
ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential



Abstract
:1. Introduction
2. Remdesivir: Chemical and Pharmacological Properties
3. ADME Properties of Remdesivir
3.1. Absorption
3.2. Distribution
3.3. Metabolism
3.4. Elimination
4. Pharmacokinetics of Remdesivir
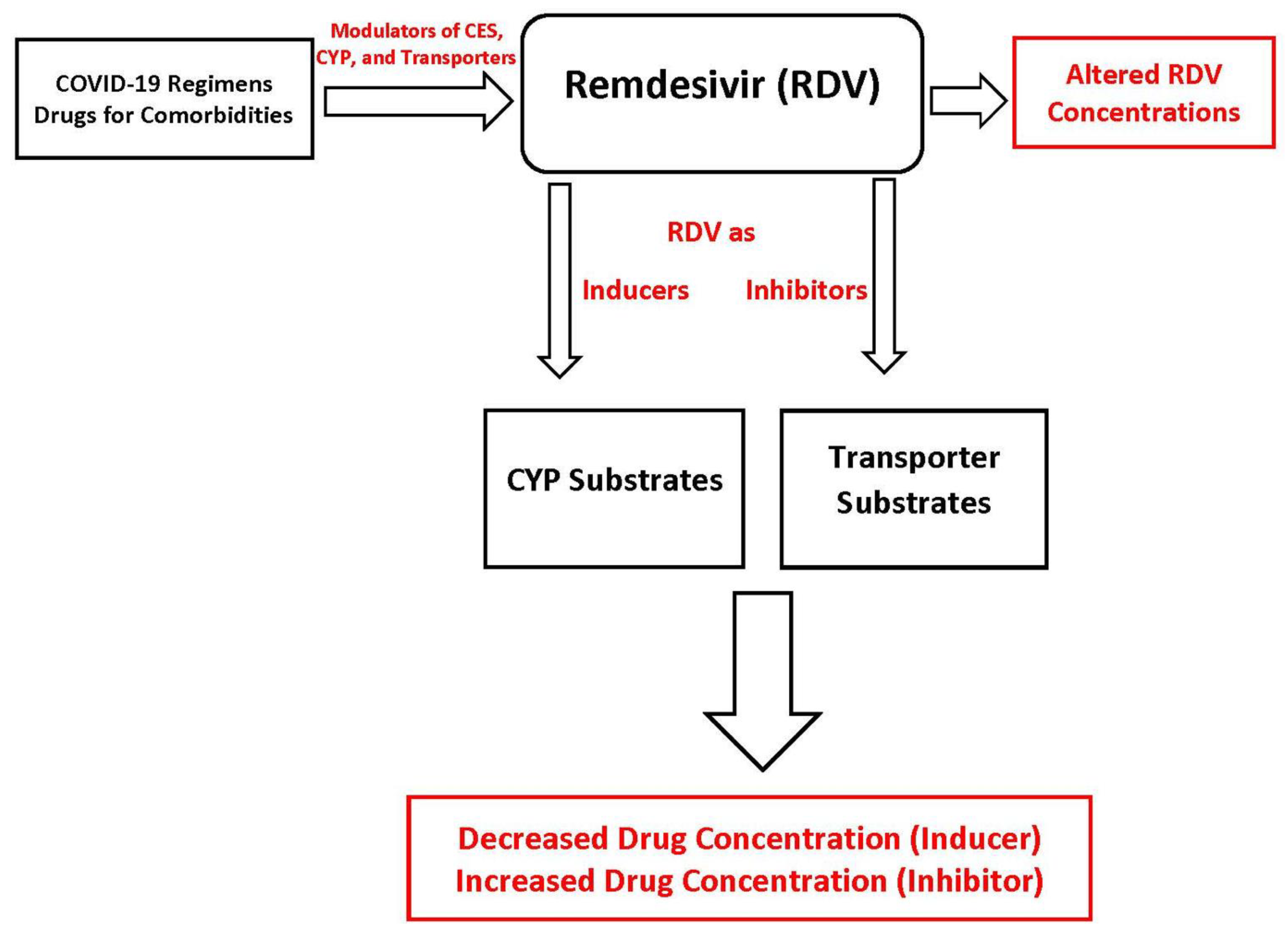
