
Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results and Discussion
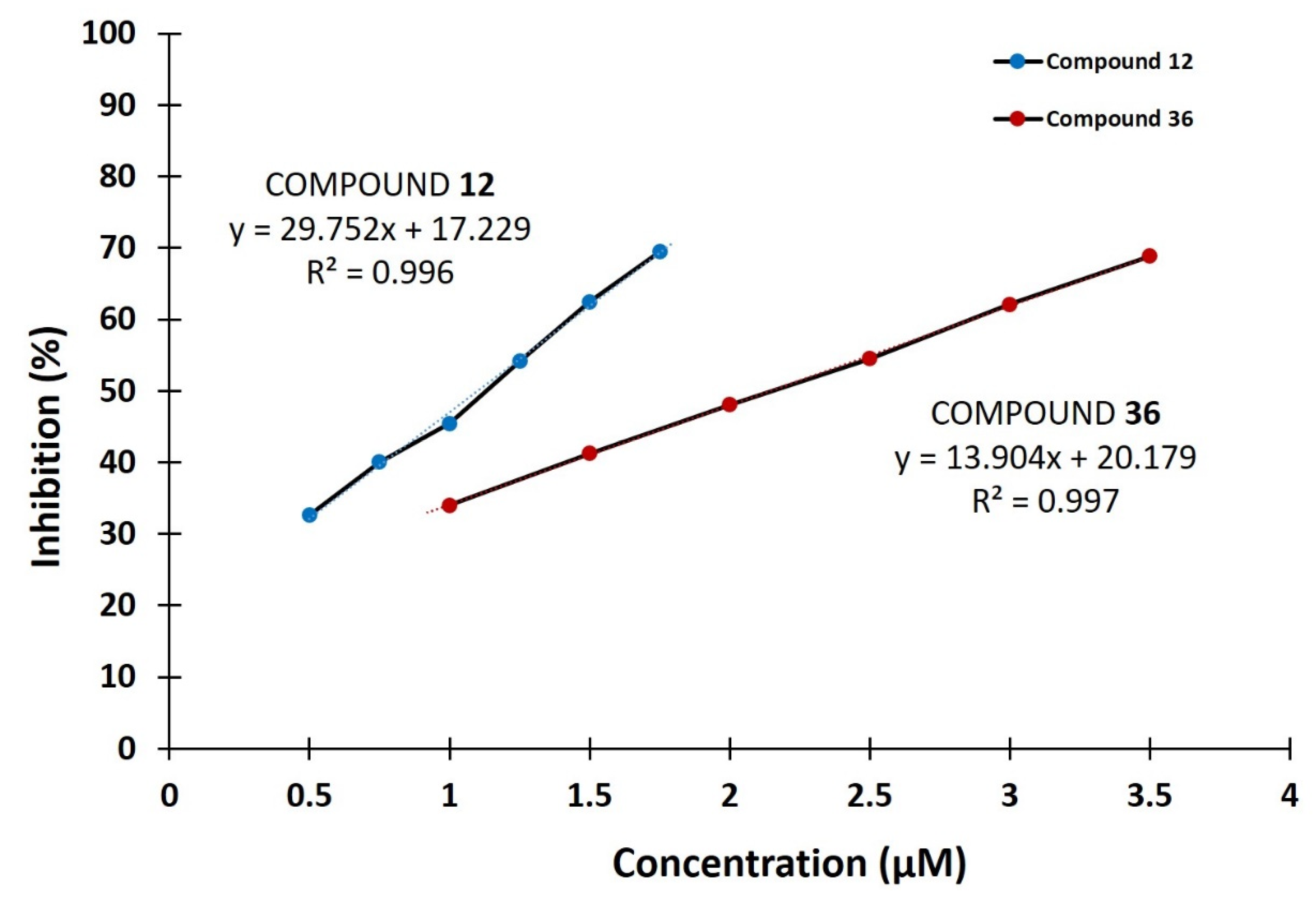
2.1. DPP III Inhibitory Activity
2.2. Results of the QSAR Analysis
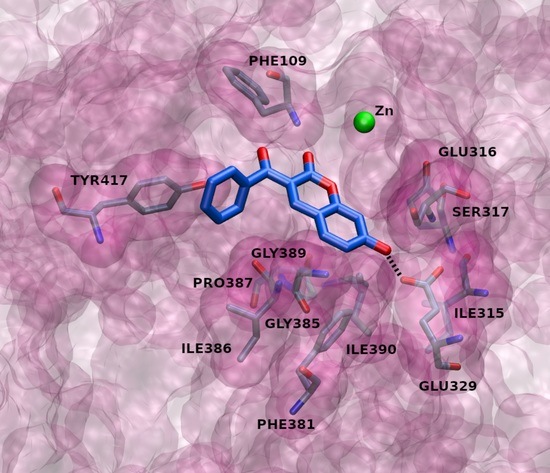
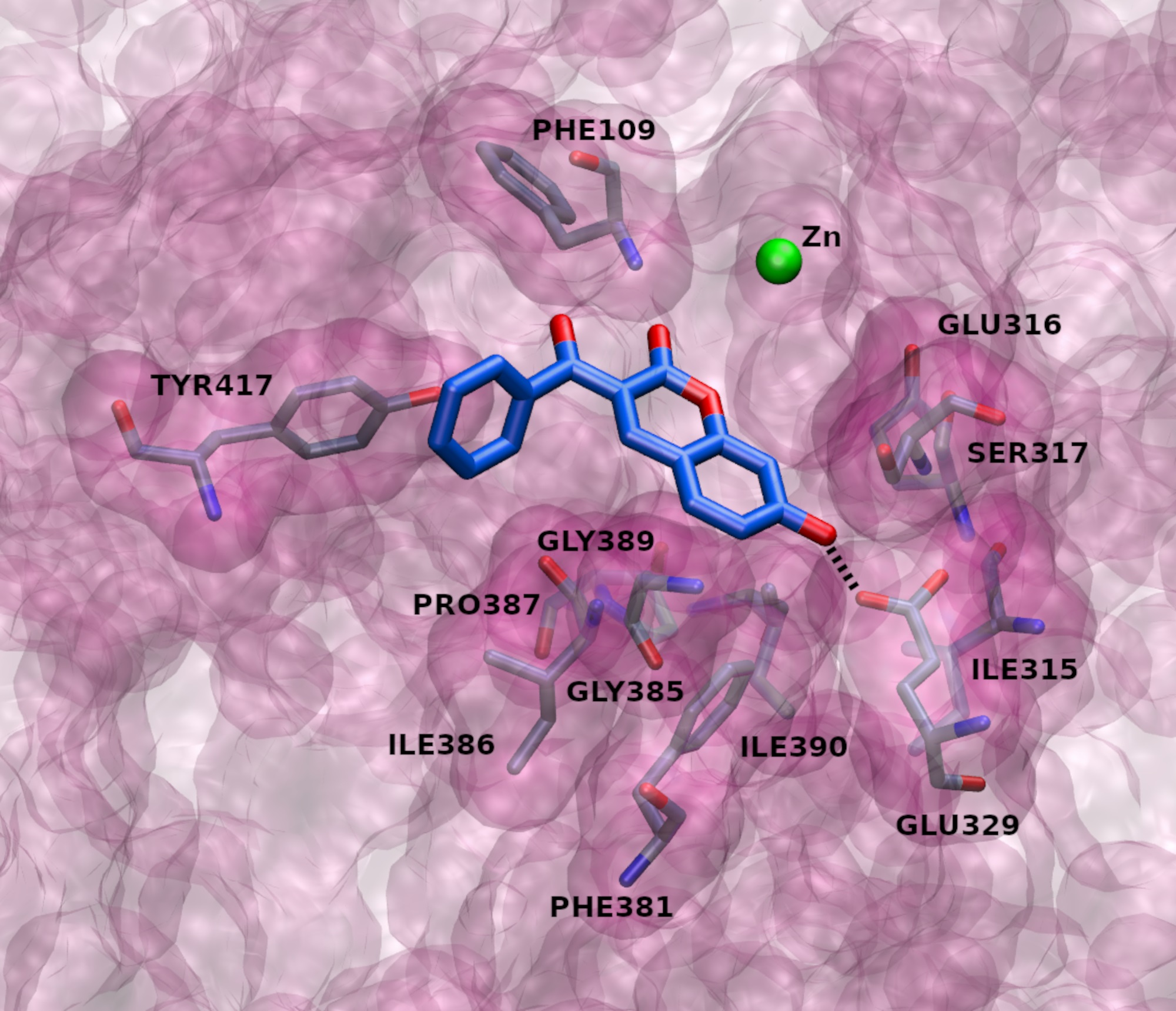
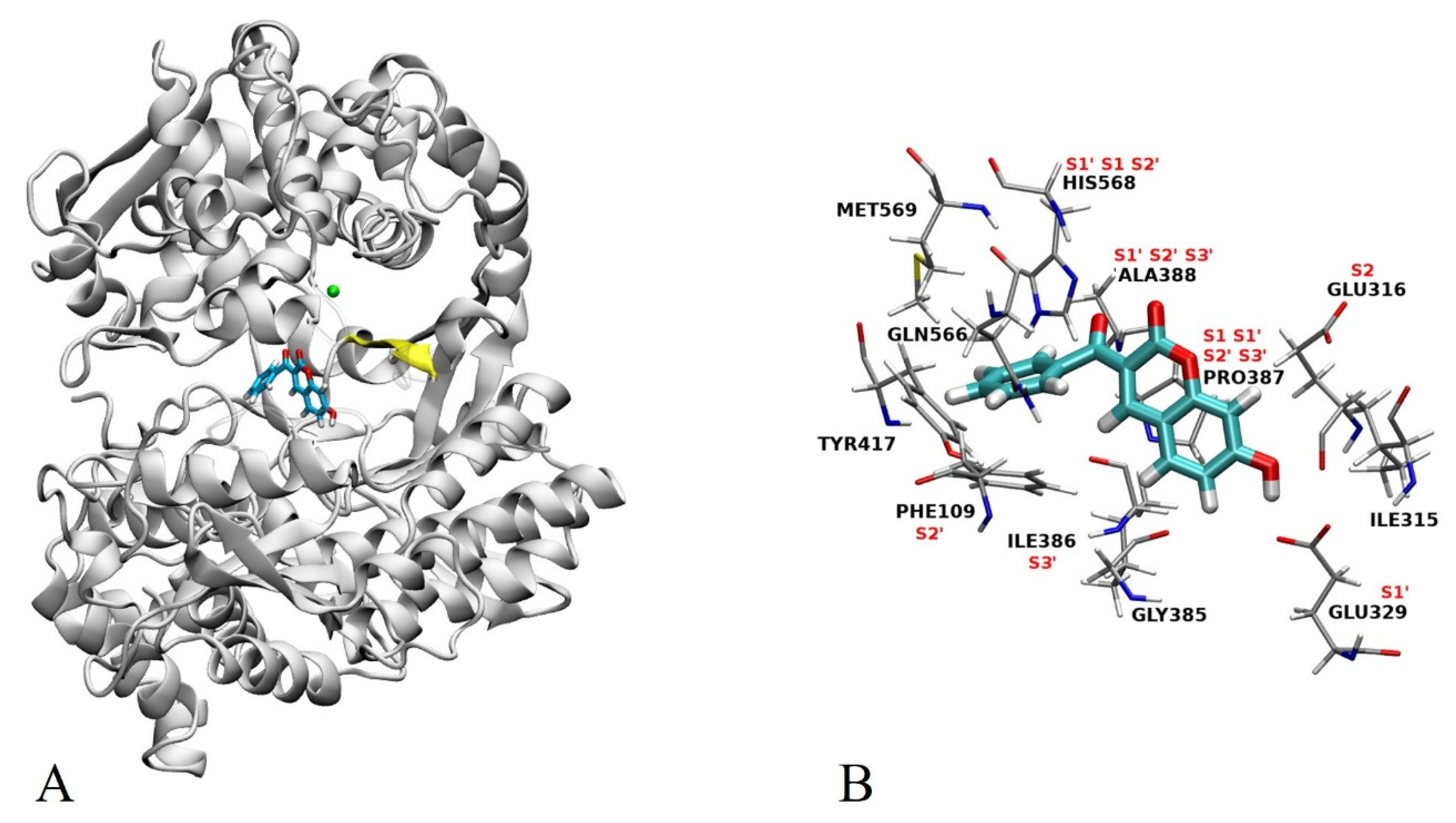
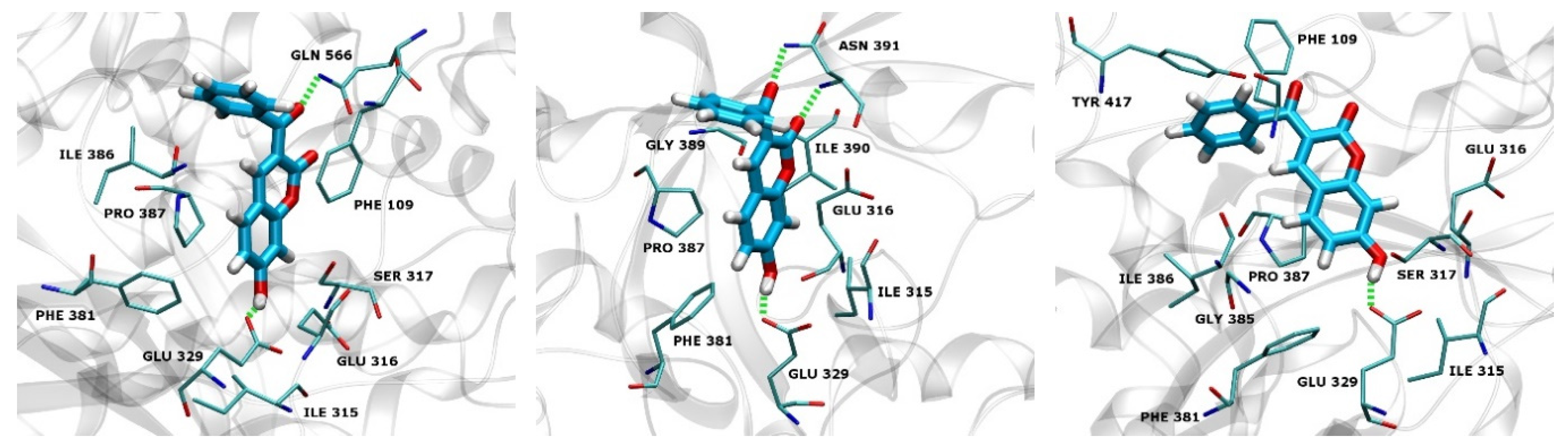
2.3. Docking
2.4. MD Simulations
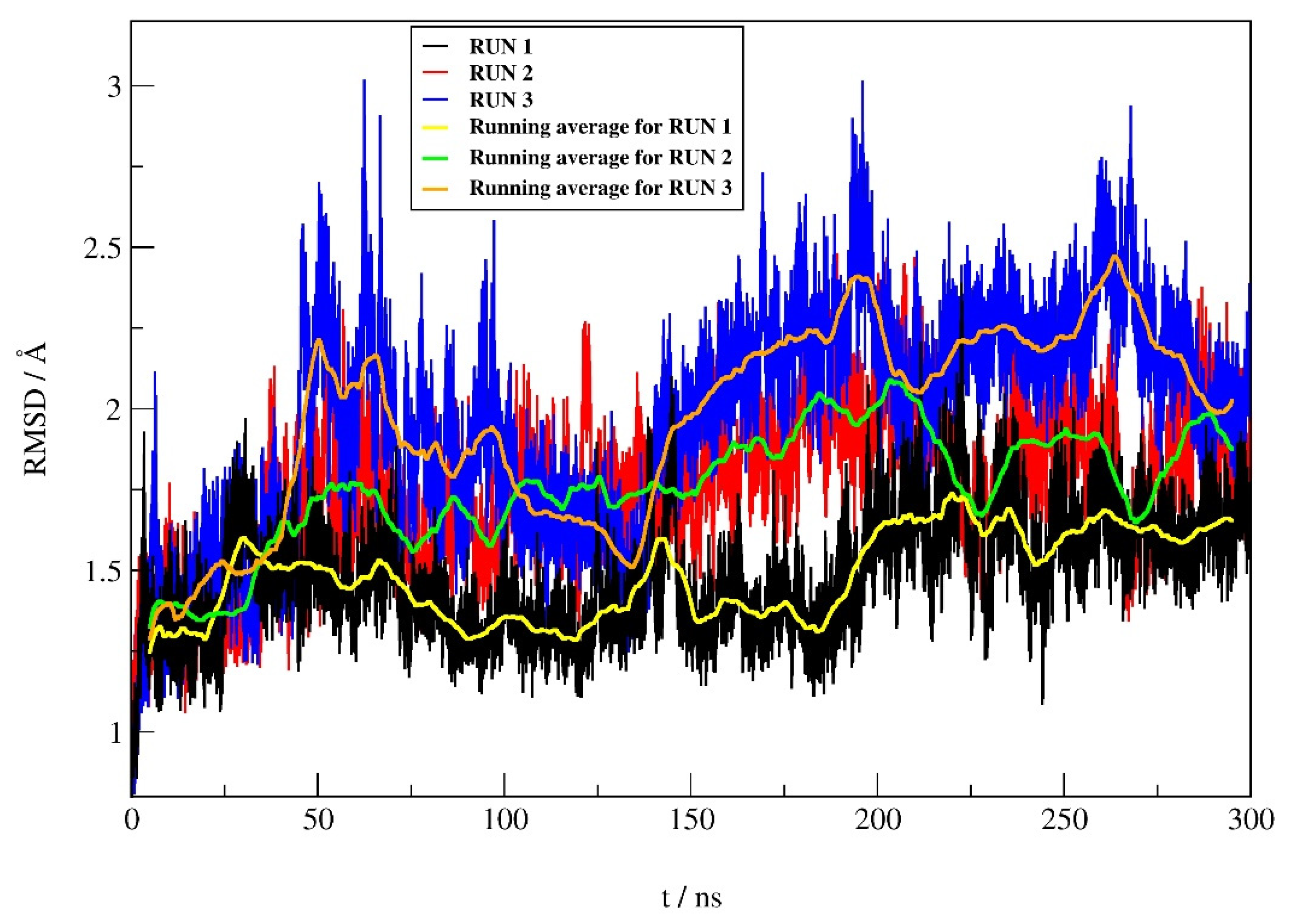
2.4.1. RMSD Profile
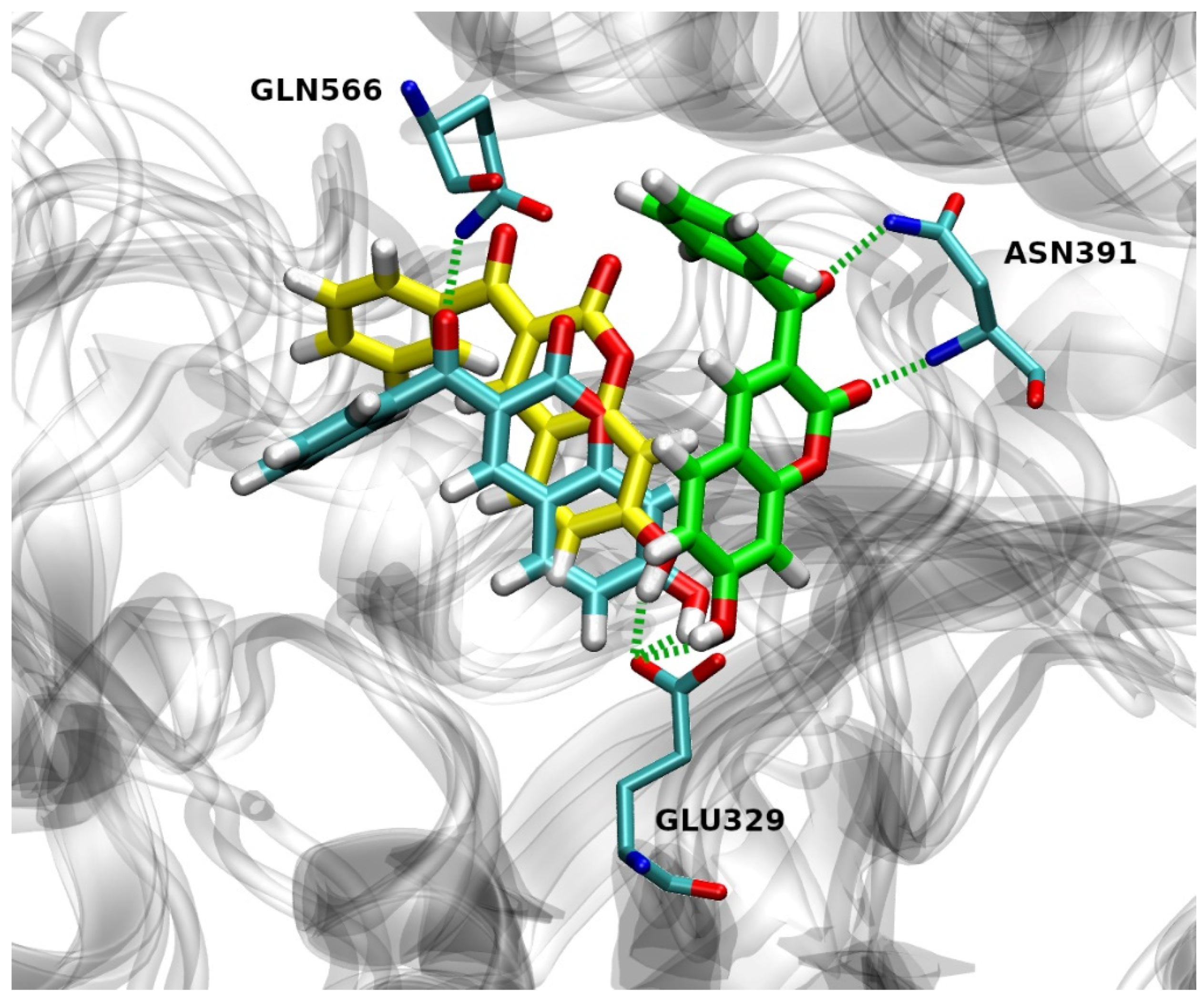
2.4.2. Hydrogen Bond Analysis
2.4.3. Native Contacts
2.4.4. Types of Intermolecular Interactions
2.4.5. MM-GBSA Free Energy Calculations
3. Material and Methods
3.1. Synthesis of Coumarin Derivatives
3.2. Heterologous Expression and Purification of Human DPP III
3.3. Assay of Human DPP III Activity
3.4. Molecular Modeling
3.4.1. QSAR Analysis
3.4.2. Preparation of the Complex Structure
3.4.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.M.; Barrett, A.J. Dipeptidyl-peptidase III. In Handbook of Proteolytic Enzymes, 2nd ed.; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2004; Volume 1, pp. 809–812. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Abramić, M.; Zubanović, M.; Vitale, L. Dipeptidyl Peptidase III from Human Erythrocytes. Biol. Chem. Hoppe Seyler 1988, 369, 29–38. [Google Scholar] [CrossRef]
- Baršun, M.; Jajčanin, N.; Vukelić, B.; Špoljarić, J.; Abramić, M. Human dipeptidyl peptidase III acts as a post-proline-cleaving enzyme on endomorphins. Biol. Chem. 2007, 388, 343–348. [Google Scholar] [CrossRef]
- Bezerra, G.A.; Dobrovetsky, E.; Viertlmayr, R.; Dong, A.; Binter, A.; Abramic, M.; Macheroux, P.; Dhe-Paganon, S.; Gruber, K. Entropy-driven binding of opioid peptides induces a large domain motion in human dipeptidyl peptidase III. Proc. Natl. Acad. Sci. USA 2012, 109, 6525–6530. [Google Scholar] [CrossRef] [PubMed]
- Karačić, Z.; Špoljarić, J.; Rozman, M.; Abramić, M. Molecular determinants of human dipeptidyl peptidase III sensitivity to thiol modifying reagents. Biol. Chem. 2012, 393, 1523–1532. [Google Scholar] [CrossRef][Green Version]
