
Capillary Liquid Chromatography for the Determination of Terpenes in Botanical Dietary Supplements

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
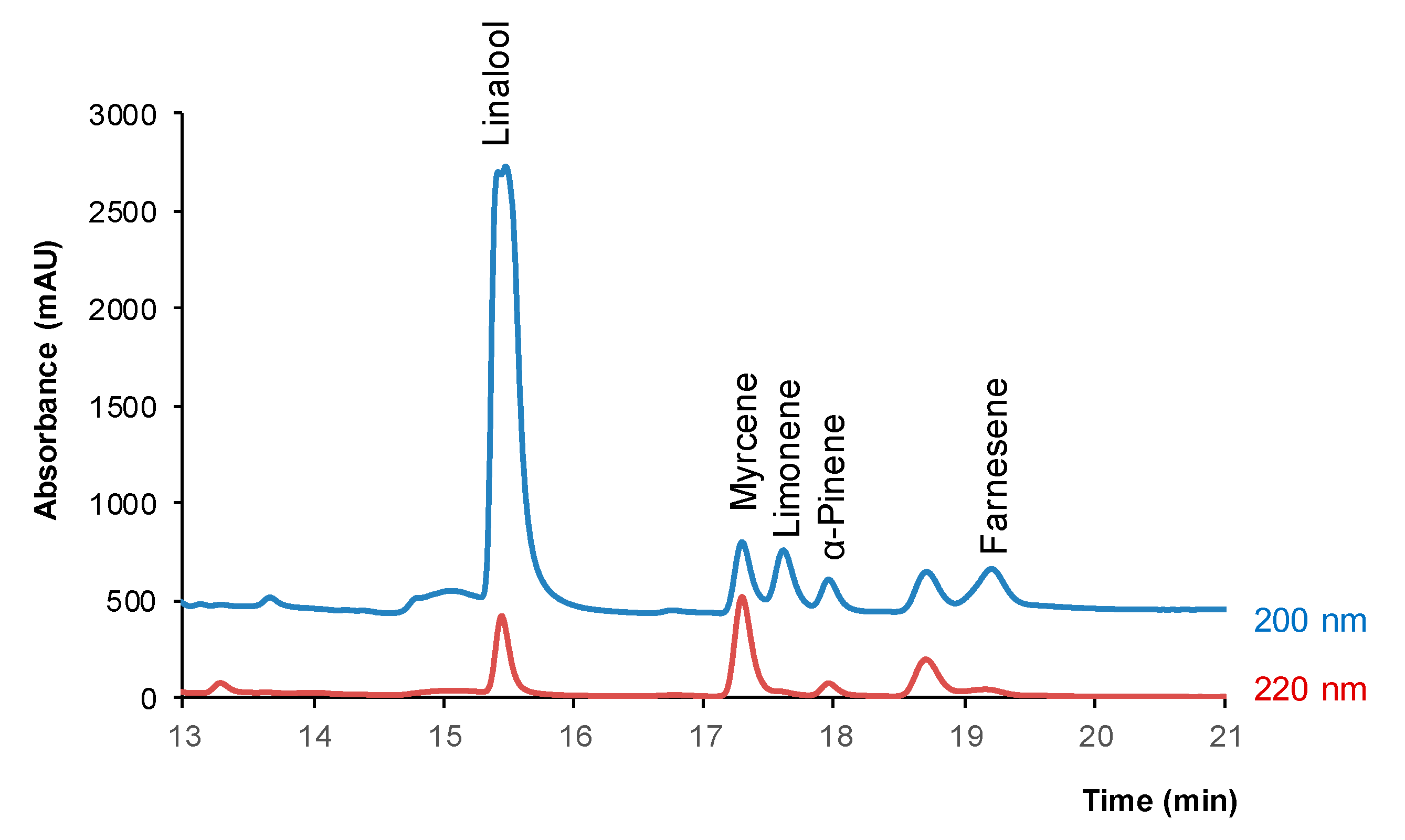
2.1. Separation and Analytical Performance
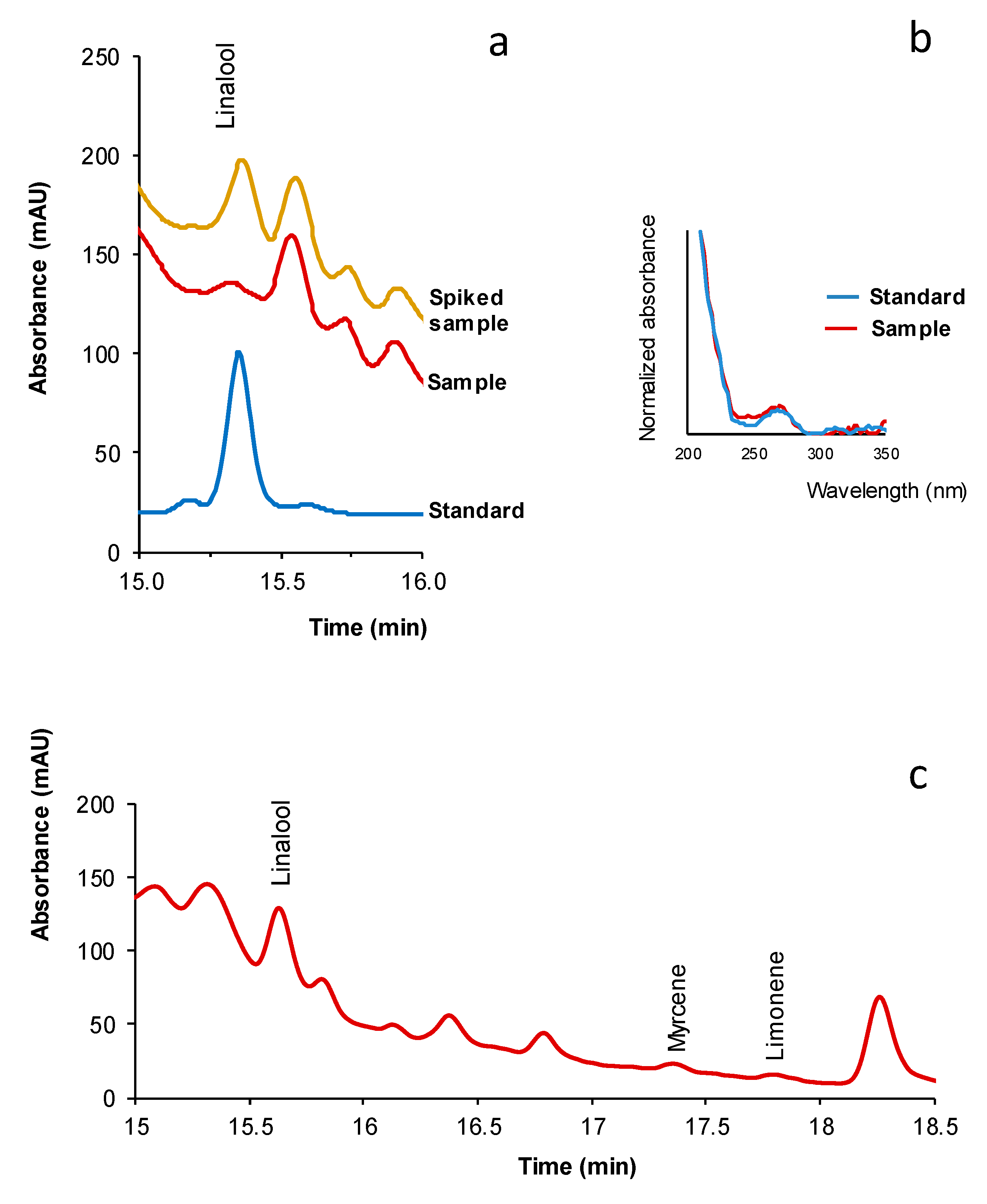
2.2. Sample Treatment
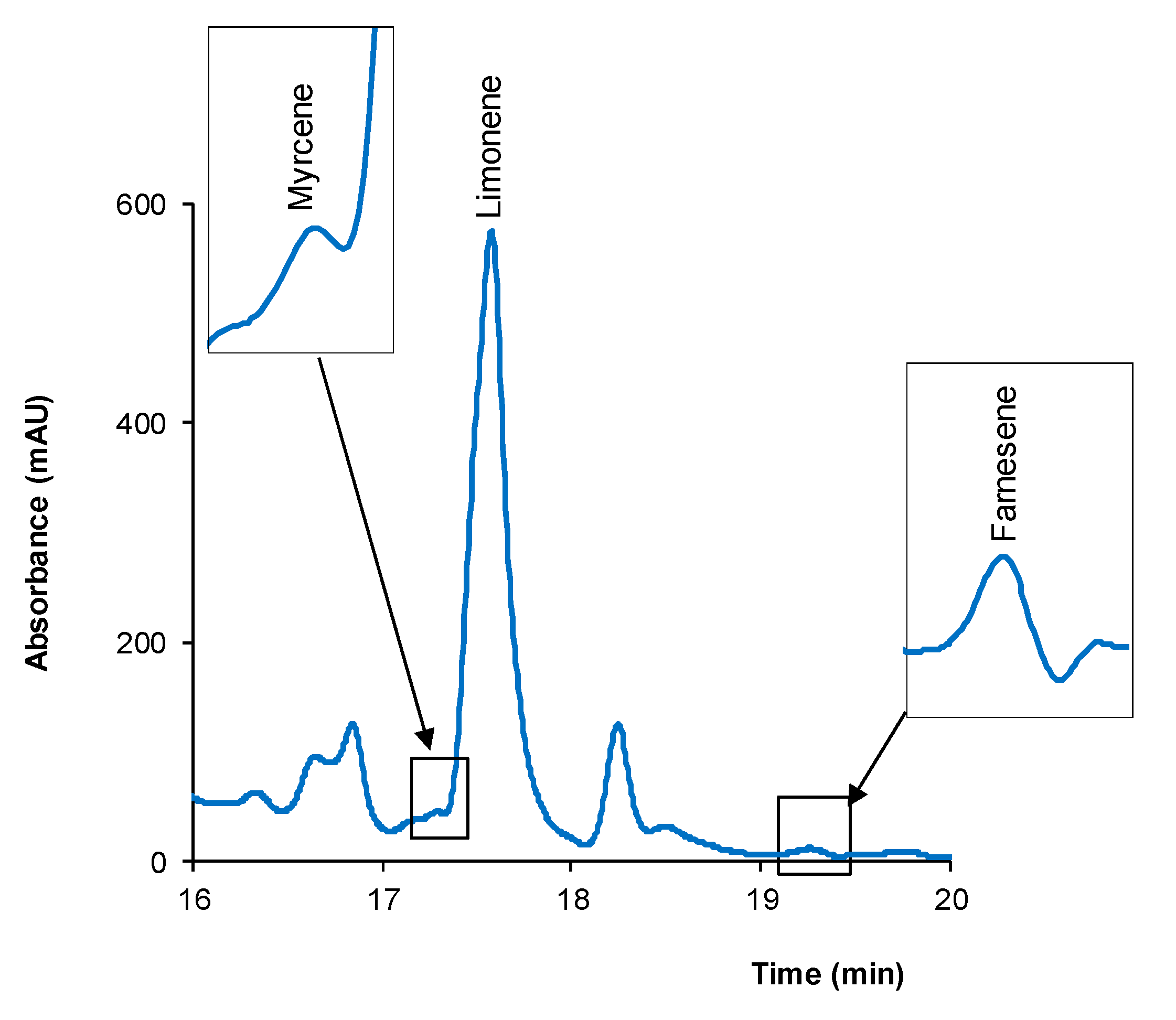
2.3. Quantification of Terpenes in Dietary Supplements
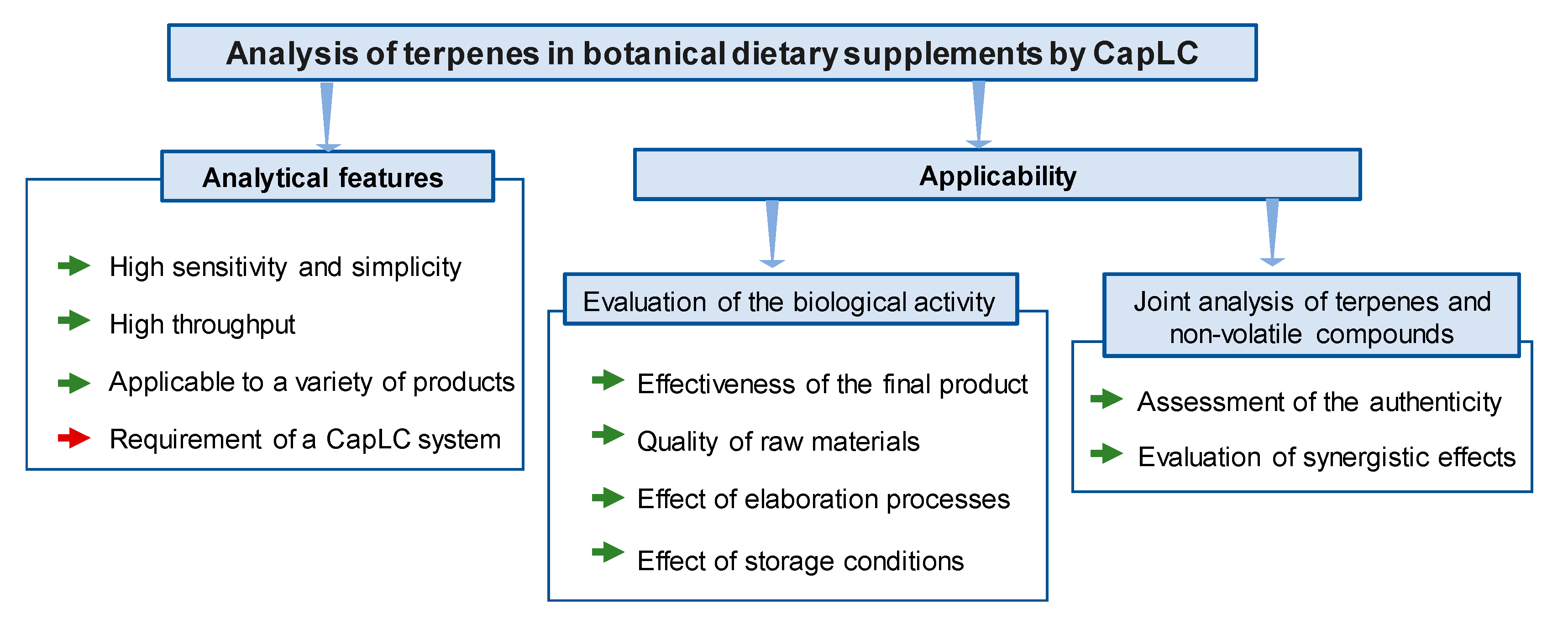
3. Discussion
4. Materials and Methods
4.1. Chemicals and Solutions
4.2. Chromatographic Conditions
4.3. Analysis of Dietary Supplements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaur, S.; Das, M. Functional Foods: An Overview. Food Sci. Biotechnol. 2011, 20, 861–875. [Google Scholar] [CrossRef]
- Eussen, S.R.B.M.; Verhagen, H.; Klungel, O.H.; Garssen, J.; van Loveren, H.; van Kranen, H.J.; Rompelberg, C.J.M. Functional Foods and Dietary Supplements: Products at the Interface Between Pharma and Nutrition. Eur. J. Pharmacol. 2011, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Sarma, N.; Giancaspro, G.; Venema, J. Dietary Supplements Quality Analysis Tools from the United States Pharmacopeia. Drug Test. Anal. 2016, 8, 418–423. [Google Scholar] [CrossRef]
