
Preclinical Studies in Anti-Trypanosomatidae Drug Development

 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
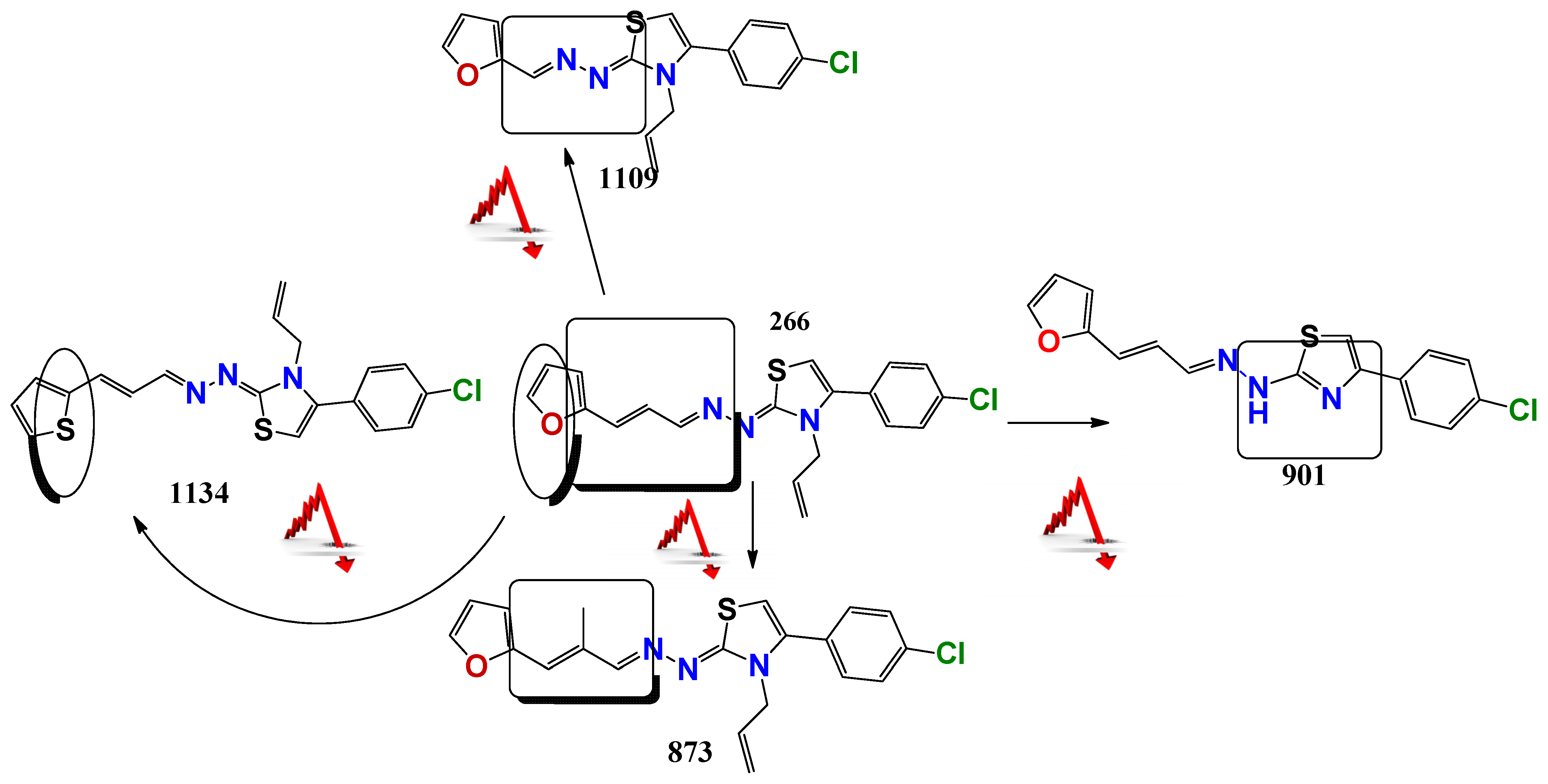
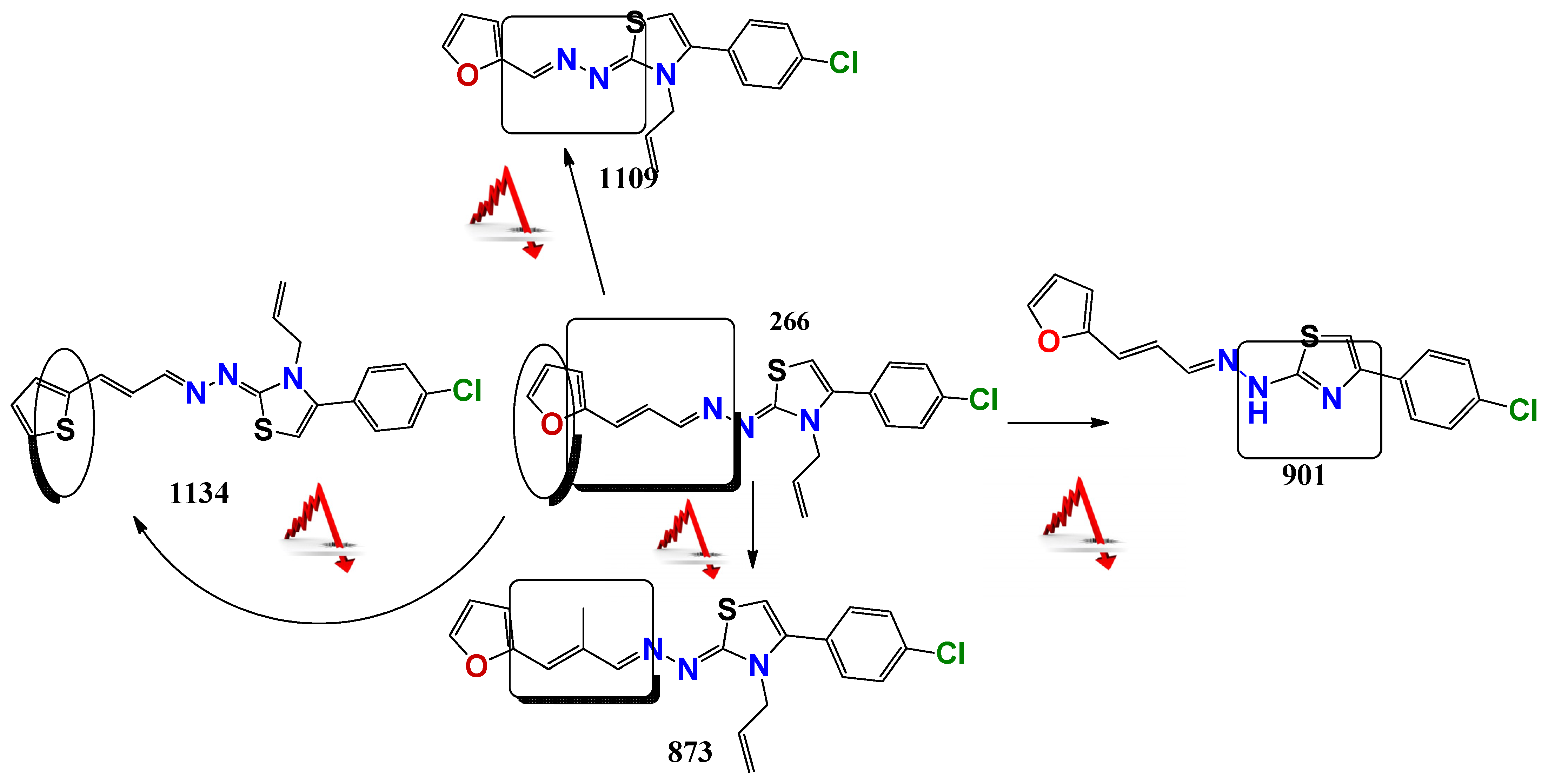
2.1. Chemistry
2.2. Antiparasitic Activity In Vitro
2.3. Toxicology
