
Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor

 ,
,  ,
,  and
and
Abstract
1. Introduction
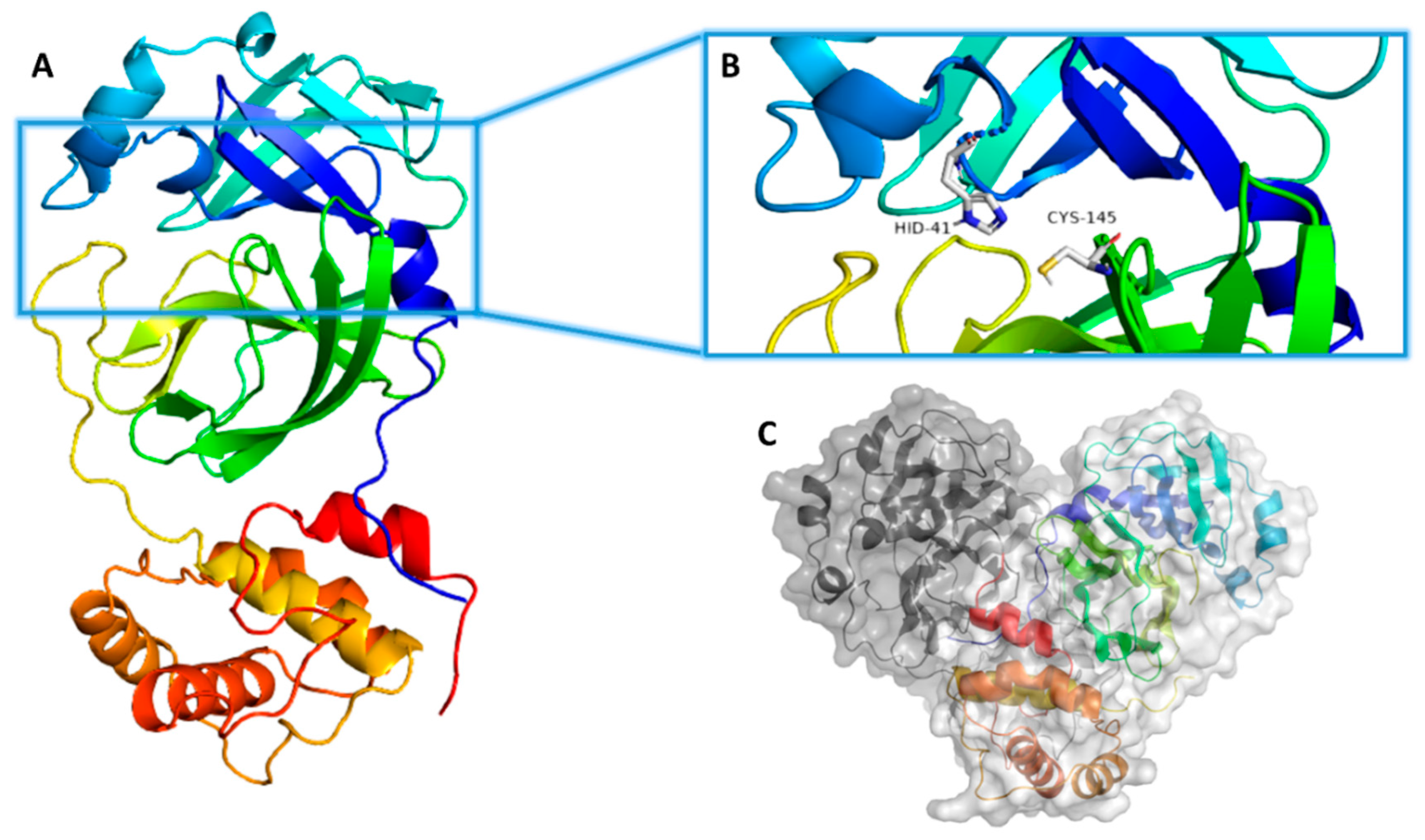
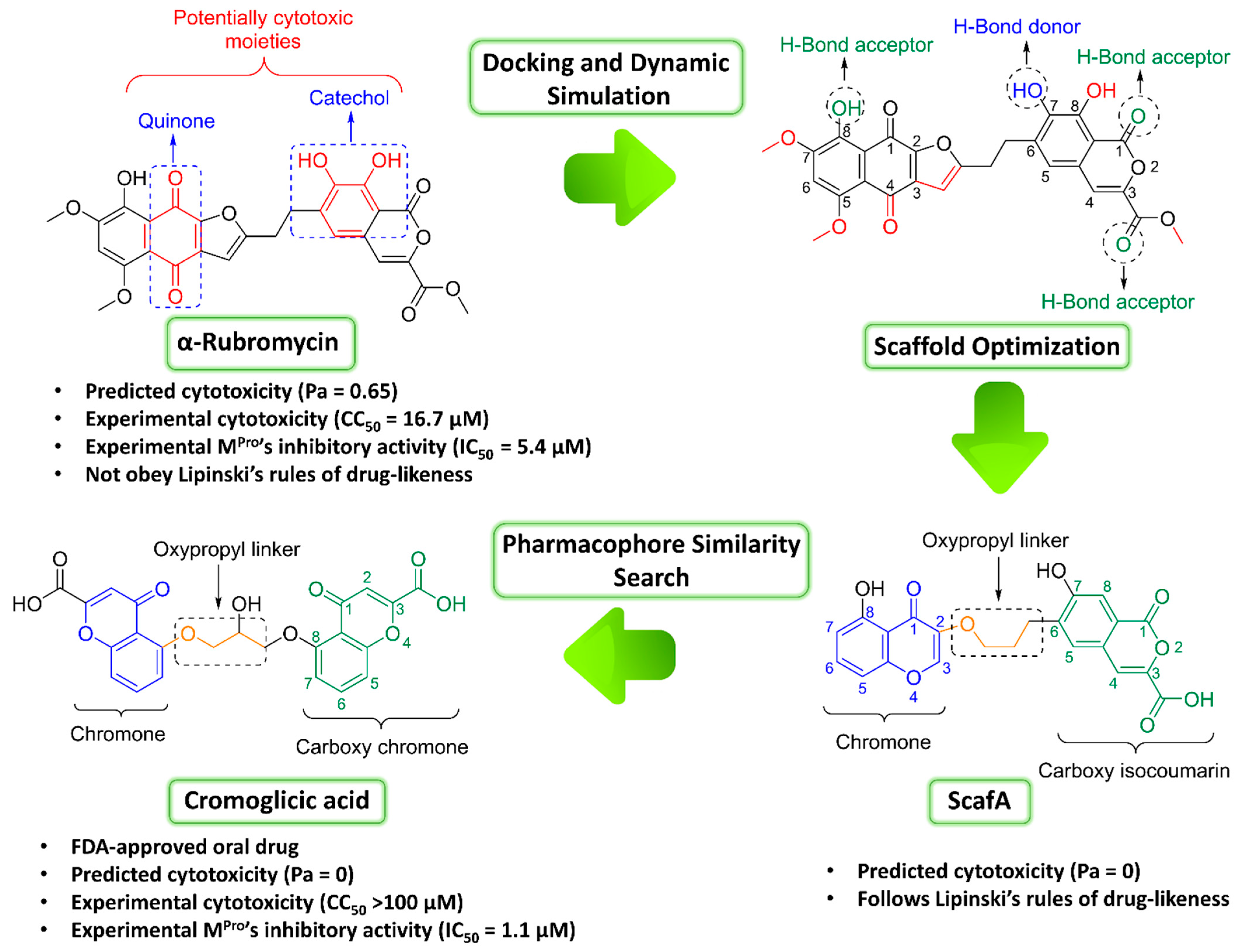
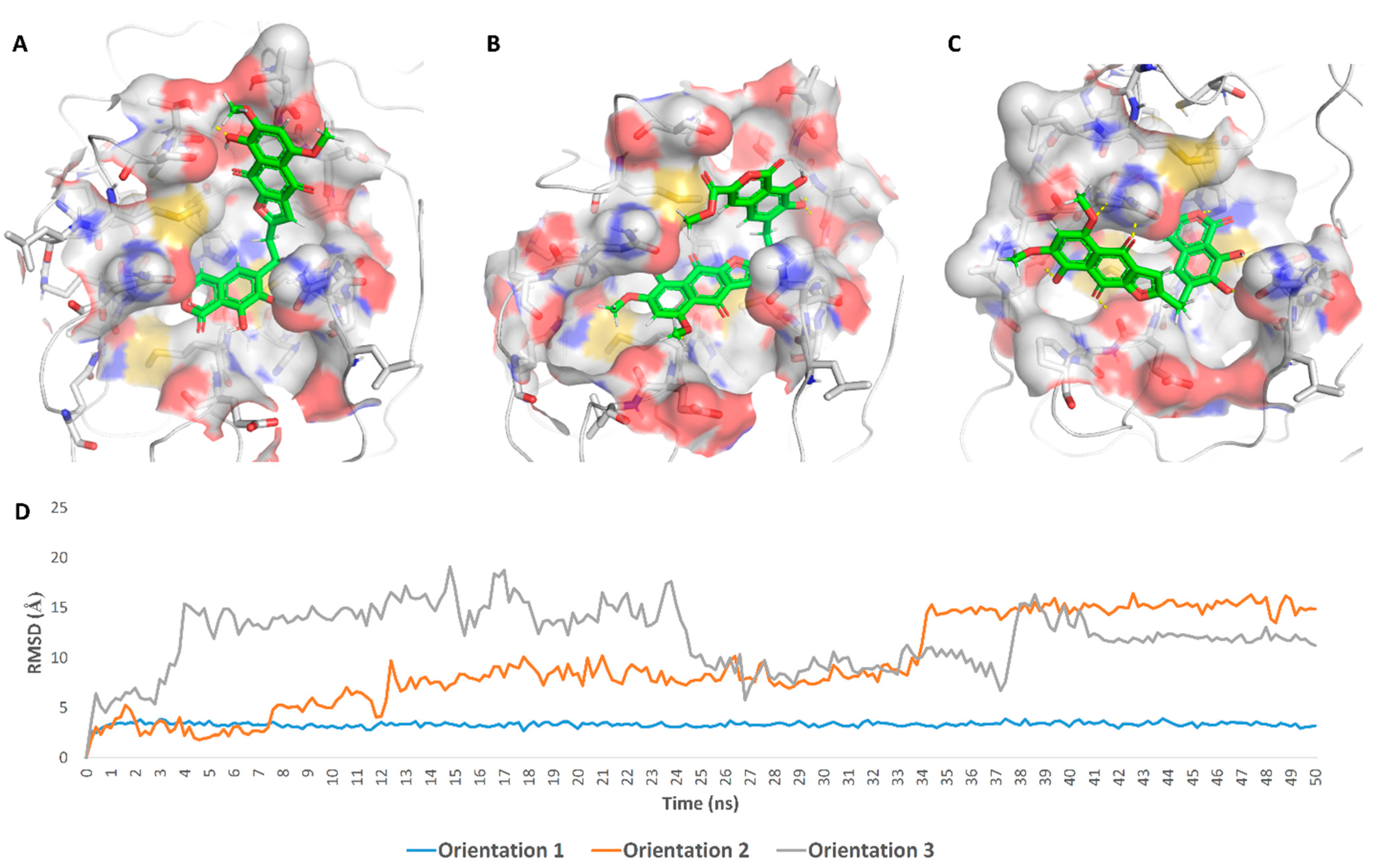
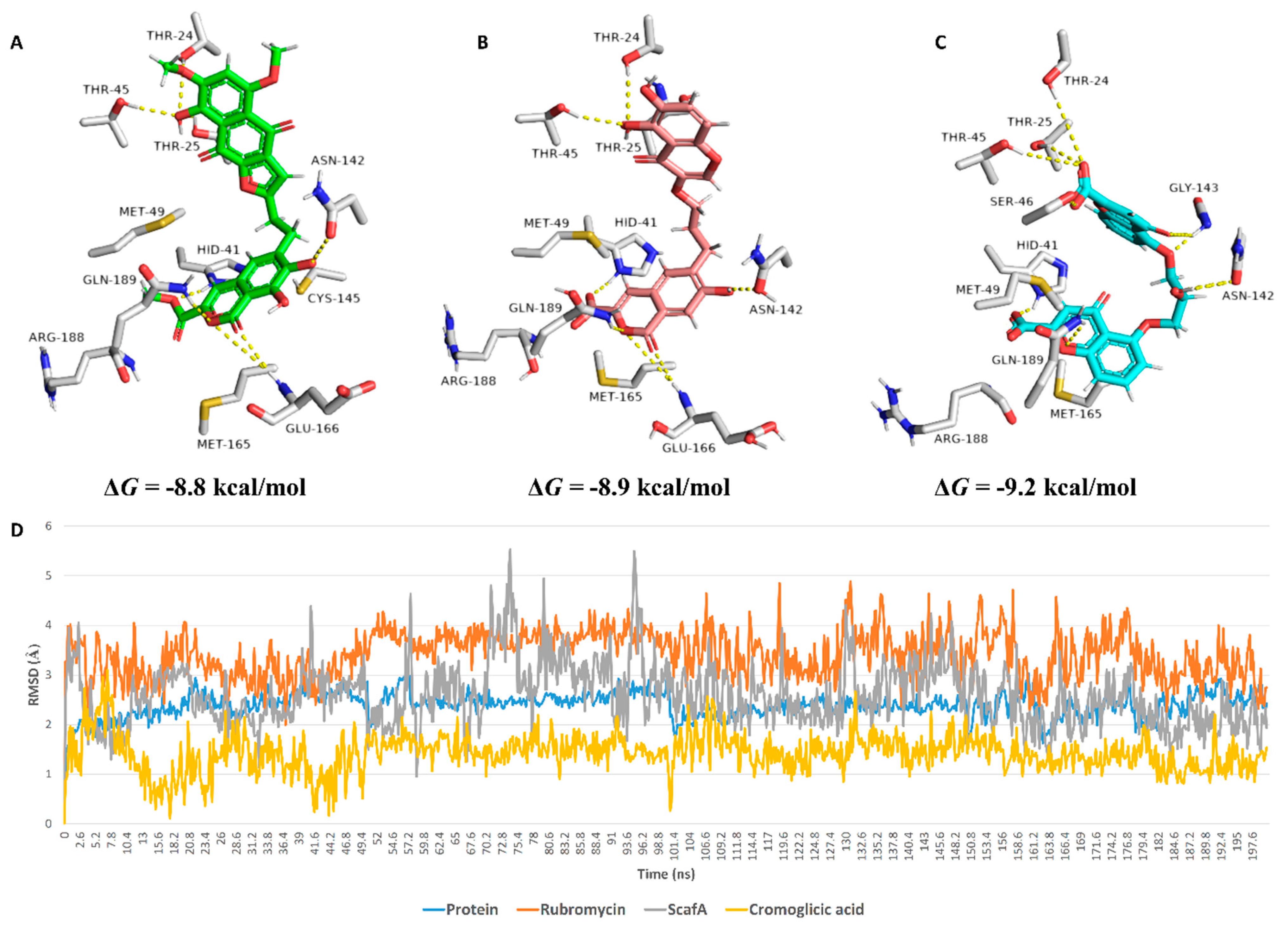
2. Results
3. Discussion
4. Materials and Methods
4.1. Isolation of α-Rubromycin
4.2. In-Vitro Assays
4.2.1. MPro Inhibition
4.2.2. MTT Cytotoxicity Assay
4.3. In Silico Investigation
4.3.1. Ensemble Docking
4.3.2. Molecular Dynamics Simulation
4.3.3. Binding Free Energy Calculations
4.3.4. Drug-Likeness Analysis
4.3.5. Toxicity Prediction
4.3.6. Pharmacophore-Based Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sayed, A.M.; Khattab, A.R.; AboulMagd, A.M.; Hassan, H.M.; Rateb, M.E.; Zaid, H.; Abdelmohsen, U.R. Nature as a treasure trove of potential anti-SARS-CoV drug leads: A structural/mechanistic rationale. RSC Adv. 2020, 10, 19790–19802. [Google Scholar] [CrossRef]
- Adamson, C.S.; Chibale, K.; Goss, R.J.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral drug discovery: Preparing for the next pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Behnam, M.A. Protein structural heterogeneity: A hypothesis for the basis of proteolytic recognition by the main protease of SARS-CoV and SARS-CoV-2. Biochimie 2021, 182, 177–184. [Google Scholar] [CrossRef]