- Andrews, K.W.; Schweitzer, A.; Zhao, C.; Holden, J.M.; Roseland, J.M.; Brandt, M.; Dwyer, J.T.; Picciano, M.F.; Saldanha, L.G.; Fisher, K.D.; et al. The Caffeine Contents of Dietary Supplements Commonly Purchased in the US: Analysis of 53 Products with Caffeine-Containing Ingredients. Anal. Bioanal. Chem. 2007, 389, 231–239. [Google Scholar] [CrossRef]
- Kowalski, R.; Baj, T.; Kowalska, G.; Pankiewicz, U. Estimation of Potential Availability of Essential Oil in Some Brands of Herbal Teas and Herbal Dietary Supplements. PLoS ONE 2015, 10, e0130714. [Google Scholar] [CrossRef] [PubMed]
- Viana, C.; Zemolin, G.M.; Müller, L.S.; Dal Molin, T.R.; Seiffert, H.; de Carvalho, L.M. Liquid Chromatographic Determination of Caffeine and Adrenergic Stimulants in Food Supplements Sold in Brazilian e-Commerce for Weight Loss and Physical Fitness. Food Addit. Contam. Part A 2016, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cox-Georgian, D.; Ramadoss, N.; Dona, C.; Basu, C. Therapeutic and Medicinal Uses of Terpenes. In Medicinal Plants. From Farm to Pharmacy, 1st ed.; Joshee, N., Dhekney, S., Parajuli, P., Eds.; Springer: Cham, Switzerland, 2019; pp. 333–359. [Google Scholar] [CrossRef]