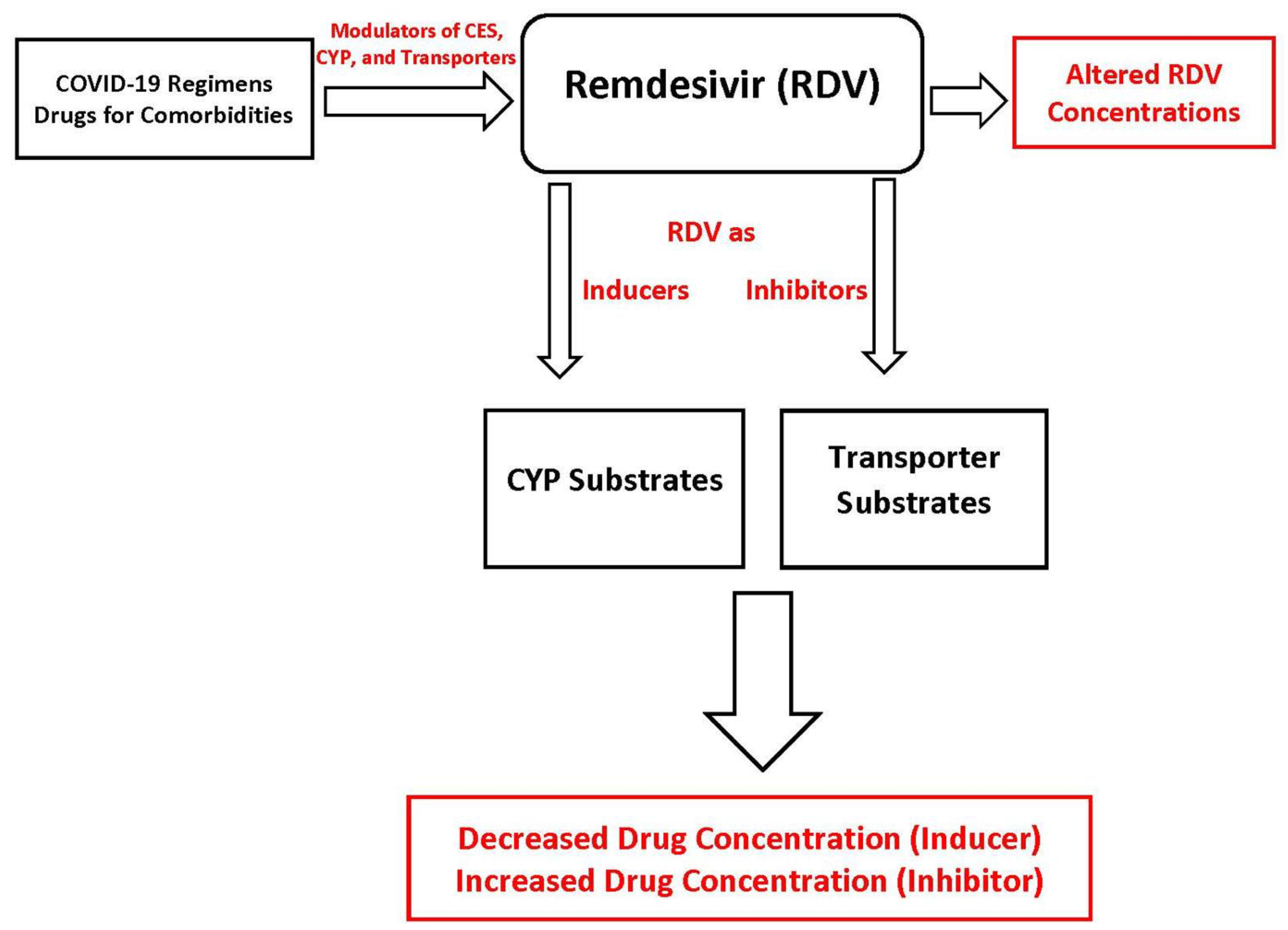
5. Potential Drug-Drug Interactions Involving Remdesivir
Remdesivir as “Victim”
6. Future Directions of Remdesivir ADME and DDI Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (WHO). WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 9 April 2021).