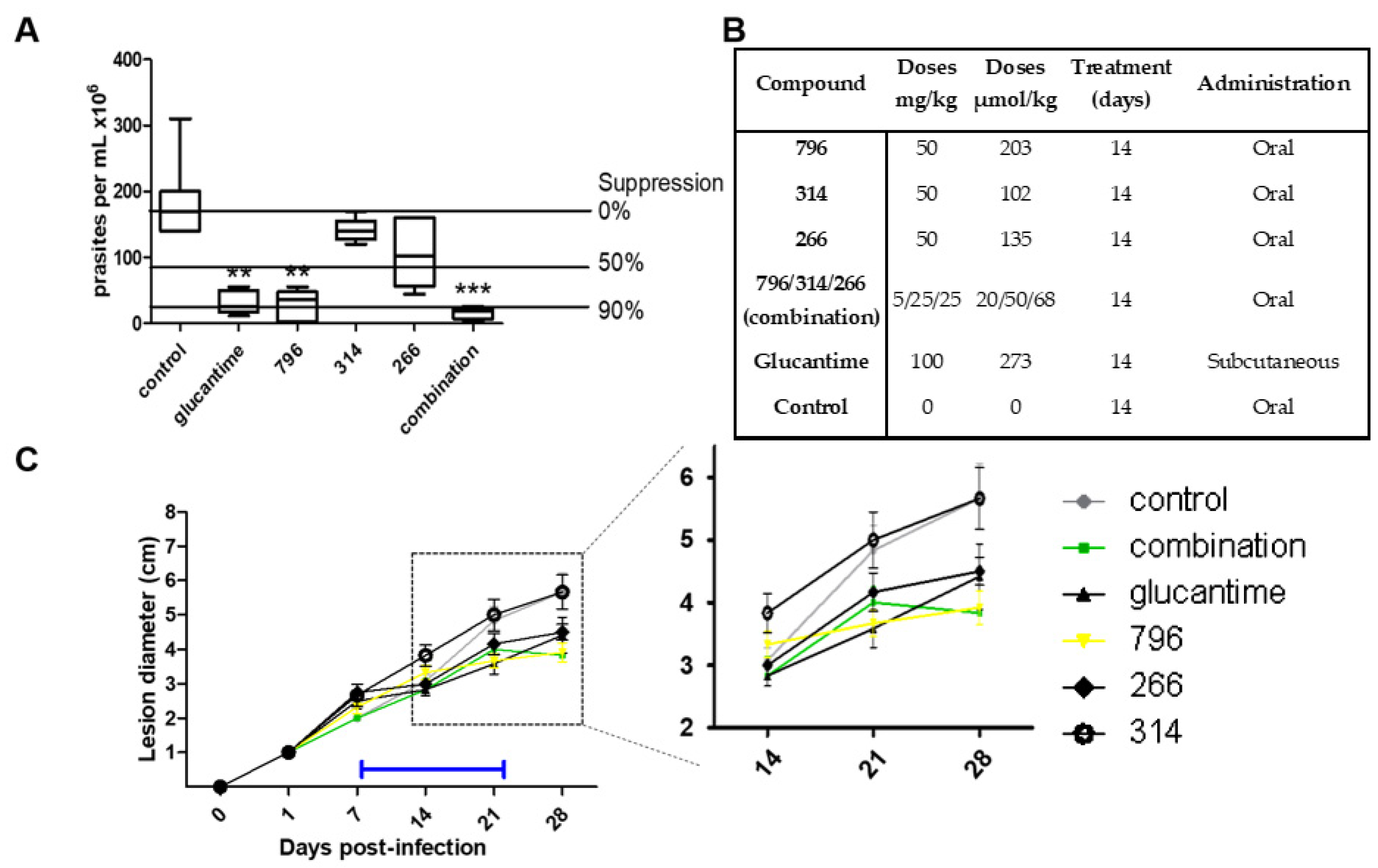
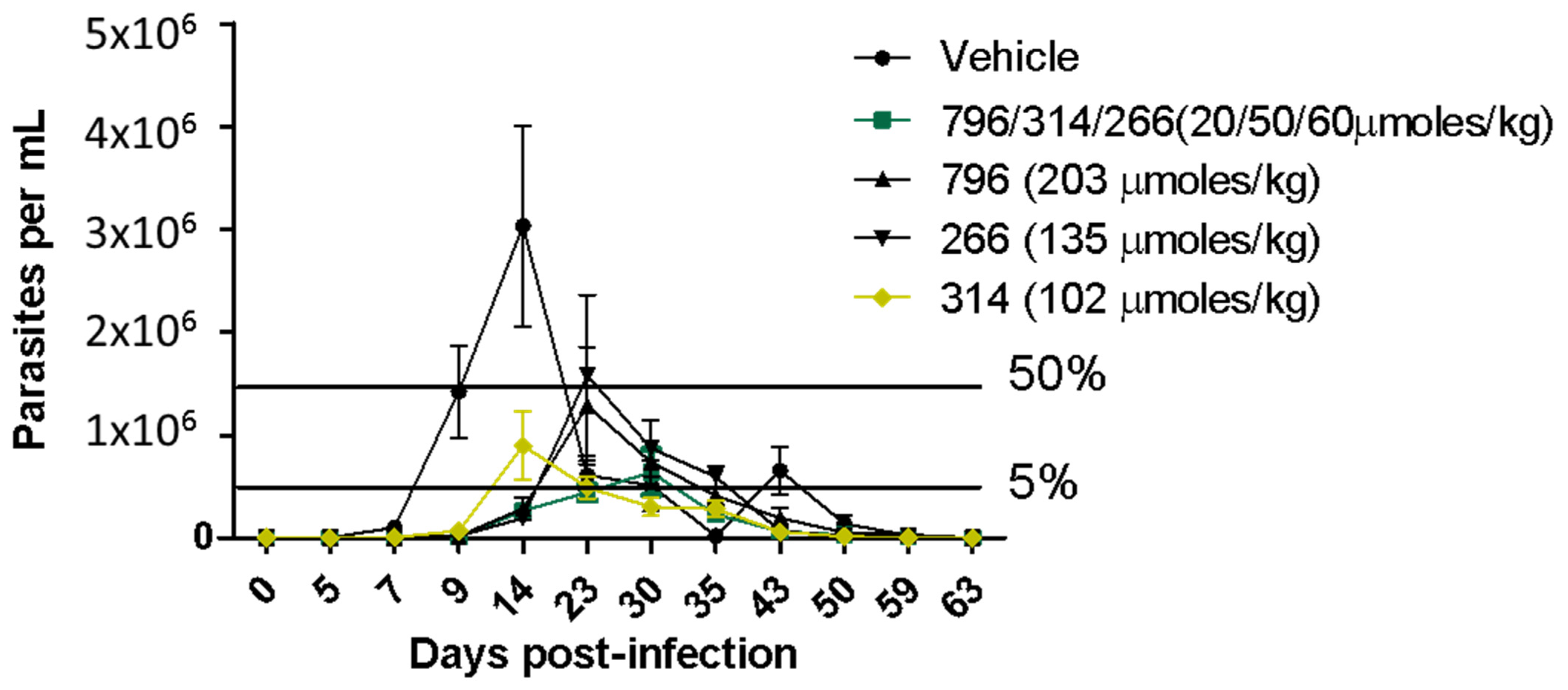
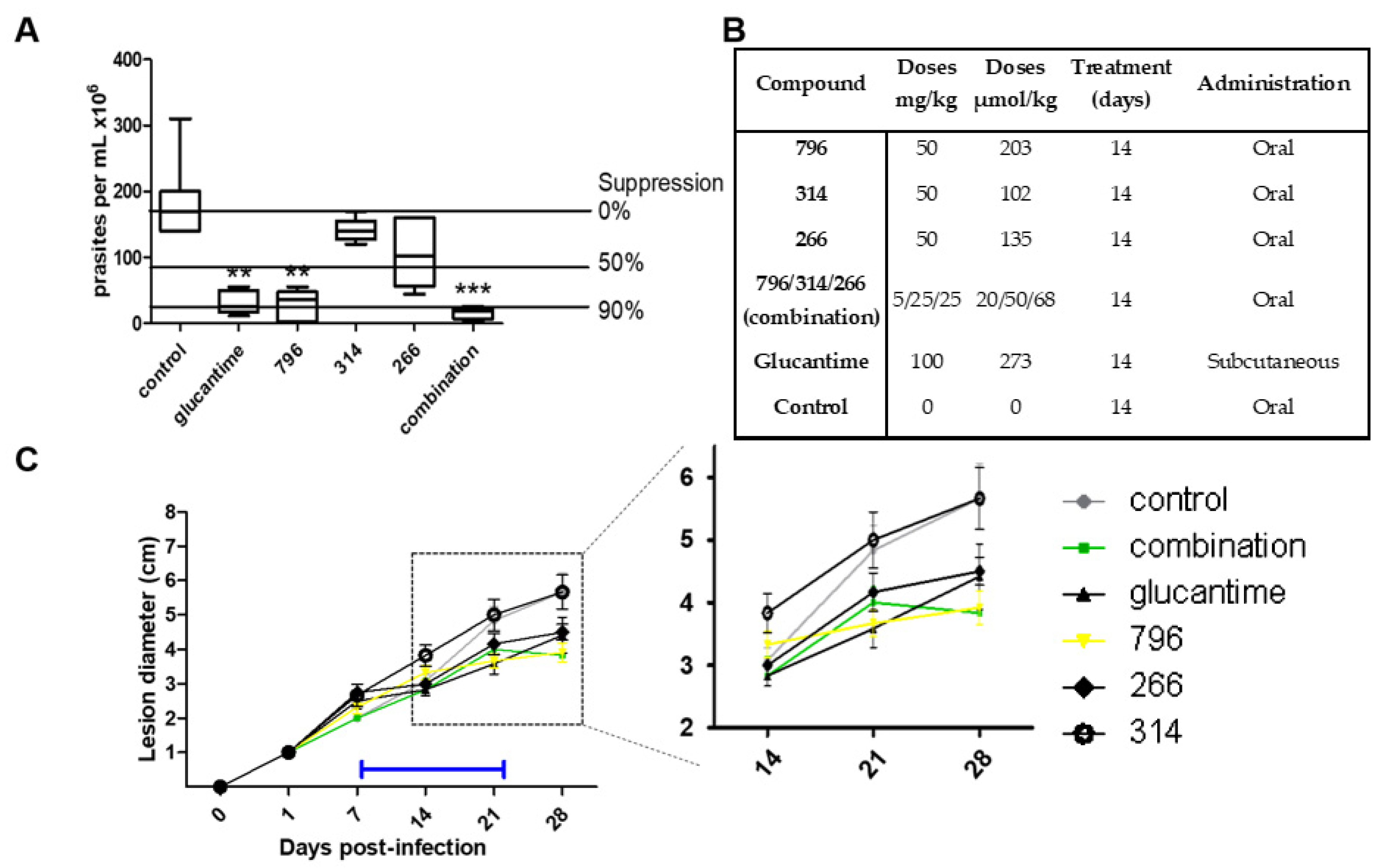
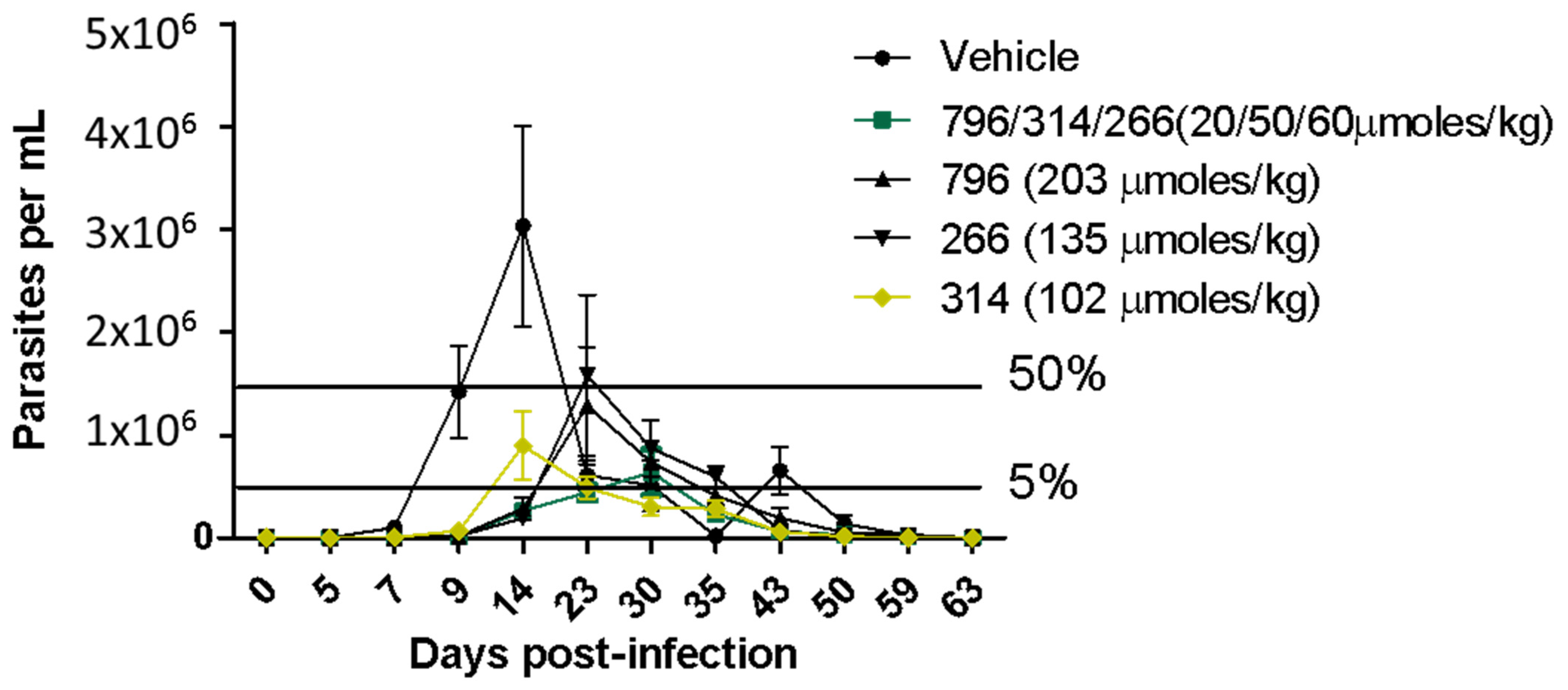
2.4. In Vivo Proof of Concept
3. Material and Methods
3.1. Cell Culturing
3.2. In Vitro Antiparasitic Activity
3.3. Nonspecific In Vitro Cytotoxicity in Mammalian Cells
3.4. Vehicles/Formulation Preparation
3.5. In Vivo Micronucleus Test
3.6. In Vivo Acute Oral Toxicity in Mice
3.7. In Vivo Anti-Leishmania Studies in Cutaneous Mice Model
3.8. In Vivo Studies in the Acute Model of Chagas Disease in Mice
3.9. Liver Fraction Stability Studies and Calculation of Pharmacokinetic Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Ending the Neglect to Attain the Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2020; Available online: https://apps.who.int/iris/handle/10665/70809 (accessed on 5 October 2018).