- Kim, T.; Song, B.; Cho, K.S.; Lee, I.-S. Therapeutic Potential of Volatile Terpenes and Terpenoids from Forests for Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 2187. [Google Scholar] [CrossRef]
- Proshkina, E.; Plyusnin, S.; Babak, T.; Lashmanova, E.; Maganova, F.; Koval, L.; Platonova, E.; Shaposhnikov, M.; Moskalev, A. Terpenoids as Potential Geroprotectors. Antioxidants 2020, 9, 529. [Google Scholar] [CrossRef]
- Zhao, J.; Ge, L.-Y.; Xiong, W.; Leong, F.; Huang, L.-Q.; Li, S.-P. Advanced Development in Phytochemicals Analysis of Medicine and Food Dual Purposes Plants Used in China (2011–2014). J. Chromatogr. A 2016, 1428, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.; Pereira, J.A.; Silva, P.; Perestrelo, R.; Câmara, J.S. Food Fingerprints–A Avaluable Tool to Monitor Food Authenticity and Safety. Food Chem. 2019, 278, 144–162. [Google Scholar] [CrossRef]
- Hąc-Wydro, K.; Flasiński, M.; Romańczuk, K. Essential Oils as Food Eco-preservatives: Model System Studies on the Effect of Temperature on Limonene Antibacterial Activity. Food Chem. 2017, 235, 127–135. [Google Scholar] [CrossRef]
