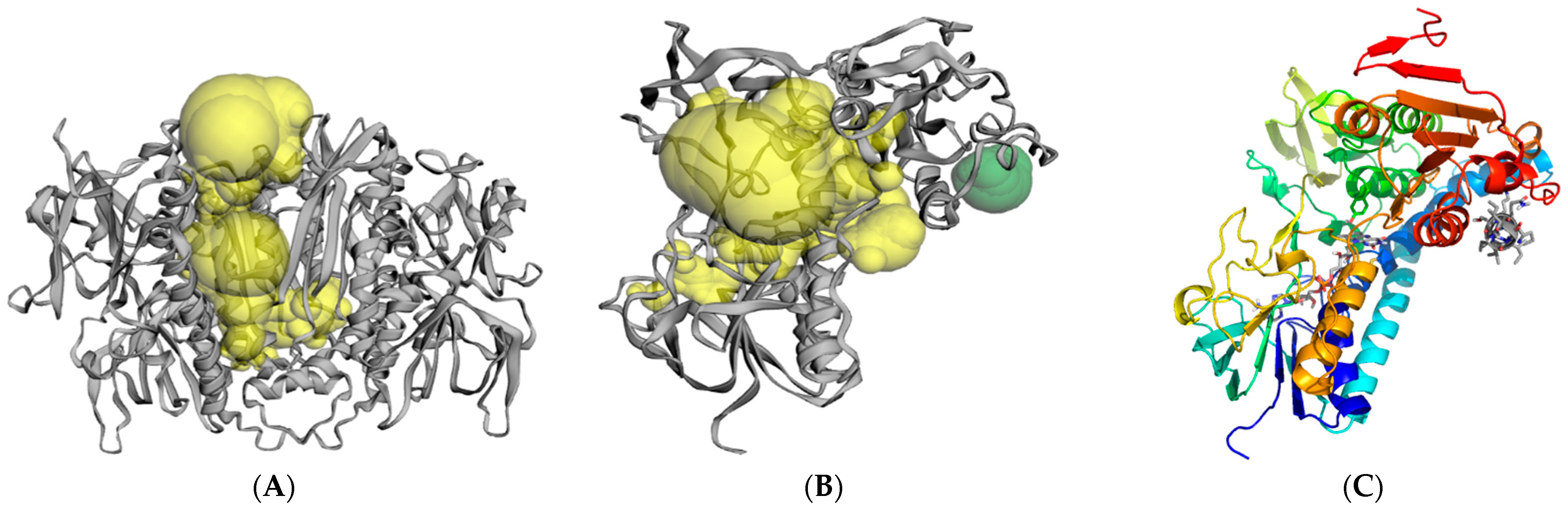
2.3. Molecular Modeling and Computer Simulations
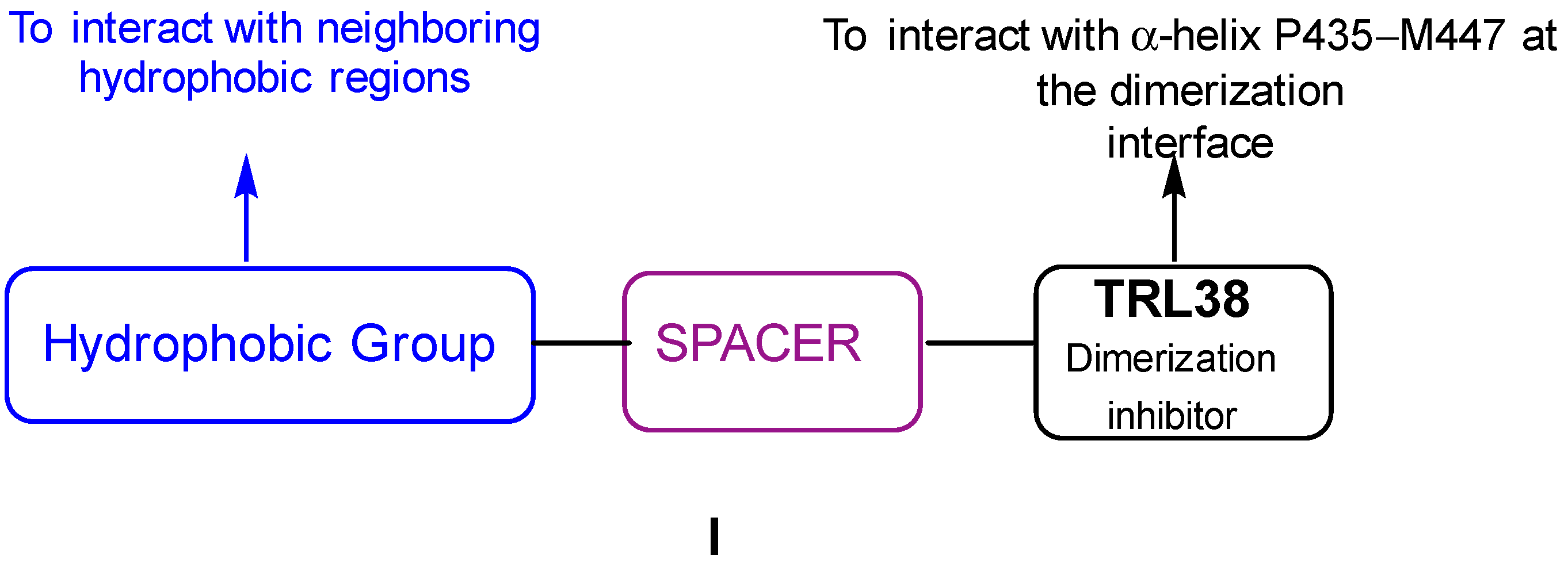
3. Discussion
4. Materials and Methods
4.1. Instrumentation and Chemicals
4.2. Syntheses
4.2.1. General Synthetic Protocols
4.2.2. Solid-Phase Synthesis of Peptide Conjugates 1a–g, 2a,d, 3a,d, 4a,d, and 7–10
4.3. Molecular Modeling and Computer Simulations
4.4. Biological Evaluation
4.4.1. LiTryR Purification
4.4.2. LiTryR Oxidoreductase Activity Assay
4.4.3. LiTryR Dimer Quantitation Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 21 June 2021).
- Saccoliti, F.; Di Santo, R.; Costi, R. Recent advancement in the search of innovative antiprotozoal agents targeting trypanothione metabolism. ChemMedChem 2020, 15, 1–17. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Ready, P.D. Epidemiology of visceral leishmaniasis. Clin. Epidemiol. 2014, 6, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, P.M.; Beaumier, C.M.; Strych, U.; Hayward, T.; Hotez, P.J.; Bottazzi, M.E. Status of vaccine research and development of vaccines for leishmanisis. Vaccine 2016, 34, 2992–2995. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, J.; Sundar, S. Current and emerging medications for the treatment of leishmaniasis. Exp. Opin. Pharmacother. 2019, 20, 1251–1265. [Google Scholar] [CrossRef]
- Alvar, J.; Arana, B.I. Appraisal of leishmaniasis chemotherapy, current status and Pipeline strategies Chapter 1 Leishmaniasis, impact and therapeutic needs. In Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–23. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Khan, F.A.K.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. Rsc. Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Ouattara, G.M.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent developments of visceral leishmaniasis treatments: Successes, pitfalls and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Bauer, H.; Schirmer, R.H. Dithiol proteins as guardians of the intracellular redox milieu in parasites: Old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew. Chem. Int. Ed. 2005, 44, 690–715. [Google Scholar] [CrossRef]
- De Rycker, M.; Baragana, B.; Duce, S.L.; Gilbert, I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature 2018, 559, 498–506. [Google Scholar] [CrossRef]
- De Gasparo, R.; Brodbeck-Persch, E.; Bryson, S.; Hentzen, N.B.; Kaiser, M.; Pai, E.F.; Krauth-Siegel, R.L.; Diederich, F. Biological evaluation and X-ray co-crystal structures of cyclohexylpyrrolidine ligands for trypanothione reductase, an enzyme from the redox metabolism of Trypanosoma. ChemMedChem 2018, 13, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, A.H.; Blackburn, P.; Ulrich, P.; Chait, B.T.; Cerami, A. Bimane cyclic esters, possible stereologues of trypanothione as antitrypanosomal agents. Bimanes 29. Science 1985, 227, 1485–1487. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Meiering, S.K.; Schmidt, H. The parasitespecific trypanothione metabolism of trypanosoma and leishmania. Biol. Chem. 2003, 384, 539–549. [Google Scholar] [CrossRef]