- Pavlova, A.; Lynch, D.L.; Daidone, I.; Zanetti-Polzi, L.; Smith, M.D.; Chipot, C.; Kneller, D.W.; Kovalevsky, A.; Coates, L.; Golosov, A.A.; et al. Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease. Chem. Sci. 2021, 12, 1513–1527. [Google Scholar] [CrossRef]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Chang, H.P.; Chou, C.Y.; Chang, G.G. Reversible unfolding of the severe acute respiratory syndrome coronavirus main protease in guanidinium chloride. Biophys. J. 2007, 92, 1374–1383. [Google Scholar] [CrossRef]
- Mondal, D.; Warshel, A. Exploring the Mechanism of Covalent Inhibition: Simulating the Binding Free Energy of α-Ketoamide Inhibitors of the Main Protease of SARS-CoV-2. Biochemistry 2020, 59, 4601–4608. [Google Scholar] [CrossRef]
- Zhou, Q.A.; Kato-Weinstein, J.; Li, Y.; Deng, Y.; Granet, R.; Garner, L.; Liu, C.; Polshakov, D.; Gessner, G.; Watkins, S. Potential therapeutic agents and associated bioassay data for COVID-19 and related human coronavirus infections. ACS Pharmacol. Transl. Sci. 2020, 3, 813–834. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Sharun, K.; Dhama, K.; Patel, S.K.; Pathak, M.; Tiwari, R.; Singh, B.R.; Sah, R.; Bonilla-Aldana, D.A.; Rodriguez-Morales, A.J.; Leblebicioglu, H. Ivermectin, a new candidate therapeutic against SARS-CoV-2/COVID-19. Ann. Clin. Microbiol. Antimicrob. 2020, 30, 19. [Google Scholar] [CrossRef]