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutiérrez-Ocampo, E.; Villamizar-Peña, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 14 October 2020).
- Siddell, S.G.; Anderson, R.; Cavanagh, D.; Fujiwara, K.; Klenk, H.D.; Macnaughton, M.R.; Pensaert, M.; Stohlman, S.A.; Sturman, L.; van der Zeijst, B.A.M. Coronaviridae. Intervirology 1983, 20, 181–189. [Google Scholar] [CrossRef]
- Chen, J. Pathogenicity and transmissibility of 2019-nCoV-A quick overview and comparison with other emerging viruses. Microbes Infect. 2020, 22, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Chen, Y.; Qin, Q. Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicate special control measures. J. Med. Virol. 2020, 92, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). People Who Are at Higher Risk for Severe Illness. Available online: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/groups-at-higher-risk.html (accessed on 15 March 2021).
- Jean, S.-S.; Lee, P.-I.; Hsueh, P.-R. Treatment options for COVID-19: The reality and challenges. J. Microbiol. Immunol. Infect. 2020, 53, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. COVID-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccines Immunother. 2020, 16, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). R&D Blueprint and COVID-19. Available online: https://www.who.int/teams/blueprint/covid-19 (accessed on 14 October 2020).
- U.S. Food and Drug Administration. Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment#:~:text=The%20emergency%20use%20authorization%20allows,children%20hospitalized%20with%20severe%20disease (accessed on 15 March 2021).
- European Medicnes Agency. Summary on Compassionate Use of Remdesivir. Available online: https://www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf (accessed on 15 March 2021).
- Doggrell, S.A. Remdesivir, a remedy or a ripple in severe COVID-19? Expert Opin. Investig. Drugs 2020, 29, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Veklury® (Remdesivir) for Pediatric Patients. Available online: https://www.fda.gov/media/137566/download (accessed on 15 March 2021).
- VEKLURY® (Remdesivir) Package Insert. 2020. Available online: https://www.vekluryhcp.com/ (accessed on 21 March 2021).
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Lazureanu, V.; Coricovac, D.; Mioc, M.; Oancea, R.; Marcovici, I.; Pinzaru, I.; Soica, C.; Tsatsakis, A.M.; Cretu, O. SARS-CoV-2: Repurposed Drugs and Novel Therapeutic Approaches-Insights into Chemical Structure-Biological Activity and Toxicological Screening. J. Clin. Med. 2020, 9, 2084. [Google Scholar] [CrossRef] [PubMed]
- Sun, D. Remdesivir for Treatment of COVID-19: Combination of Pulmonary and IV Administration May Offer Aditional Benefit. AAPS J. 2020, 22, 77. [Google Scholar] [CrossRef]
- Sahakijpijarn, S.; Moon, C.; Koleng, J.J.; Christensen, D.J.; Williams, R.O., III. Development of Remdesivir as a Dry Powder for Inhalation by Thin Film Freezing. Pharmaceutics 2020, 12, 1002. [Google Scholar] [CrossRef] [PubMed]
- Humeniuk, R.; Mathias, A.; Cao, H.; Osinusi, A.; Shen, G.; Chng, E.; Ling, J.; Vu, A.; German, P. Safety, Tolerability, and Pharmacokinetics of Remdesivir, An Antiviral for Treatment of COVID-19, in Healthy Subjects. Clin. Transl. Sci. 2020, 13, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Yang, K. What Do We Know About Remdesivir Drug Interactions? Clin. Transl. Sci. 2020, 13, 842–844. [Google Scholar] [CrossRef]
- Tempestilli, M.; Caputi, P.; Avataneo, V.; Notari, S.; Forini, O.; Scorzolini, L.; Marchioni, L.; Bartoli, T.A.; Castilletti, C.; Lalle, E.; et al. Pharmacokinetics of remdesivir and GS-441524 in two critically ill patients who recovered from COVID-19. J. Antimicrob. Chemother. 2020, 75, 2977–2980. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Kebriaei, R.; Dresser, L.D. Remdesivir: Review of Pharmacology, Pre-clinical Data, and Emerging Clinical Experience for COVID-19. Pharmacotherapy 2020, 40, 659–671. [Google Scholar] [CrossRef]