- Frearson, J.A.; Wyatt, P.A.; Gilbert, I.H.; Fairlamb, A.H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 23, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Leroux, A.E.; Krauth-Siegel, R.L. Thiol redox biology of trypanosomatids and potential targets for chemotherapy. Mol. Biochem. Parasitol. 2016, 206, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Dumas, C.; Oullette, M.; Tovar, J.; Cunningham, M.L.; Fairlamb, A.H.; Tamar, S.; Olivier, M.; Papadopoulou, B. Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J. 1997, 16, 2590–2598. [Google Scholar] [CrossRef] [Green Version]
- Fairlamb, A.H.; Ridley, R.G.; Vial, H.J. Drugs against Parasitic Diseases: R&D Methodologies and Issues, Discoveries and Drug Development; TDR/PRD/03.1; WHO: Geneva, Switzerland, 2003; pp. 107–118. [Google Scholar]
- Krieger, S.; Schwarz, W.; Ariyanayagam, M.R.; Fairlamb, A.H.; Krauth-Siegel, R.L.; Clayton, C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000, 35, 542–552. [Google Scholar] [CrossRef]
- Wyllie, S.; Cunningham, M.L.; Fairlamb, A.H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004, 279, 39925–39932. [Google Scholar] [CrossRef] [Green Version]
- Toro, M.A.; Sánchez-Murcia, P.A.; Moreno, D.; Ruiz-Santaquiteria, M.; Alzate, J.F.; Negri, A.; Camarasa, M.J.; Gago, F.; Velázquez, S.; Jiménez-Ruiz, A. Probing the dimerization interface of Leishmania infantum trypanothione reductase with site-directed mutagenesis and short peptides. ChemBioChem 2013, 14, 1212–1217. [Google Scholar] [CrossRef]
- Sánchez-Murcia, P.A.; Ruiz-Santaquiteria, M.; Toro, M.A.; De Lucio, H.; Jiménez, M.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; Velázquez, S. Comparison of hydrocarbon- and lactam-bridged cyclic peptides as dimerization inhibitors of Leishmania infantum trypanothione reductase. RSC Adv. 2015, 5, 55784–55794. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Santaquiteria, M.; De Castro, S.; Toro, M.A.; De Lucio, H.; Gutiérrez, K.J.; Sánchez-Murcia, P.A.; Jiménez, M.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; et al. Trypanothione reductase inhibition and anti-leishmanial activity of all-hydrocarbon stapled α-helical peptides with improved proteolytic stability. Eur. J. Med. Chem. 2018, 149, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Santaquiteria, M.; Sánchez-Murcia, P.A.; Toro, M.A.; De Lucio, H.; Gutiérrez, K.J.; De Castro, S.; Carneiro, F.A.C.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; et al. First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem. 2017, 135, 49–59. [Google Scholar] [CrossRef] [PubMed]