- Kupska, M.; Chmiel, T.; Jędrkiewicz, R.; Wardencki, W.; Namieśnik, J. Comprehensive Two-Dimensional Gas Chromatography for Determination of the Terpenes Profile of Blue Honeysuckle Berries. Food Chem. 2014, 152, 88–93. [Google Scholar] [CrossRef]
- D’Onofrio, C.; Matarese, F.; Cuzzola, A. Study of the Terpene Profile at Harvest and During Berry Development of Vitis vinifera L. Aromatic Varieties Aleatico, Brachetto, Malvasia di Candia Aromatica and Moscato Bianco. J. Sci. Food Agric. 2017, 97, 2898–2907. [Google Scholar] [CrossRef]
- Diaconeasa, Z.; Iuhas, C.I.; Ayvaz, H.; Rugină, D.; Stanilă, A.; Dulf, F.; Bunea, A.; Socaci, S.A.; Socaciu, C.; Pintea, A. Phytochemical Characterization of Commercial Processed Blueberry, Blackberry, Blackcurrant, Cranberry, and Raspberry and Their Antioxidant Activity. Antioxidants 2019, 8, 540. [Google Scholar] [CrossRef]
- Pollon, M.; Torchio, F.; Giacosa, S.; Segade, S.R.; Rolle, L. Use of Density Sorting for the Selection of Aromatic Grape Berries with Different Volatile Profile. Food Chem. 2019, 276, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.M.; Amorim, T.L.; Grazul, M.L.; de Oliveira, M.A.L. Differentiation of aromatic, bittering