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Ozgoren, A.A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Lescure, F.-X.; Le Loup, G.; Freilij, H.; Develoux, M.; Paris, L.; Brutus, L.; Pialoux, G. Chagas disease: Changes in knowledge and management. Lancet Infect. Dis. 2010, 10, 556–570. [Google Scholar] [CrossRef]
- Aguilera, E.; Álvarez, G.; Cerecetto, H.; Gonzalez, M. Polypharmacology in the Treatment of Chagas Disease. Curr. Med. Chem. 2019, 26, 4476–4489. [Google Scholar] [CrossRef] [PubMed]
- Esch, K.J.; Petersen, C.A. Transmission and Epidemiology of Zoonotic Protozoal Diseases of Companion Animals. Clin. Microbiol. Rev. 2013, 26, 58–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, S.R.; Kelly, J.M. Trypanocidal drugs: Mechanisms, resistance and new targets. Expert Rev. Mol. Med. 2009, 11, e31. [Google Scholar] [CrossRef]
- De Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.D.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in Development of New Treatment for Leishmaniasis. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Rojas, R.; Valderrama, L.; Valderrama, M.; Varona, M.X.; Ouellette, M.; Saravia, N.G. Resistance to Antimony and Treatment Failure in Human Leishmania (Viannia) Infection. J. Infect. Dis. 2006, 193, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Oudhia, K.; Gazanion, E.; Vergnes, B.; Oury, B.; Sereno, D. Leishmania antimony resistance: What we know what we can learn from the field. Parasitol. Res. 2011, 109, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, A.; Horn, D. Melarsoprol Resistance in African Trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, G.; Ma, P.; Elena, C.; Rivas, A.; Cuchilla, K.; Echeverr, G.; Piro, O.E.; Chorilli, M.; Leal, S.M.; Escobar, P.; et al. Optimization of Antitrypanosomatid Agents: Identification of Nonmutagenic Drug Candidates with in Vivo Activity. J. Med. Chem. 2014, 57, 3984–3999. [Google Scholar] [CrossRef]
- Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; De Bilbao, N.V.; Aguirre-López, B.; Cabrera, N.; Díaz Mazariegos, S.; et al. Potent and Selective Inhibitors of Trypanosoma cruzi Triosephosphate Isomerase with Concomitant Inhibition of Cruzipain: Inhibition of Parasite Growth through Multitarget Activity. ChemMedChem 2015, 11, 1328–1338. [Google Scholar] [CrossRef]
- Álvarez, G.; Varela, J.; Cruces, E.; Fernández, M.; Gabay, M.; Leal, S.M.; Escobar, P.; Sanabria, L.; Serna, E.; Torres, S.; et al. Identification of a New Amide-Containing Thiazole as a Drug Candidate for Treatment of Chagas’ Disease. Antimicrob. Agents Chemother. 2014, 59, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.; Perdomo, C.; Espindola, A.; Corvo, I.; Faral-Tello, P.; Robello, C.; Serna, E.; Benítez, F.; Riveros, R.; Torres, S.; et al. A Nature-Inspired Design Yields a New Class of Steroids Against Trypanosomatids. Molecules 2019, 24, 3800. [Google Scholar] [CrossRef] [Green Version]
- Veale, C.G.L. Unpacking the Pathogen Box—An Open Source Tool for Fighting Neglected Tropical Disease. ChemMedChem 2019, 14, 386–453. [Google Scholar] [CrossRef]
- Faral-Tello, P.; Greif, G.; Satragno, D.; Basmadjián, Y.; Robello, C. Leishmania infantum isolates exhibit high infectivity and reduced susceptibility to amphotericin B. RSC Med. Chem. 2020, 11, 913–918. [Google Scholar] [CrossRef]
- Cruz, A.K.; Titust, R.; Beverley, S.M. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting (tetraploid/population bioogy/aneuploidy/dihydrofolate reductase-thymidylate synthase/protozoan parasite). Proc. Natl. Acad. Sci. USA 1993, 90, 1599–1603. [Google Scholar] [CrossRef] [Green Version]