- Kumar, P.; Reithofer, V.; Reisinger, M.; Wallner, S.; Pavkov-Keller, T.; Macheroux, P.; Gruber, K. Substrate complexes of human dipeptidyl peptidase III reveal the mechanism of enzyme inhibition. Sci. Rep. 2016, 6, 23787. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; González, M.; Tomić, S. The Large Scale Conformational Change of the Human DPP III–Substrate Prefers the “Closed” Form. J. Chem. Inf. Model. 2012, 52, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Tomić, S. Hunting the human DPP III active conformation: Combined thermodynamic and QM/MM calculations. Dalton Trans. 2014, 43, 15503–15514. [Google Scholar] [CrossRef]
- Tomić, A.; Kovačević, B.; Tomić, S. Concerted nitrogen inversion and hydrogen bonding to Glu451 are responsible for protein-controlled suppression of the reverse reaction in human DPP III. Phys. Chem. Chem. Phys. 2016, 18, 27245–27256. [Google Scholar] [CrossRef]
- Liu, Y.; Kern, J.T.; Walker, J.R.; Johnson, J.A.; Schultz, P.G.; Luesch, H. A genomic screen for activators of the antioxidant response element. Proc. Natl. Acad. Sci. USA 2007, 104, 5205–5210. [Google Scholar] [CrossRef]
- Matić, S.; Kekez, I.; Tomin, M.; Bogár, F.; Šupljika, F.; Kazazić, S.; Hanić, M.; Jha, S.; Brkić, H.; Bourgeois, B.; et al. Binding of dipeptidyl peptidase III to the oxidative stress cell sensor Kelch-like ECH-associated protein 1 is a two-step process. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Sato, H.; Kimura, K.; Yamamoto, Y.; Hazato, T. Activity of DPP III in human cerebrospinal fluid derived from patients with pain. Masui. Jpn. J. Anesthesiol. 2003, 52, 257–263. [Google Scholar]
- Šimaga, Š.; Babić, D.; Osmak, M.; Ilić-Forko, J.; Vitale, L.; Miličić, D.; Abramić, M. Dipeptidyl peptidase III in malignant and non-malignant gynaecological tissue. Eur. J. Cancer 1998, 34, 399–405. [Google Scholar] [CrossRef]