- Deb, S.; Reeves, A.A. Simulation of Remdesivir Pharmacokinetics and Its Drug Interactions. J. Pharm. Pharm. Sci. 2021, 24, 277–291. [Google Scholar] [CrossRef]
- Goldman, J.D.; Lye, D.C.B.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.Y.; Nahass, R.G.; et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N. Engl. J. Med. 2020, 383, 1827–1837. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Sörgel, F.; Malin, J.J.; Hagmann, H.; Kinzig, M.; Bilal, M.; Eichenauer, D.A.; Scherf-Clavel, O.; Simonis, A.; El Tabei, L.; Fuhr, U.; et al. Pharmacokinetics of remdesivir in a COVID-19 patient with end-stage renal disease on intermittent haemodialysis. J. Antimicrob. Chemother. 2021, 76, 825–827. [Google Scholar] [CrossRef]
- Kiser, T.H.; Fish, D.N.; Aquilante, C.L.; Rower, J.E.; Wempe, M.F.; MacLaren, R.; Teitelbaum, I. Evaluation of sulfobutylether-beta-cyclodextrin (SBECD) accumulation and voriconazole pharmacokinetics in critically ill patients undergoing continuous renal replacement therapy. Crit. Care 2015, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, R.K.; Alcorn, H., Jr.; Lawrence, L.; Paulson, S.K.; Quintas, M.; Luke, D.R.; Cammarata, S.K. Clinical Pharmacokinetics of Sulfobutylether-beta-Cyclodextrin in Patients With Varying Degrees of Renal Impairment. J. Clin. Pharmacol. 2018, 58, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, D.R.; Tomaszewski, K.; Damle, B.; Schlamm, H.T. Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J. Pharm. Sci. 2010, 99, 3291–3301. [Google Scholar] [CrossRef]
- Li, Y.-N.; Su, Y. Remdesivir attenuates high fat diet (HFD)-induced NAFLD by regulating hepatocyte dyslipidemia and inflammation via the suppression of STING. Biochem. Biophys. Res. Commun. 2020, 526, 381–388. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. Lond B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef]
- Nelson, D.R.; Zeldin, D.C.; Hoffman, S.M.G.; Maltais, L.J.; Wain, H.M.; Nebert, D.W. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 2004, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Deb, S.; Arrighi, S. Potential Effects of COVID-19 on Cytochrome P450-Mediated Drug Metabolism and Disposition in Infected Patients. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 185–203. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Dick, R.A. Refinement of In Vitro Methods for Identification of Aldehyde Oxidase Substrates Reveals Metabolites of Kinase Inhibitors. Drug Metab. Dispos. 2018, 46, 846–859. [Google Scholar] [CrossRef] [Green Version]
- COVID-19 Treatment Guidelines Panel, National Institutes of Health (NIH). Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 15 March 2021).
- Emami, A.; Javanmardi, F.; Pirbonyeh, N.; Akbari, A. Prevalence of Underlying Diseases in Hospitalized Patients with COVID-19: A Systematic Review and Meta-Analysis. Arch. Acad. Emerg. Med. 2020, 8, e35. [Google Scholar]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Varis, T.; Kivisto, K.T.; Backman, J.T.; Neuvonen, P.J. The cytochrome P450 3A4 inhibitor itraconazole markedly increases the plasma concentrations of dexamethasone and enhances its adrenal-suppressant effect. Clin. Pharmacol. Ther. 2000, 68, 487–494. [Google Scholar] [CrossRef]
- Kim, K.-A.; Park, J.-Y.; Lee, J.-S.; Lim, S. Cytochrome P450 2C8 and CYP3A4/5 are involved in chloroquine metabolism in human liver microsomes. Arch. Pharm. Res. 2003, 26, 631–637. [Google Scholar] [CrossRef]
- Sugie, M.; Asakura, E.; Zhao, Y.L.; Torita, S.; Nadai, M.; Baba, K.; Kitaichi, K.; Takagi, K.; Takagi, K.; Hasegawa, T. Possible involvement of the drug transporters P glycoprotein and multidrug resistance-associated protein Mrp2 in disposition of azithromycin. Antimicrob. Agents Chemother. 2004, 48, 809–814. [Google Scholar] [CrossRef] [Green Version]
- KALETRA (Lopinavir and Ritonavir) Package Insert. Available online: https://www.rxabbvie.com/pdf/kaletratabpi.pdf (accessed on 15 March 2021).