- De Lucio, H.; Gamo, A.M.; Ruiz-Santaquiteria, M.; De Castro, S.; Sánchez-Murcia, P.A.; Toro, M.A.; Gutiérrez, K.J.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; et al. Improved proteolytic stability and potent activity against Leishmania infantum trypanothione reductase of α/β-peptide foldamers conjugated to cell-penetrating peptides. Eur. J. Med. Chem. 2017, 140, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Revuelto, A.; Ruiz-Santaquiteria, M.; De Lucio, H.; Gamo, A.; Carriles, A.A.; Gutiérrez, K.J.; Sánchez-Murcia, P.A.; Hermoso, J.A.; Gago, F.; Camarasa, M.J.; et al. Pyrrolopyrimidine vs imidazole-phenyl-thiazole scaffolds in nonpeptidic dimerization inhibitors of Leishmania infantum trypanothione reductase. ACS Infect. Dis. 2019, 5, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revuelto, A.; de Lucio, H.; García-Soriano, J.C.; Sánchez-Murcia, P.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; Velázquez, S. Efficient dimerization disruption of leishmania infantum trypanothione reductase by triazole-phenyl-thiazoles. J. Med. Chem. 2021, 64, 6137–6160. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.L.; Denny, P.W. Antimicrobial peptides for leishmaniasis. Curr. Opin. Investig. Drugs 2010, 11, 868–875. [Google Scholar] [CrossRef]
- Robles-Loaiza, A.A.; Pinos-Tamayo, E.A.; Mendes, B.; Teixeira, C.; Cláudia Alves, C.; Gomes, P.; Almeida, J.R. Peptides to tackle leishmaniasis: Current status and future directions. Int. J. Mol. Sci. 2021, 22, 4400. [Google Scholar] [CrossRef]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Marder, O.; Shvo, Y.; Albericio, F. HCTU and TCTU. New coupling reagents: Development and industrial aspects. Chim. Oggi. 2002, 20, 37–41. [Google Scholar] [CrossRef]
- Gellerman, G.; Gaisin, V.; Brider, T. One-pot derivatization of medicinally important 9-aminoacridines by reductive amination and SNAr reaction. Tetrahedrom Lett. 2010, 51, 836–839. [Google Scholar] [CrossRef]
- Hamilton, C.J.; Saravanamuthu, A.; Eggleston, I.M.; Fairlamb, A.H. Ellman’s-reagent-mediated regeneration of trypanothione in situ: Substrate-economical microplate and time-dependent inhibition assays for trypanothione reductase. Biochem. J. 2003, 369, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Lucio, H.; Toro, M.A.; Camarasa, M.J.; Velázquez, S.; Gago, F.; Jiménez-Ruiz, A. Pseudoirreversible slow-binding inhibition of trypanothione reductase by a protein-protein interaction disruptor. Br. J. Pharmacol. 2020, 177, 5163–5176. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | R | Spacer Type | ||
|---|---|---|---|---|
| TRL38 b | >25 (65.1%) e,* | >25 (61.4%) e,& | ||
| Amide Link | ||||
| 1a |  | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 22.0 ± 0.7 | 9.4 ± 0.1 |