- Sterkers, Y.; Lachaud, L.; Bourgeois, N.; Crobu, L.; Bastien, P.; Pagès, M. Novel insights into genome plasticity in Eukaryotes: Mosaic aneuploidy in Leishmania. Mol. Microbiol. 2012, 86, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Laffitte, M.-C.N.; Leprohon, P.; Papadopoulou, B.; Ouellette, M. Plasticity of the Leishmania genome leading to gene copy number variations and drug resistance. F1000Research 2016, 5, 2350. [Google Scholar] [CrossRef] [Green Version]
- Tadele, M.; Abay, S.M.; Makonnen, E.; Hailu, A. Leishmania donovani Growth Inhibitors from Pathogen Box Compounds of Medicine for Malaria Venture. Drug Des. Dev. Ther. 2020, 14, 1307–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amlabu, W.E.; Antwi, C.A.; Awandare, G.; Gwira, T.M. Elucidating the possible mechanism of action of some pathogen box compounds against Leishmania donovani. PLoS Negl. Trop. Dis. 2020, 14, e0008188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, G.; Perdomo, C.; Coronel, C.; Aguilera, E.; Varela, J.; Aparicio, G.; Zolessi, F.R.; Cabrera, N.; Vega, C.; Rolón, M.; et al. Multi-Anti-Parasitic Activity of Arylidene Ketones and Thiazolidene Hydrazines against Trypanosoma cruzi and Leishmania spp. Molecules 2017, 22, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L. Reactive Metabolites in the Biotransformation of Molecules Containing a Furan Ring. Chem. Res. Toxicol. 2013, 26, 6–25. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Mäser, P.; Tadoori, L.P.; Ioset, J.-R.; Brun, R. Antiprotozoal Activity Profiling of Approved Drugs: A Starting Point toward Drug Repositioning. PLoS ONE 2015, 10, e0135556. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chen, C.; Zhang, X.; Zhang, C.; Zhong, Q.; Chen, G.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. Structure–Activity Relationship and Pharmacokinetic Studies of 1,5-Diheteroarylpenta-1,4-dien-3-ones: A Class of Promising Curcumin-Based Anticancer Agents. J. Med. Chem. 2015, 58, 4713–4726. [Google Scholar] [CrossRef] [Green Version]
- Saramago, L.; Gomes, H.; Aguilera, E.; Cerecetto, H.; González, M.; Cabrera, M.; Alzugaray, M.F.; Junior, I.D.S.V.; Da Fonseca, R.N.; Aguirre-López, B.; et al. Novel and Selective Rhipicephalus microplus Triosephosphate Isomerase Inhibitors with Acaricidal Activity. Vet. Sci. 2018, 5, 74. [Google Scholar] [CrossRef] [Green Version]
- Sierra, N.; Folio, C.; Robert, X.; Long, M.; Guillon, C.; Álvarez, G. Looking for Novel Capsid Protein Multimerization Inhibitors of Feline Immunodeficiency Virus. Pharmaceuticals 2018, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Pérez Tort, G.; Marchesi, D. Miltefosina: Una Nueva Alternativa para el Tratamiento de la Leishmaniasis Canina. Perfil Farmacológico. Rev. Vet. Argent. 2009, XXVI, 1–8. [Google Scholar]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorganic Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef]
- Macherey, A.C.; Dansette, P.M. Biotransformations Leading to Toxic Metabolites. Chemical Aspect. In The Practice of Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 2008; pp. 674–696. [Google Scholar]
- Aguilera, E.; Varela, J.; Serna, E.; Torres, S.; Yaluff, G.; De Bilbao, N.V.; Cerecetto, H.; Alvarez, G.; González, M. Looking for combination of benznidazole and Trypanosoma cruzi-triosephosphate isomerase inhibitors for Chagas disease treatment. Mem. Inst. Oswaldo Cruz 2018, 113, 153–160. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous Cultivation of Trypanosoma brucei Blood Stream Forms in a Medium Containing a Low Concentration of Serum Protein without Feeder Cell Layers Continuous Cultivation of Trypanosoma brucei Blood Stream Forms in a Medium Containing a Low Concentrati. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, F.; Corvo, I.; Bergalli, L.; Ilarraz, A.; Cabrera, M.; Gil, J.; Susuki, B.M.; Caffrey, C.R.; Timson, D.J.; Robert, X.; et al. Novel and selective inactivators of Triosephosphate isomerase with anti-trematode activity. Sci. Rep. 2020, 10, 1–13. [Google Scholar]