- Neodo, A.; Schulz, J.D.; Huwyler, J.; Keiser, J. In Vitro and In Vivo Drug-Drug Interaction Study of the Effects of Ivermectin and Oxantel Pamoate on Tribendimidine. Antimicrob. Agents Chemother. 2018, 63, e00762-18. [Google Scholar] [CrossRef] [Green Version]
- Skálová, L.; Szotáková, B.; Machala, M.; Neča, J.; Souček, P.; Havlasová, J.; Wsól, V.; Křídová, L.; Kvasničková, E.; Lamka, J. Effect of ivermectin on activities of cytochrome P450 isoenzymes in mouflon (Ovis musimon) and fallow deer (Dama dama). Chem. Biol. Interact. 2001, 137, 155–167. [Google Scholar] [CrossRef]
- Kineret® (Anakinra) Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103950s5136lbl.pdf (accessed on 15 March 2021).
- Islam, M.; Frye, R.F.; Richards, T.J.; Sbeitan, I.; Donnelly, S.S.; Glue, P.; Agarwala, S.S.; Kirkwood, J.M. Differential effect of IFNalpha-2b on the cytochrome P450 enzyme system: A potential basis of IFN toxicity and its modulation by other drugs. Clin. Cancer Res. 2002, 8, 2480–2487. [Google Scholar]
- Renton, K.W.; Knickle, L.C. Regulation of hepatic cytochrome P-450 during infectious disease. Can. J. Physiol. Pharmacol. 1990, 68, 777–781. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 15 March 2021).
- Amiodarone Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/018972s038s039lbl.pdf (accessed on 15 March 2021).
- Leegwater, E.; Strik, A.; Wilms, E.B.; Bosma, L.B.E.; Burger, D.M.; Ottens, T.H.; van Nieuwkoop, C. Drug-induced liver injury in a COVID-19 patient: Potential interaction of remdesivir with P-glycoprotein inhibitors. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Crowe, A.; Tan, A.M. Oral and inhaled corticosteroids: Differences in P-glycoprotein (ABCB1) mediated efflux. Toxicol. Appl. Pharmacol. 2012, 260, 294–302. [Google Scholar] [CrossRef]
- Rijpma, S.R.; van den Heuvel, J.J.M.W.; van der Velden, M.; Sauerwein, R.W.; Russel, F.G.; Koenderink, J.B. Atovaquone and quinine anti-malarials inhibit ATP binding cassette transporter activity. Malar. J. 2014, 13, 359. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, M.J.; Potter, P.M. Carboxylesterase inhibitors. Expert Opin. Ther. Pat. 2011, 21, 1159–1171. [Google Scholar] [CrossRef]
- Sánchez Pascua, T.S. Carboxylesterase 1 Genetic Variability, Expression and Potential for Drug-Drug Interaction. Doctor in Philosophy Thesis, University of Liverpool, Liverpool, UK, 2014. [Google Scholar]
- Deb, S.; Pandey, M.; Adomat, H.; Tomlinson Guns, E.S. Cytochrome P450 3A-mediated microsomal biotransformation of 1alpha,25-dihydroxyvitamin D3 in mouse and human liver: Drug-related induction and inhibition of catabolism. Drug Metab. Dispos. 2012, 40, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Markowitz, J.S. Natural Products as Modulators of CES1 Activity. Drug Metab. Dispos. 2020, 48, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Song, L.; Zhang, H.; Matoney, L.; LeCluyse, E.; Yan, B. Dexamethasone differentially regulates expression of carboxylesterase genes in humans and rats. Drug Metab. Dispos. 2000, 28, 186–191. [Google Scholar]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.-W.; Jin, Q.; Wang, D.-D.; Qian, Q.-K.; Hao, D.-C.; Ge, G.-B.; Yang, L. Carboxylesterase Inhibitors: An Update. Curr. Med. Chem. 2018, 25, 1627–1649. [Google Scholar] [CrossRef] [PubMed]