- Šimaga, Š.; Babić, D.; Osmak, M.; Šprem, M.; Abramić, M. Tumor cytosol dipeptidyl peptidase III activity is increased with histological aggressiveness of ovarian primary carcinomas. Gynecol. Oncol. 2003, 91, 194–200. [Google Scholar] [CrossRef]
- Šafranko, Ž.M.; Sobočanec, S.; Šarić, A.; Jajčanin-Jozić, N.; Krsnik, Ž.; Aralica, G.; Balog, T.; Abramić, M. The effect of 17β-estradiol on the expression of dipeptidyl peptidase III and heme oxygenase 1 in liver of CBA/H mice. J. Endocrinol. Investig. 2015, 38, 471–479. [Google Scholar] [CrossRef][Green Version]
- Prajapati, S.C.; Chauhan, S.S. Human dipeptidyl peptidase III mRNA variant I and II are expressed concurrently in multiple tumor derived cell lines and translated at comparable efficiency in vitro. Mol. Biol. Rep. 2016, 43, 457–462. [Google Scholar] [CrossRef]
- Allard, M.; Simonnet, G.; Dupouy, B.; Vincent, J.D. Angiotensin II Inactivation Process in Cultured Mouse Spinal Cord Cells. J. Neurochem. 1987, 48, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Shimizu, A.; Kurita, S.; Zankov, D.P.; Takeuchi, K.; Yasuda-Yamahara, M.; Kume, S.; Ishida, T.; Ogita, H. Novel Therapeutic Role for Dipeptidyl Peptidase III in the Treatment of Hypertension. Hypertension 2016, 68, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Taschler, U.; Domenig, O.; Poglitsch, M.; Bourgeois, B.; Pollheimer, M.; Pusch, L.M.; Malovan, G.; Frank, S.; Madl, T.; et al. Dipeptidyl peptidase 3 modulates the renin–angiotensin system in mice. J. Biol. Chem. 2020, 295, 13711–13723. [Google Scholar] [CrossRef] [PubMed]
- Matovina, M.; Agić, D.; Abramić, M.; Matić, S.; Karačić, Z.; Tomić, S. New findings about human dipeptidyl peptidase III based on mutations found in cancer. RSC Adv. 2017, 7, 36326–36334. [Google Scholar] [CrossRef]
- Matić, J.; Šupljika, F.; Tir, N.; Piotrowski, P.; Schmuck, C.; Abramić, M.; Piantanida, I.; Tomić, S. Guanidiniocarbonyl-pyrrole-aryl conjugates as inhibitors of human dipeptidyl peptidase III: Combined experimental and computational study. RSC Adv. 2016, 6, 83044–83052. [Google Scholar] [CrossRef]