and dual-purpose commercial hops from their terpenic profiles: An Approach Involving Batch Extraction, GC–MS and Multivariate Analysis. Food Res. Int. 2020, 138, 109768. [Google Scholar] [CrossRef]
- He, F.; Qian, Y.L.; Qian, M.C. Flavor and Chiral Stability of Lemon-Flavored Hard Tea during Storage. Food Chem. 2018, 239, 622–630. [Google Scholar] [CrossRef]
- Kim, M.K.; Jang, H.W.; Lee, K.-G. Sensory and Instrumental Volatile Flavor Analysis of Commercial Orange Juices Prepared by Different Processing Methods. Food Chem. 2018, 267, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Mukazayire, M.J.; Tomani, J.C.; Stévigny, C.; Chalchat, J.C.; Conforti, F.; Menichini, F.; Duez, P. Essential Oils of Four Rwandese Hepatoprotective Herbs: Gas Chromatography–Mass Spectrometry Analysis and Antioxidant Activities. Food Chem. 2011, 129, 753–760. [Google Scholar] [CrossRef]
- Nazario, C.D.E.; Silva, M.R.; Franco, M.S.; Lanças, F.M. Evolution in Miniaturized Column Liquid Chromatography Instrumentation and Applications: An Overview. J. Chromatogr. A 2015, 1421, 18–37. [Google Scholar] [CrossRef]