- Behera, P.; Patro, B.K.; Singh, A.K.; Chandanshive, P.D.; Pradhan, S.K.; Pentapati, S.S.K.; Batmanabane, G.; Mohapatra, P.R.; Padhy, B.M.; Bal, S.K. Role of ivermectin in the prevention of SARS-CoV-2 infection among healthcare workers in India: A matched case-control study. PLoS ONE 2021, 16, e0247163. [Google Scholar] [CrossRef]
- Cao, R.; Hu, H.; Li, Y.; Wang, X.; Xu, M.; Liu, J.; Zhong, W. Anti-SARS-CoV-2 potential of artemisinin in vitro. ACS Infect. Dis. 2020, 6, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Saund, S.; Windlass, H.; Trieu, V. Repurposing Anti-Malaria Phytomedicine Artemisinin as a COVID-19 Drug. Front. Pharmacol. 2021, 12, 407. [Google Scholar] [CrossRef]
- Atkinson, D.J.; Brimble, M.A. Isolation, biological activity, biosynthesis and synthetic studies towards the rubromycin family of natural products. Nat. Prod. Rep. 2015, 32, 811–840. [Google Scholar] [CrossRef]
- Goldman, M.E.; Salituro, G.S.; Bowen, J.A.; Williamson, J.M.; Zink, D.L.; Schleif, W.A.; Emini, E.A. Inhibition of human immunodeficiency virus-1 reverse transcriptase activity by rubromycins: Competitive interaction at the template. primer site. Mol. Pharmacol. 1990, 38, 20–25. [Google Scholar]