- Šmidlehner, T.; Karačić, Z.; Tomić, S.; Schmuck, C.; Piantanida, I. Fluorescent cyanine-guanidiniocarbonyl-pyrrole conjugate with pH-dependent DNA/RNA recognition and DPP III fluorescent labelling and inhibition properties. Mon. Chem. Chem. Mon. 2018, 149, 1307–1313. [Google Scholar] [CrossRef]
- Ćehić, M.; Sajko, J.S.; Karačić, Z.; Piotrowski, P.; Šmidlehner, T.; Jerić, I.; Schmuck, C.; Piantanida, I.; Tomić, S. The guanidiniocarbonylpyrrole–fluorophore conjugates as theragnostic tools for dipeptidyl peptidase III monitoring and inhibition. J. Biomol. Struct. Dyn. 2019, 38, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Agić, D.; Brkić, H.; Tomić, S.; Karačić, Z.; Špoljarević, M.; Lisjak, M.; Bešlo, D.; Abramić, M. Validation of flavonoids as potential dipeptidyl peptidase III inhibitors: Experimental and computational approach. Chem. Biol. Drug Des. 2017, 89, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Blagojević, B.; Agić, D.; Serra, A.T.; Matić, S.; Matovina, M.; Bijelić, S.; Popović, B.M. An in vitro and in silico evaluation of bioactive potential of cornelian cherry (Cornus mas L.) extracts rich in polyphenols and iridoids. Food Chem. 2021, 335, 127619. [Google Scholar] [CrossRef]
- Detsi, A.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Expert Opin. Ther. Pat. 2017, 27, 1201–1226. [Google Scholar] [CrossRef]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple Coumarins and Analogues in Medicinal Chemistry: Occurrence, Synthesis and Biological Activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Lončarić, M.; Gašo-Sokač, D.; Jokić, S.; Molnar, M. Recent Advances in the Synthesis of Coumarin Derivatives from Different Starting Materials. Biomolecules 2020, 10, 151. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Parellada, J.; Suárez, G.; Guinea, M. Inhibition of Zinc Metallopeptidases by Flavonoids and Related Phenolic Compounds: Structure-Activity Relationships. J. Enzym. Inhib. 1998, 13, 347–359. [Google Scholar] [CrossRef]