- Sushko, I.; Novotarskyi, S.; Ko, R.; Pandey, A.K.; Cherkasov, A.; Liu, H.; Yao, X.; Tomas, O.; Hormozdiari, F.; Dao, P.; et al. Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Ferreira, M.E.; De Arias, A.R.; De Ortiz, S.T.; Fuentes, S.; Nakayama, H.; Schinini, A.; Hocquemiller, R. In vivo efficacy of oral and intralesional administration of 2-substituted quinolines in experimental treatment of new world cutaneous leishmaniasis caused by Leishmania amazonensis. Antimicrob. Agents Chemother. 1996, 40, 2447–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boiani, M.; Merlino, A.; Gerpe, A.; Porcal, W.; Croce, F.; Depaula, S.; Rodríguez, M.A. o-Nitroanilines as major metabolic products of in microsomal and cytosolic fractions of rat hepatocytes and in whole parasitic cells. Xenobiotica 2009, 39, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Compound a | |||
|---|---|---|---|
| 314 | 1019 | 796 | 266 |
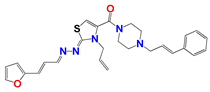 | 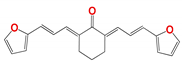 | 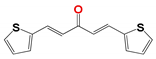 | 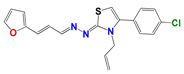 |
| Activity in vitro anti-T. cruzi (multiple strains) | |||
| bEC50 0.72 µM Amastigotes | EC50 0.60 µM epimastigotes | EC50 5.0 µM epimastigotes | EC50 > 0.25 µM amastigotes |
| Selectivity index > 100 (EC50 mammalian cell/EC50 T. cruzi) | |||
| Mechanism of action | |||
| Cruzipain cIC50 4.3 µM | triosephosphate isomerase IC50 86 nM | unknown | unknown |
| Stability in vitro (microsomal, plasma, other solutions) | |||
| High | low | moderate | high |
| Toxicology and Efficacy | |||
| Ames Test (mutagenicity) | |||
| No | no | unknown | no |
| Micronucleus test in mice (Genotoxicity) | |||
| No | unknown | unknown | no |
| Acute oral toxicity (up and down test) | |||
| d LD50 > 2000 mg/kg in mice | unknown | unknown | LD50 > 2000 mg/kg in mice |
| Full control of the parasitemia in vivo at 50 mg/kg in the murine model of Chagas disease | |||
| Compounds | EC50 (μM) | Selectivity Index b | |||
|---|---|---|---|---|---|
| L. infantuma | MΦ Cytotoxicity a | MΦ/Reference | MΦ/Veterinary | ||
| Reference | Veterinary | ||||
| MMV272144 | 2.4 ± 0.3 | 1.2 ± 0.1 | >50 | >21 | >42 |
| MMV688761 | 4.9 ± 0.1 | 3.9 ± 0.3 | >50 | >10 | >13 |
| MMV688768 | 9.8 ± 0.5 | 6.8 ± 0.1 | >50 | >5 | >7 |
| MMV688763 | 2.1 ± 0.2 | 0.9 ± 0.1 | >50 | >24 | >56 |
| MMV021013 | 0.4 ± 0.1 | 0.3 ± 0.1 | >50 | >125 | >167 |
| Miltefosine c | 5.3 ± 0.1 | 4.1 ± 0.1 | 50 ± 7 | 10 | 12 |
| Chemical Code | EC50 ± SD (μM) a;b | |||||
|---|---|---|---|---|---|---|
| Lin–Ref | Lin–Vet | L. amaz | L. brazilsc | T. cruzic | T. brucei | |
| Nifurtimox d | 6.0 ± 1.0 | 10.0 ± 2.0 | 6.0 ± 2.0 | 7.0 ± 2.0 | 1.44.0 e | |
| Glucantime d | 26.0 ± 1.0 | 18.0 ± 2.0 | 20.0 ± 9.0 | |||
| Miltefosine d | 5.3 ± 0.1 f | 4.1 ± 0.1 f | 5 ± 2 | 8 ± 2 | ||
| 1385 | 9.0 ± 1.0 | 19.0 ± 3.0 | ||||
| 1109 | ||||||
| 266 | 2.0 ± 0.2 | 9.0 ± 1.0 | 7.0 ± 1.0 | 20.0 ± 2.0 | 1.6 ± 0.5 | 5.0 ± 1.0 |
| 872 | 10.0 ± 5.0 | 10.0 ± 1.0 | 8.0 ± 2.0 | 3.0 ± 0.5 | 6.0 ± 1.0 | |
| 873 | 0.09 ± 0.02 | 14.0 ± 3.0 | ||||
| 1153 | <3.0 | 6.0 ± 1.0 | ||||
| 295 | 3.5 ± 0.2 | |||||
| 133 | ||||||
| 877 | 15 ± 3 | 10 ± 1 | ||||
| 1134 | ||||||
| 314 | 1.3 ± 0.5 | 2.5 ± 1.0 | 12.0 ± 5.0 | 4.0 ± 1.0 | 3.1 ± 0.2 | 5.0 ± 1.0 |
| 1112 | 1.5 ± 0.2 | 1.2 ± 0.2 | 12.0 ± 3.0 | |||
| 1115 | 11.0 ± 4.0 | 7.0 ± 2.0 | ||||
| 901 | 1.2 ± 0.3 | 5.0 ± 1.0 | ||||
| 1119 | <6.0 | 10.0 ± 2.0 | 12.0 ± 1.0 | |||
| 1102 | 5.0 ± 2.0 | 16.0 ± 4.0 | 14.0 ± 2.0 | 1.6 ± 0.3 | 14.0 ± 2.0 | |
| 1140 | ||||||
| 912 | <0.4 | 18.0 ± 5.0 | ||||