- Ponce-Rodríguez, H.D.; Verdú-Andrés, J.; Herráez-Hernández, R.; Campíns-Falcó, P. Innovations in Extractive Phases for In-Tube Solid-Phase Microextraction Coupled to Miniaturized Liquid Chromaography: A Critical Review. Molecules 2020, 25, 2460. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Donati, E.; Touloupakis, E.; Aturki, Z. Miniaturized Separation Techniques as Analytical Methods to Ensure Quality and Safety of Dietary Supplements. Trends Anal. Chem. 2018, 103, 156–183. [Google Scholar] [CrossRef]
- Ponce-Rodríguez, H.D.; Herráez-Hernández, R.; Verdú-Andrés, J.; Campíns-Falcó, P. Quantitative Analysis of Terpenic Compounds in Microsamples of Resins by Capillary Liquid Chromatography. Molecules 2019, 24, 4068. [Google Scholar] [CrossRef] [PubMed]
- Mirghaed, A.T.; Ghelichpour, M.; Hoseini, S.M. Myrcene and Linalool as New Anesthetic and Sedative Agents in Common Carp, Cyprinus carpio-Comparison with Eugenol. Aquaculture 2016, 464, 165–170. [Google Scholar] [CrossRef]
- Kiyama, R. Nutritional Implications of Ginger: Chemistry, Biological Activities and Signaling Pathways. J. Nutr. Biochem. 2020, 86, 108486. [Google Scholar] [CrossRef]
- PubChem Database. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 14 June 2021).