- Ali, Y.; Seong, S.H.; Jung, H.A.; Choi, J.S. Angiotensin-I-Converting Enzyme Inhibitory Activity of Coumarins from Angelica decursiva. Molecules 2019, 24, 3937. [Google Scholar] [CrossRef]
- Xu, X.-M.; Zhang, Y.; Qu, D.; Feng, X.-W.; Chen, Y.; Zhao, L. Osthole suppresses migration and invasion of A549 human lung cancer cells through inhibition of matrix metalloproteinase-2 and matrix metallopeptidase-9 in vitro. Mol. Med. Rep. 2012, 6, 1018–1022. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed.; Wiley VCH: Weinheim, Germany, 2000; pp. 91–92, 131–132. [Google Scholar]
- Estrada, E.; Ramírez, A. Edge Adjacency Relationships and Molecular Topographic Descriptors. Definition and QSAR Applications. J. Chem. Inf. Comput. Sci. 1996, 36, 837–843. [Google Scholar] [CrossRef]
- Rastija, V.; Agić, D.; Tomić, S.; Nikolic, S.; Hranjec, M.; Karminski-Zamola, G.; Abramić, M. Synthesis, QSAR, and Molecular Dynamics Simulation of Amidino-substituted Benzimidazoles as Dipeptidyl Peptidase III Inhibitors. Acta Chim. Slov. 2015, 62, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Schuur, J.H.; Selzer, P.; Gasteiger, J. The Coding of the Three-Dimensional Structure of Molecules by Molecular Transforms and Its Application to Structure-Spectra Correlations and Studies of Biological Activity. J. Chem. Inf. Comput. Sci. 1996, 36, 334–344. [Google Scholar] [CrossRef]
- Abramić, M.; Špoljarić, J.; Šimaga, Š. Prokaryotic homologs help to define consensus sequences in peptidase family M49. Period. Biol. 2004, 106, 161–168. [Google Scholar]
- Lončarić, M.; Sušjenka, M.; Molnar, M. An Extensive Study of Coumarin Synthesis via Knoevenagel Condensation in Choline Chloride Based Deep Eutectic Solvents. Curr. Org. Synth. 2020, 17, 98–108. [Google Scholar] [CrossRef]
- Špoljarić, J.; Salopek-Sondi, B.; Makarević, J.; Vukelić, B.; Agić, D.; Šimaga, Š.; Jajčanin-Jozić, N.; Abramić, M. Absolutely conserved tryptophan in M49 family of peptidases contributes to catalysis and binding of competitive inhibitors. Bioorg. Chem. 2009, 37, 70–76. [Google Scholar] [CrossRef]