- Metsä-Ketelä, M.; Ylihonko, K.; Mäntsälä, P. Partial activation of a silent angucycline-type gene cluster from a rubromycin β producing Streptomyces sp. PGA64. J. Antibiot. 2004, 57, 502–510. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Bukowska, B.; Kowalska, S. Phenol and catechol induce prehemolytic and hemolytic changes in human erythrocytes. Toxicol. Lett. 2004, 152, 73–84. [Google Scholar] [CrossRef]
- Metz, J.T.; Huth, J.R.; Hajduk, P.J. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups. J. Comput. Aided Mol. Des. 2007, 21, 139–144. [Google Scholar] [CrossRef]
- Huth, J.R.; Song, D.; Mendoza, R.R.; Black-Schaefer, C.L.; Mack, J.C.; Dorwin, S.A.; Ladror, U.S.; Severin, J.M.; Walter, K.A.; Bartley, D.M. Toxicological evaluation of thiol-reactive compounds identified using a la assay to detect reactive molecules by nuclear magnetic resonance. Chem. Res. Toxicol. 2007, 20, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Monks, T.J.; Jones, D.C. The metabolism and toxicity of quinones, quinonimines, quinone methides, and quinone-thioethers. Curr. Drug Metabol. 2002, 3, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Sok, D.E. Identification of alkylation-sensitive target chaperone proteins and their reactivity with natural products containing Michael acceptor. Arch. Pharmacal Res. 2003, 26, 1047–1054. [Google Scholar] [CrossRef]