- Kraujalytė, V.; Pelvan, E.; Alasalvar, C. Volatile Compounds and Sensory Characteristics of Various Instant Teas Produced from Black Tea. Food Chem. 2016, 194, 864–872. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Activity |
|---|---|---|
| Linalool |  | |
| Myrcene |  | |
| Limonene |  | |
| α-Pinene |  | |
| Farnesene |  |
| Compound | tr (min) | Concentration Interval (µg mL−1) | Linearity, n = 10 /R2 | LOD 1 (µg mL−1) | LOQ 2 (µg mL−1) | RSD 3, n = 3 (%) |
|---|---|---|---|---|---|---|
| Linalool | 15.5 | 0.02–1.0 | y = (20,200 ± 200) x + (67 ± 90) /0.999 | 0.005 | 0.02 | 11 |
| Myrcene | 17.3 | 0.05–2.5 | y = (530 ± 20) x +(−20 ± 20) /0.992 | 0.01 | 0.04 | 2 |
| Limonene | 17.6 | 1.0–10.0 | y = (920 ± 40) x +(−600 ± 300) /0.993 | 0.25 | 1.0 | 3 |
| α-Pinene | 18.0 | 1.0–10.0 | y = (166 ± 6) x +(−60 ± 30) /0.993 | 0.25 | 1.0 | 11 |
| Farnesene | 19.3 | 1.0–5.0 | y = (900 ± 40) x +(−200 ± 100) /0.991 | 0.25 | 1.0 | 11 |
| Compound | Mean Recovery (%) | Precision, RDS (%) | |
|---|---|---|---|
| Intra-Day (n = 3) | Inter-Day (n = 6) | ||
| Linalool | 96 ± 5 | 5 | 4 |
| Myrcene | 106 ± 3 | 3 | 10 |
| Limonene | 95 ± 10 | 11 | 13 |
| α-Pinene | 98 ± 9 | 10 | 13 |
| Farnesene | 105 ± 7 | 7 | 6 |
| Sample | Found Compounds | Found Amounts 1 | |
|---|---|---|---|
| mg per unit (n = 3) | mg per g (n = 6) | ||
| GCE-1 | Limonene Myrcene | 0.350 ± 0.004 0.059 ± 0.003 | 0.318 ± 0.003 0.054 ± 0.003 |
| GCE-2 | - | - | - |
| GCE-3 | Limonene Myrcene | 0.84 ± 0.01 1.3 ± 0.2 | 1.68 ± 0.01 2.6 ± 0.3 |
| FB-1 | Limonene Linalool Myrcene | 0.129 ± 0.001 0.0031 ± 0.0001 0.0200 ± 0.001 | 0.258 ± 0.002 0.006 ± 0.001 0.039 ± 0.003 |
| FB-2 | Limonene Linalool Myrcene | 0.130 ± 0.002 0.003 ± 0.001 0.0030 ± 0.0005 | 0.520 ± 0.005 0.012 ± 0.001 0.012 ± 0.001 |
| HP | Limonene Linalool Myrcene Farnesene | 2.60 ± 0.05 <LOQ 0.150 ± 0.001 0.100 ± 0.003 | 1.90 ± 0.04 <LOQ 0.110 ± 0.001 0.070 ± 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponce-Rodríguez, H.D.; Verdú-Andrés, J.; Campíns-Falcó, P.; Herráez-Hernández, R. Capillary Liquid Chromatography for the Determination of Terpenes in Botanical Dietary Supplements. Pharmaceuticals 2021, 14, 580. https://doi.org/10.3390/ph14060580
Ponce-Rodríguez HD, Verdú-Andrés J, Campíns-Falcó P, Herráez-Hernández R. Capillary Liquid Chromatography for the Determination of Terpenes in Botanical Dietary Supplements. Pharmaceuticals. 2021; 14(6):580. https://doi.org/10.3390/ph14060580
Chicago/Turabian StylePonce-Rodríguez, Henry Daniel, Jorge Verdú-Andrés, Pilar Campíns-Falcó, and Rosa Herráez-Hernández. 2021. "Capillary Liquid Chromatography for the Determination of Terpenes in Botanical Dietary Supplements" Pharmaceuticals 14, no. 6: 580. https://doi.org/10.3390/ph14060580
APA StylePonce-Rodríguez, H. D., Verdú-Andrés, J., Campíns-Falcó, P., & Herráez-Hernández, R. (2021). Capillary Liquid Chromatography for the Determination of Terpenes in Botanical Dietary Supplements. Pharmaceuticals, 14(6), 580. https://doi.org/10.3390/ph14060580