- Hocquet, A.; Langgård, M. An Evaluation of the MM+ Force Field. J. Mol. Model. 1998, 4, 94–112. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Hanwell, M.D.; E Curtis, D.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Chemin. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.; Radchenko, E.; Zefirov, N.S.; Makarenko, A.; et al. Virtual Computational Chemistry Laboratory–Design and Description. J. Comput. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-D.; Lee, K.-H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models. Part 2. New Intercomparable Thresholds for Different Validation Criteria and the Need for Scatter Plot Inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Tomić, A.; Berynskyy, M.; Wade, R.C.; Tomić, S. Molecular simulations reveal that the long range fluctuations of human DPP III change upon ligand binding. Mol. BioSyst. 2015, 11, 3068–3080. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Horvat, G.; Ramek, M.; Agić, D.; Brkić, H.; Tomić, S. New Zinc Ion Parameters Suitable for Classical MD Simulations of Zinc Metallopeptidases. J. Chem. Inf. Model. 2019, 59, 3437–3453. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Case, D.; Betz, R.; Cerutti, D.; Cheatham, T.E., III; Darden, T.; Duke, R.E.; Giese, T.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer; Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound No. | Substituents | DPP III Inh. (%) | Log (% hDPP III Inh.) exp. | Log (% DPP III Inh.) Calc. * |
| 1 | 3-acetyl; 6-bromo | 28.5 | 1.45 | 1.29 |
| 2 | 3-acetyl; 6-hydroxy | 12.8 | 1.11 | Excl. |
| 3 | 3-acetyl; 7-diethylamino | NA | 0.00 | - |
| 4 | 3-acetyl; 7-hydroxy | 16.2 | 1.21 | 1.70 |
| 5 | 3-acetyl; 8-ethoxy | NA | 0.00 | 0.35 |
| 6 | 3-acetyl; 8-hydroxy | NA | 0.00 | - |
| 7 | 3-acetyl | 7.8 | 0.89 | 1.01 |
| 8 | 3-benzoyl; 6-chloro | 4.4 | 0.64 | 1.01 |
| 9 | 3-benzoyl; 6,8-dibromo | NA | 0.00 | - |
| 10 | 3-benzoyl; 6-hydroxy | 67.5 | 1.83 | 1.95 |