| 1b |  | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 18.2 ± 1.0 | 8.7 ± 0.5 |
| 1c |  | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 16.6 ± 3.8 | 22.9 ± 1.4 |
| 1d | 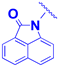 | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 6.7 ± 0.4 | 7.8 ± 0.1 |
| 1e | 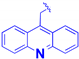 | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 14.6 ± 3.8 | 5.5 ± 0.5 |
| 1f | 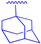 | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 7.6 ± 1.2 | 8.1 ± 0.2 |
| 3g | H | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 13.4 ± 1.7 | 21.1 ± 1.6 |
| 2a |  | -CH2-CH2-O-CH2-CO- | 22.3 ± 1.5 | 15.0 ± 0.6 |
| 2d | 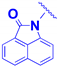 | -CH2-CH2-O-CH2-CO- | 6.9 ± 1.9 | 7.1 ± 0.1 |
| 3a |  | -CH2-CH2-CO- | 20.6 ± 1.6 | 12.7 ± 1.1 |
| 3d | 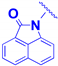 | -CH2-CH2-CO- | 10.1 ± 1.5 | 5.3 ± 0.9 |
| 4a |  | No spacer | 10.3 ± 1.8 | 11.4 ± 0.1 |
| 4d | 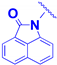 | No spacer | 8.0 ± 1.5 | 7.1 ± 0.2 |
| Aminoalkyl link | ||||
| 7 | Ph-CH2N | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 8.3 ± 1.4 | 12.6 ± 0.6 |
| 8 | (Ph-CH2)2N | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 9.8 ± 2.3 | 7.1 ± 0.5 |
| 9 | Ph-CH2N+(Me)2 | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 18.3 ± 2.6 | >25 (53.3 %) & |
| 10 | (Ph-CH2)2N+Me | -CH2-CH2-O-CH2-CH2-O-CH2-CO- | 9.2 ± 1.1 | 21.2 ± 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revuelto, A.; López-Martín, I.; de Lucio, H.; García-Soriano, J.C.; Zanda, N.; de Castro, S.; Gago, F.; Jiménez-Ruiz, A.; Velázquez, S.; Camarasa, M.-J. Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase. Pharmaceuticals 2021, 14, 689. https://doi.org/10.3390/ph14070689
Revuelto A, López-Martín I, de Lucio H, García-Soriano JC, Zanda N, de Castro S, Gago F, Jiménez-Ruiz A, Velázquez S, Camarasa M-J. Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase. Pharmaceuticals. 2021; 14(7):689. https://doi.org/10.3390/ph14070689
Chicago/Turabian StyleRevuelto, Alejandro, Isabel López-Martín, Héctor de Lucio, Juan Carlos García-Soriano, Nicola Zanda, Sonia de Castro, Federico Gago, Antonio Jiménez-Ruiz, Sonsoles Velázquez, and María-José Camarasa. 2021. "Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase" Pharmaceuticals 14, no. 7: 689. https://doi.org/10.3390/ph14070689
APA StyleRevuelto, A., López-Martín, I., de Lucio, H., García-Soriano, J. C., Zanda, N., de Castro, S., Gago, F., Jiménez-Ruiz, A., Velázquez, S., & Camarasa, M.-J. (2021). Small Molecule–Peptide Conjugates as Dimerization Inhibitors of Leishmania infantum Trypanothione Disulfide Reductase. Pharmaceuticals, 14(7), 689. https://doi.org/10.3390/ph14070689