| 903 | 3.0 ± 1.0 | 12.0 ± 2.0 | 17.0 ± 5.0 | |||
| 263 | <0.3 | 5.0 ± 1.0 | ||||
| 909 | 10.0 | 6.0 ± 1.0 | 19.0 ± 1.0 | |||
| 1366 | 12.0 ± 2.0 | |||||
| 1367 | ||||||
| 1369 | 8.0 ± 2.0 | 15.0 ± 5.0 | ||||
| 1222 | ||||||
| 1219 | ||||||
| 1147 | 5.0 ± 1.0 | 9.0 ± 2.0 | <12.0 | |||
| 1097 | ||||||
| Chemical Code | EC50 ± SD (μM) a;b | |||||
|---|---|---|---|---|---|---|
| Lin–Ref | Lin–Vet | L. amaz | L. brazilsc | T. cruzic | T. brucei | |
| Glucantime d | 26.0 ± 1.0 | 18.0 ± 2.0 | 20.0 ± 9.0 | |||
| Curcumin | 5.0 ± 1.0 | 6.0 ± 1.0 | 6.0 ± 1.0 | 8.0 ± 1.0 | ||
| 797 | 11.0 ± 2.0 | |||||
| 799 | <0.3 | 12.0 ± 3.0 | 5.0 ± 1.0 | 4.2 ± 0.9 | 5.0 ± 1.0 | 1.7 ± 0.5 |
| 800 | 14.0 | 15.0 ± 1.0 | ||||
| 795 | 5.0 ± 1.0 | 10.0 ± 2.0 | 10.0 ± 6.0 | 6.0 ± 2.0 | 24.0 ± 2.0 | 1.8 ± 0.5 |
| 793 | <0.3 | 5.0 ± 0.7 | 17.0 ± 2.0 | |||
| 809 | 3.0 ± 1.0 | 16.0 ± 2.0 | 6.0 ± 1.0 | 8.0 ± 2.0 | ||
| 1223 | <0.3 | 18.0 ± 4.0 | 16.0 ± 4.0 | 5.0 ± 2.0 | ||
| 1019 | <0.3 | 7.0 ± 1.0 | 13.0 ± 7.0 | 0.6 ± 0.2 | ||
| 1282 | ||||||
| 796 | 4.0 ± 0.5 | 10.0 ± 2.0 | 8.0 ± 2.0 | 4.0 ± 0.5 | 5.0 ± 0.8 | 0.6 ± 0.1 |
| 1387 | 16.0 ± 2.0 | 0.7 ± 0.1 | ||||
| 1414 | 12.0 ± 1.0 | 16.0 | ||||
| 798 | 3.0 ± 0.5 | 19.0 ± 5.0 | 13 ± 1 | |||
| 1018 | <0.3 | 11.0 ± 3.0 | 0.04 ± 0.01 | 15.0 ± 2.0 | ||
| 1245 | <0.3 | 16.0 ± 1.0 | 0.6 ± 0.2 | 16.0 ± 3.0 | ||
| Chemical Code | EC50 ± SD (µM) a | Selectivity Index b | |||
|---|---|---|---|---|---|
| MΦ f | Fibroblasts | MΦ/L. amaz | MΦ/Lin–Ref | MΦ/Lin–Vet | |
| Nifurtimox c | 200 ± 9 | nd | 33 | 33 | 20 |
| Glucantime c | 15 ± 1 | nd | 0.83 | 0.57 | Nd |
| Miltefosine c | 50 ± 7 | nd | 10 | 56 | 10 |
| 266 d | 60 ± 6 | 405 ± 10 | 9 | 30 | 7 |
| 872 d | 66 ± 7 | 319 ± 16 | 7 | 8 | 2 |
| 314 d | 30 ± 5 | 346 ± 9 | 3 | 23 | 12 |
| Curcumin e | 10 ± 2 | nd | 2 | 2 | Nd |
| 795 e | 115 ± 2 | 114 ± 6 | 11 | 23 | 11 |
| 809 e | 33 ± 8 | 543 ± 6 | 2 | 6 | Nd |
| 796 e | 38 ± 7 | 158 ± 5 | 5 | 10 | 4 |
| 799 e | 115 ± 8 | nd | 23 | >63 | 10 |
| Treatment a | Number EPMn b | Number EPC c | Media Mn/Mouse ± SD d |
|---|---|---|---|
| Control | 19 | 5000 | 4 ± 1 |
| 796 | 21 | 5000 | 5 ± 1 |
| Cyclophosphamide | 180 | 5000 | 36 ± 2 |
| Compound | Solubility (mg/mL) | Gastrointestinal Absorption | BBB Permeability | Penetrability of Skin (cm/s) | Bioavailability | Lipophilicity (LogP) |
|---|---|---|---|---|---|---|
| Miltefosine | 1.9 × 10−3 | Low | no | −4.0 | 0.55 | 3.8 |
| Benznidazole | 2.3 | High | no | −7.2 | 0.55 | 0.5 |
| 314 | 3.5 × 10−3 | High | no | −6.3 | 0.55 | 4.2 |
| 266 | 2.2 × 10−3 | High | no | −5.2 | 0.55 | 4.8 |
| 796 | 3.9 × 10−2 | High | yes | −5.3 | 0.55 | 3.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdomo, C.; Aguilera, E.; Corvo, I.; Faral-Tello, P.; Serna, E.; Robello, C.; Wilkinson, S.R.; Yaluff, G.; Alvarez, G. Preclinical Studies in Anti-Trypanosomatidae Drug Development. Pharmaceuticals 2021, 14, 644. https://doi.org/10.3390/ph14070644
Perdomo C, Aguilera E, Corvo I, Faral-Tello P, Serna E, Robello C, Wilkinson SR, Yaluff G, Alvarez G. Preclinical Studies in Anti-Trypanosomatidae Drug Development. Pharmaceuticals. 2021; 14(7):644. https://doi.org/10.3390/ph14070644
Chicago/Turabian StylePerdomo, Cintya, Elena Aguilera, Ileana Corvo, Paula Faral-Tello, Elva Serna, Carlos Robello, Shane R. Wilkinson, Gloria Yaluff, and Guzmán Alvarez. 2021. "Preclinical Studies in Anti-Trypanosomatidae Drug Development" Pharmaceuticals 14, no. 7: 644. https://doi.org/10.3390/ph14070644
APA StylePerdomo, C., Aguilera, E., Corvo, I., Faral-Tello, P., Serna, E., Robello, C., Wilkinson, S. R., Yaluff, G., & Alvarez, G. (2021). Preclinical Studies in Anti-Trypanosomatidae Drug Development. Pharmaceuticals, 14(7), 644. https://doi.org/10.3390/ph14070644