- Merali, Z.; Ross, S.; Paré, G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metab. Drug Interact. 2014, 29, 143–151. [Google Scholar] [CrossRef]
- Takahashi, T.; Luzum, J.A.; Nicol, M.R.; Jacobson, P.A. Pharmacogenomics of COVID-19 therapies. NPJ Genom. Med. 2020, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Mele, F.; Florio, L.L.; Bertolino, L.; Andini, R.; Galdo, M.; De Rosa, R.; Corcione, A.; Durante-Mangoni, E. Liver injury in remdesivir-treated COVID-19 patients. Hepatol. Int. 2020, 14, 881–883. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Substrate | CYP Inhibitor | CYP Inducer |
|---|---|---|---|
| Remdesivir [15,18,24] | CES, CYP3A4, CYP2C8, CYP2D6, P-gp, OATP1B1 | CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 | CYP1A2, CYP2B6 |
| Dexamethasone [43] | CYP3A4, P-gp | CYP3A4 | CYP3A4 |
| Chloroquine/Hydroxychloroquin [44] | CYP2C8, CYP3A4/5, CYP2D6 | CYP2D6 | None reported |
| Azithromycin [45] | CYP3A4, P-gp | None reported | None reported |
| Lopinavir/Ritonavir [46] | CYP1A2, CYP2B6, CYP2D6, CYP3A4, P-gp | CYP2D6, CYP3A4 | CYP1A2, CYP2B6, CYP2C19, CYP2C9 |
| Ivermectin [47,48] | CYP3A4, P-gp | CYP2C9, CYP2D6, CYP2C19, CYP3A4 | CYP1A, 2B and 3A |
| Anakinra [49] | None reported | None reported | None reported |
| Interferon beta [50,51] | None reported | CYP1A2 | None reported |
| Drug | Process Modified | Potential Outcome |
|---|---|---|
| Drugs Affecting Remdesivir ADME | ||
| Amiodarone [52,53,54] | Inhibition of P-gp and CYP | Increased remdesivir concentration |
| Chloroquine [55,56] | Inhibition of P-gp | Increased remdesivir concentration |
| Fluoxetine, Grapefruit juice [57] | Inhibition of CES | Increased remdesivir concentration |
| Dexamethasone, Rifampin [58] | Induction of CES | Decreased remdesivir concentration |
| Phenytoin, St. John’s Wort [52,59] | Induction of CYP3A4 | Decreased remdesivir concentration |
| Lopinavir-ritonavir [46] | Inhibition of CYP3A4 | Increased remdesivir concentration |
| Drugs Affected by Remdesivir | ||
| Theophylline [15,18,24] | Induction of CYP1A2 | Decreased theophylline concentration |
| Olmesartan [15,18,24] | Inhibition of OATP 1B1/1B3 transporters | Increased olmesartan concentration |
| Warfarin [15,18,24] | Inhibition of CYP2C9 | Increased warfarin concentration |
| Irinotecan [15,18,24] | Inhibition of OATP 1B1 | Increased irinotecan concentration |
| Atorvastatin [15,18,24] | Inhibition of CYP3A4 | Increased atorvastatin concentration |
| Metformin [15,18,24] | Inhibition of MATE1 | Increased metformin concentration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deb, S.; Reeves, A.A.; Hopefl, R.; Bejusca, R. ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential. Pharmaceuticals 2021, 14, 655. https://doi.org/10.3390/ph14070655
Deb S, Reeves AA, Hopefl R, Bejusca R. ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential. Pharmaceuticals. 2021; 14(7):655. https://doi.org/10.3390/ph14070655
Chicago/Turabian StyleDeb, Subrata, Anthony Allen Reeves, Robert Hopefl, and Rebecca Bejusca. 2021. "ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential" Pharmaceuticals 14, no. 7: 655. https://doi.org/10.3390/ph14070655
APA StyleDeb, S., Reeves, A. A., Hopefl, R., & Bejusca, R. (2021). ADME and Pharmacokinetic Properties of Remdesivir: Its Drug Interaction Potential. Pharmaceuticals, 14(7), 655. https://doi.org/10.3390/ph14070655