| 11 | 3-benzoyl; 7-benzoyl | 22.8 | 1.36 | 1.18 |
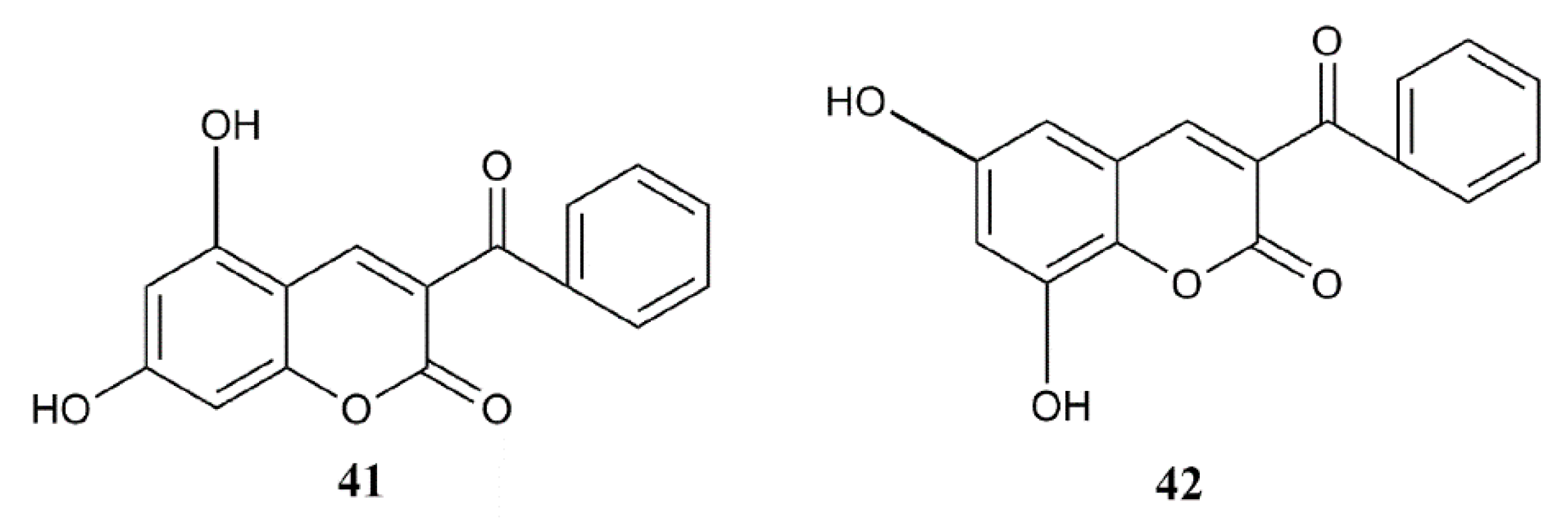
| 12 | 3-benzoyl; 7-hydroxy | 100.0 (1.10 ± 0.05 µM) | 2.00 | 1.89 |
| 13 | 3-benzoyl; 7-methoxy | 16.5 | 1.22 | 0.78 |
| 14 | 3-benzoyl; 8-ethoxy | NA | 0.00 | - |
| 15 | 3-benzoyl | 9.6 | 0.98 | 0.65 |
| 16 | 3-cyano; 6-bromo | 7.9 | 0.90 | 0.98 |
| 17 | 3-cyano; 6-methoxy | 19.8 | 1.30 | 0.83 |
| 18 | 3-cyano; 6-hydroxy | 44.6 | 1.65 | 1.81 |
| 19 | 3- cyano; 7-benzoyl | 7.1 | 0.85 | 1.04 |
| 20 | 3-cyano; 7-methoxy | NA | 0.00 | - |
| 21 | 3- cyano; 8-hydroxy | 62.6 | 1.80 | 1.67 |
| 22 | 3-cyano; 8-ethoxy | NA | 0.00 | 0.17 |
| 23 | 3-cyano | NA | 0.00 | - |
| 24 | 3-ethoxycarbonyl; 6-bromo | NA | 0.00 | - |
| 25 | 3-ethoxycarbonyl; 6-chloro | 20.1 | 1.30 | 0.93 |
| 26 | 3-ethoxycarbonyl; 6-dihydroxyamino | 59.7 | 1.78 | 1.49 |
| 27 | 3-ethoxycarbonyl; 6-hydroxy | 66.0 | 1.82 | 1.76 |
| 28 | 3- ethoxycarbonyl; 6,8-dibromo | 29.4 | 1.47 | 1.82 |
| 29 | 3-ethoxycarbonyl; 7-methoxy | NA | 0.00 | 0.26 |
| 30 | 3-ethoxycarbonyl; 8-ethoxy | NA | 0.00 | 0.37 |
| 31 | 3- ethoxycarbonyl | NA | 0.00 | - |
| 32 | 3- methoxycarbonyl; 6-bromo | 6.5 | 0.81 | 0.78 |
| 33 | 3-methoxycarbonyl; 6-dihydroxyamino | 21.2 | 1.33 | 1.46 |
| 34 | 3-methoxycarbonyl; 6-hydroxy | 23.5 | 1.37 | 1.35 |
| 35 | 3-methoxycarbonyl; 6-methoxy | 9.9 | 1.00 | 0.64 |
| 36 | 3-methoxycarbonyl; 7-hydroxy | 100.0 (2.14 ± 0.06 µM) | 2.00 | 1.50 |
| 37 | 3-methoxycarbonyl; 7-methoxy | NA | 0.00 | 0.56 |
| 38 | 3-methoxycarbonyl | 2.3 | 0.35 | 0.59 |
| 39 | coumarin | NA | 0.00 | −0.37 |
| 40 | 7-hydroxycoumarin | 2.1 | 0.33 | 0.49 |
| Statistical Parameters | Model 1 | Model 2 |
|---|---|---|
| Ntr | 27 | 26 |
| Nex | 5 | 5 |
| R2 | 0.746 | 0.796 |
| R2adj | 0.713 | 0.768 |
| s | 0.352 | 0.323 |
| F | 22.565 | 28.572 |
| Kxx | 0.210 | 0.190 |
| ΔK | 0.201 | 0.215 |
| RMSEtr | 0.325 | 0.297 |
| MAEtr | 0.276 | 0.253 |
| CCCtr | 0.855 | 0.886 |
| Q2LOO | 0.650 | 0.710 |
| RMSEcv | 0.381 | 0.354 |
| MAEcv | 0.326 | 0.302 |
| PRESScv | 3.923 | 3.258 |