- Li, W.W.; Heinze, J.; Haehnel, W. Site-specific binding of quinones to proteins through thiol addition and addition− elimination reactions. J. Amer. Chem. Soc. 2005, 127, 6140–6141. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Rarey, M.; Dixon, J.S. Feature trees: A new molecular similarity measure based on tree matching. J. Comput. Aided Mol. Des. 1998, 12, 471–490. [Google Scholar] [CrossRef]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A web tool for low to ultra-high throughput ligand-based virtual screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Flower, R.J.; Henderson, G.; Loke, Y.K.; MacEwan, D.; Rang, H.P. Rang & Dale’s Pharmacology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Vinetz, J.M. Dexamethasone in the management of covid-19. BMJ 2020, 370. [Google Scholar] [CrossRef]
- Sayed, A.M.; Khalaf, A.M.; Abdelrahim, M.E.; Elgendy, M.O. Repurposing of some anti-infective drugs for COVID-19 treatment: A surveillance study supported by an in silico investigation. Int. J. Clin. Pract. 2020, 75, e13877. [Google Scholar] [CrossRef]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.; Scott, N.W.; Mustafa, M.Z.; Mustafa, M.S.; Azuara-Blanco, A. Topical antihistamines and mast cell stabilisers for treating seasonal and perennial allergic conjunctivitis. Cochr. Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Sanchez-Gonzalez, M.A.; Moskowitz, D.; Issuree, P.D.; Yatzkan, G.; Rizvi, S.A.; Day, K. A Pathophysiological Perspective on COVID-19’s Lethal Complication: From Viremia to Hypersensitivity Pneumonitis-like Immune Dysregulation. Infect. Chemother. 2020, 52, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C. Potential association of mast cells with coronavirus disease 2019. Ann. Aller. Asth. Immunol. 2021, 126, 217–218. [Google Scholar] [CrossRef]
- Afrin, L.B.; Weinstock, L.B.; Molderings, G.J. Covid-19 hyperinflammation and post-Covid-19 illness may be rooted in mast cell activation syndrome. Inter. J. Infect. Dis. 2020, 100, 327–332. [Google Scholar] [CrossRef]
- Pfizer Investor Day Features Significant Number of Pipeline Advances for COVID-19 Programs and Across Numerous Therapeutic Areas; Pfizer: Brooklyn, NY, USA, 2020; Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-investor-day-features-significant-numberpipeline (accessed on 25 September 2020).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: New York, NY, USA, 2006; p. 43. [Google Scholar]
- Release, S. 3: Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, 2017; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Schrodinger LLC. Maestro, Version 9.0; Schrodinger LLC: New York, NY, USA, 2009. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Kim, S.; Oshima, H.; Zhang, H.; Kern, N.R.; Re, S.; Lee, J.; Rous, B.; Sugita, Y.; Jiang, W.; Im, W. CHARMM-GUI free energy calculator for absolute and relative ligand solvation and binding free energy simulations. J. Chem. Theory Comput. 2020, 16, 7207–7218. [Google Scholar] [CrossRef]
- Ngo, S.T.; Tam, N.M.; Quan, P.M.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 2302–2312. [Google Scholar] [CrossRef]
- Tutone, M.; Perricone, U.; Almerico, A.M. Conf-VLKA: A structure-based revisitation of the Virtual Lock-and-key Approach. J. Mol. Graph. Model. 2017, 71, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inform. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
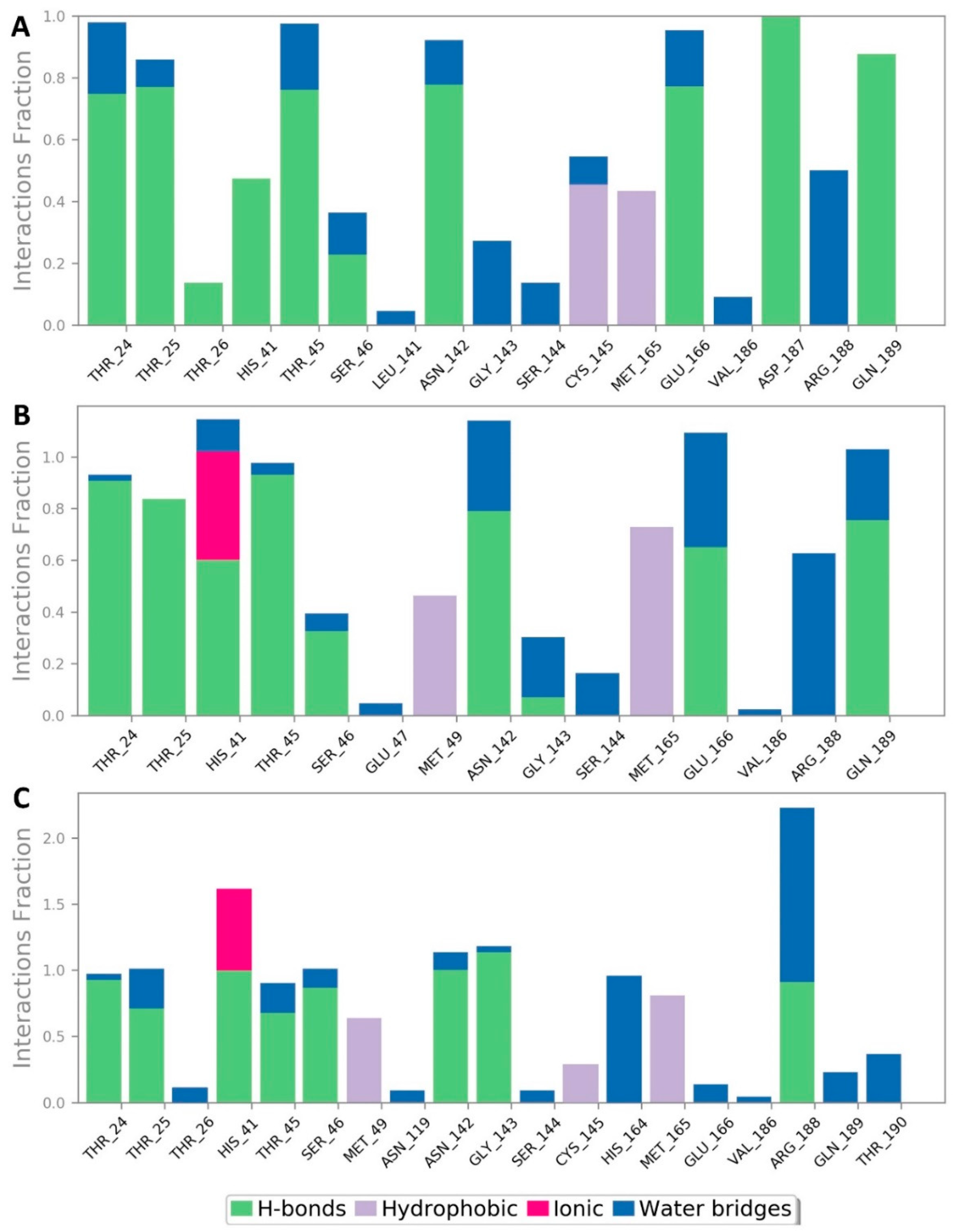
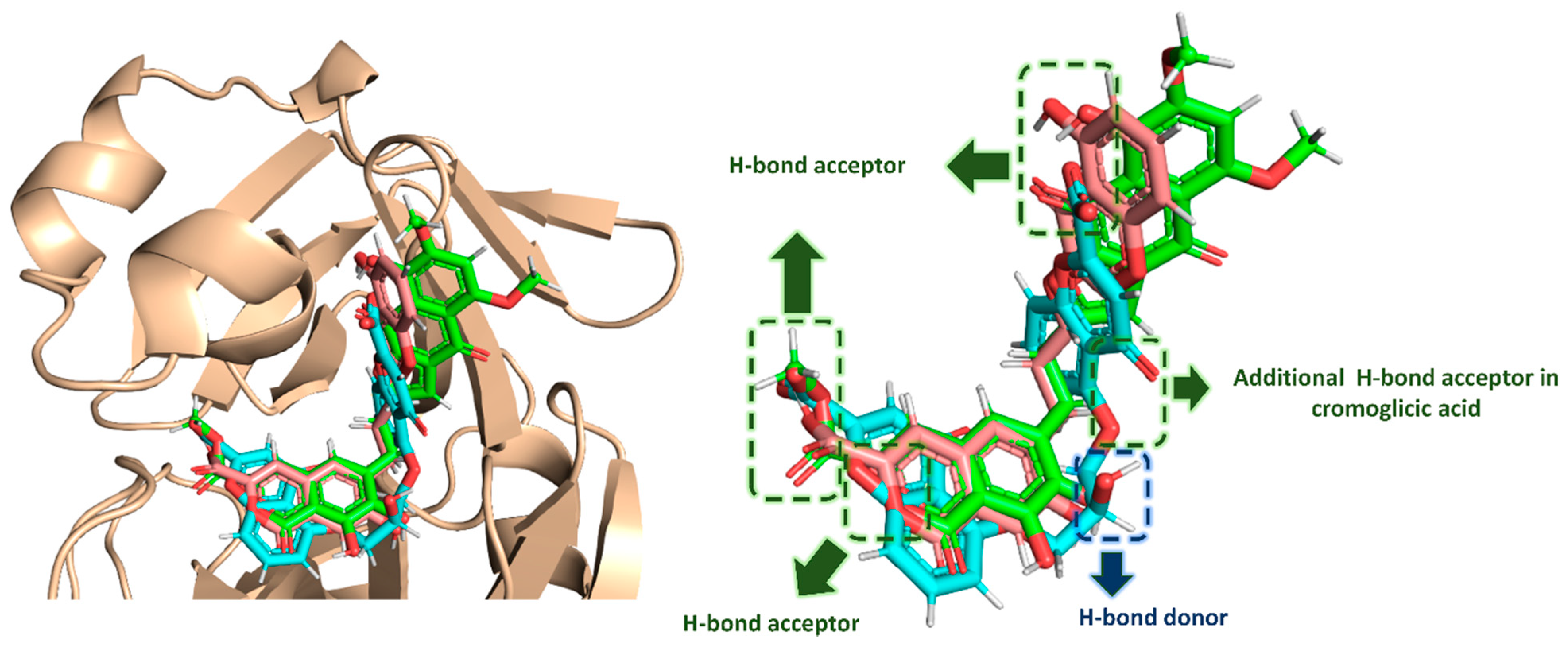
| Compound | Docking SCORE | ΔG * | Interactions | ||
|---|---|---|---|---|---|
| H-Bonding | Water Bridges | Hydrophobic | |||