| CCCcv | 0.807 | 0.844 |
| R2Yscr | 0.115 | 0.121 |
| Q2Yscr | −0.236 | −0.244 |
| RMSEext | 0.292 | 0.295 |
| MAEext | 0.255 | 0.275 |
| R2ext | 0.795 | 0.785 |
| CCCext | 0.868 | 0.873 |
| Q2F1 | 0.783 | 0.778 |
| Q2F2 | 0.780 | 0.776 |
| Q2F3 | 0.794 | 0.798 |
| r2m average | 0.614 | 0.653 |
| r2m difference | 0.186 | 0.174 |
| Applicability domain | ||
| N outliers | 1 (2) | - |
| N out of app. domain | - | - |
| Descriptor | EEig05x | Mor10u | nArOH |
|---|---|---|---|
| EEig05x | 1.000 | ||
| Mor10u | −0.264 | 1.000 | |
| nArOH | −0.129 | 0.489 | 1.000 |
| Energy Component | Run 1 | Run 2 | Run 3 |
|---|---|---|---|
| Evdw | −28.33 | −27.44 | −26.16 |
| Eele | −30.55 | −30.76 | −23.34 |
| EGB | 35.94 | 41.26 | 34.69 |
| ESA | −3.44 | −4.06 | −3.41 |
| ΔGgas | −58.88 | −58.20 | −49.50 |
| ΔGsolv | 32.50 | 37.20 | 31.28 |
| ΔGbind | −26.36 | −20.98 | −18.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agić, D.; Karnaš, M.; Šubarić, D.; Lončarić, M.; Tomić, S.; Karačić, Z.; Bešlo, D.; Rastija, V.; Molnar, M.; Popović, B.M.; et al. Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study. Pharmaceuticals 2021, 14, 540. https://doi.org/10.3390/ph14060540
Agić D, Karnaš M, Šubarić D, Lončarić M, Tomić S, Karačić Z, Bešlo D, Rastija V, Molnar M, Popović BM, et al. Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study. Pharmaceuticals. 2021; 14(6):540. https://doi.org/10.3390/ph14060540
Chicago/Turabian StyleAgić, Dejan, Maja Karnaš, Domagoj Šubarić, Melita Lončarić, Sanja Tomić, Zrinka Karačić, Drago Bešlo, Vesna Rastija, Maja Molnar, Boris M. Popović, and et al. 2021. "Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study" Pharmaceuticals 14, no. 6: 540. https://doi.org/10.3390/ph14060540
APA StyleAgić, D., Karnaš, M., Šubarić, D., Lončarić, M., Tomić, S., Karačić, Z., Bešlo, D., Rastija, V., Molnar, M., Popović, B. M., & Lisjak, M. (2021). Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study. Pharmaceuticals, 14(6), 540. https://doi.org/10.3390/ph14060540