| α-Rubromycin | −8.2 kcal/mol | −8.8 kcal/mol | THR-24, THR-25, HIS-41, ASN-142, GLU-166, GLN-189 | ARG-188 | MET-49, CYS-145, MET-165 |
| ScafA | −8.2 kcal/mol | −8.9 kcal/mol | THR-24, THR-25, HIS-41 **, ASN-142, GLU-166, GLN-189 | GLU-166, ASN-142, ARG-188 | MET-49, CYS-145, MET-165 |
| Cromoglicic acid | −8.3 kcal/mol | −9.2 kcal/mol | THR-24, THR-25, HIS-41 **, SER-46, ASN-142, GLY-143, GLN-189 | HIS-164, ARG-188 | MET-49, CYS-145, MET-165 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhadrami, H.A.; Sayed, A.M.; Al-Khatabi, H.; Alhakamy, N.A.; Rateb, M.E. Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor. Pharmaceuticals 2021, 14, 541. https://doi.org/10.3390/ph14060541
Alhadrami HA, Sayed AM, Al-Khatabi H, Alhakamy NA, Rateb ME. Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor. Pharmaceuticals. 2021; 14(6):541. https://doi.org/10.3390/ph14060541
Chicago/Turabian StyleAlhadrami, Hani A., Ahmed M. Sayed, Heba Al-Khatabi, Nabil A. Alhakamy, and Mostafa E. Rateb. 2021. "Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor" Pharmaceuticals 14, no. 6: 541. https://doi.org/10.3390/ph14060541
APA StyleAlhadrami, H. A., Sayed, A. M., Al-Khatabi, H., Alhakamy, N. A., & Rateb, M. E. (2021). Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor. Pharmaceuticals, 14(6), 541. https://doi.org/10.3390/ph14060541