
Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease


Abstract
1. Introduction
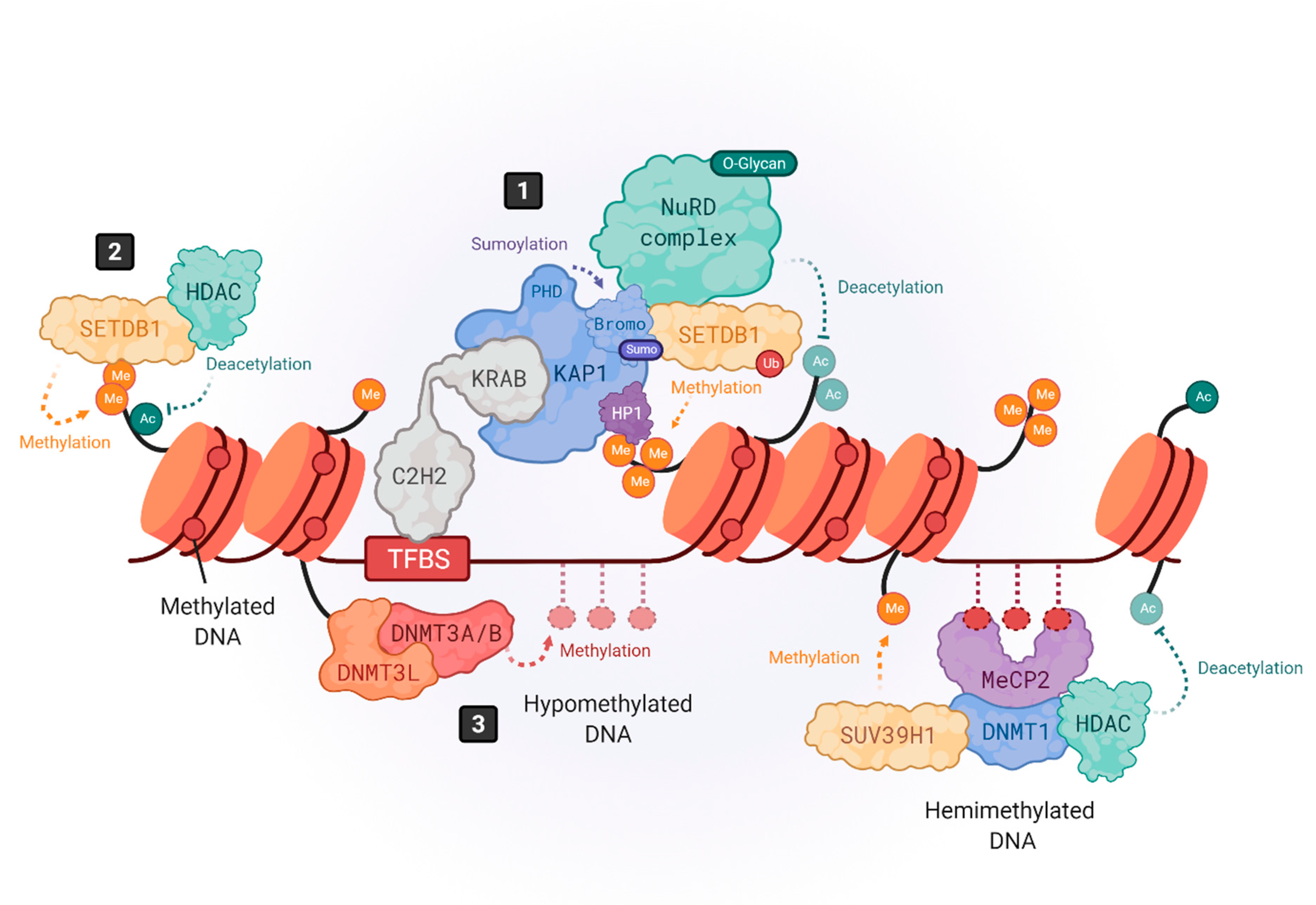
2. Epigenetic Control of HERVs

3. Transactivation of HERVs
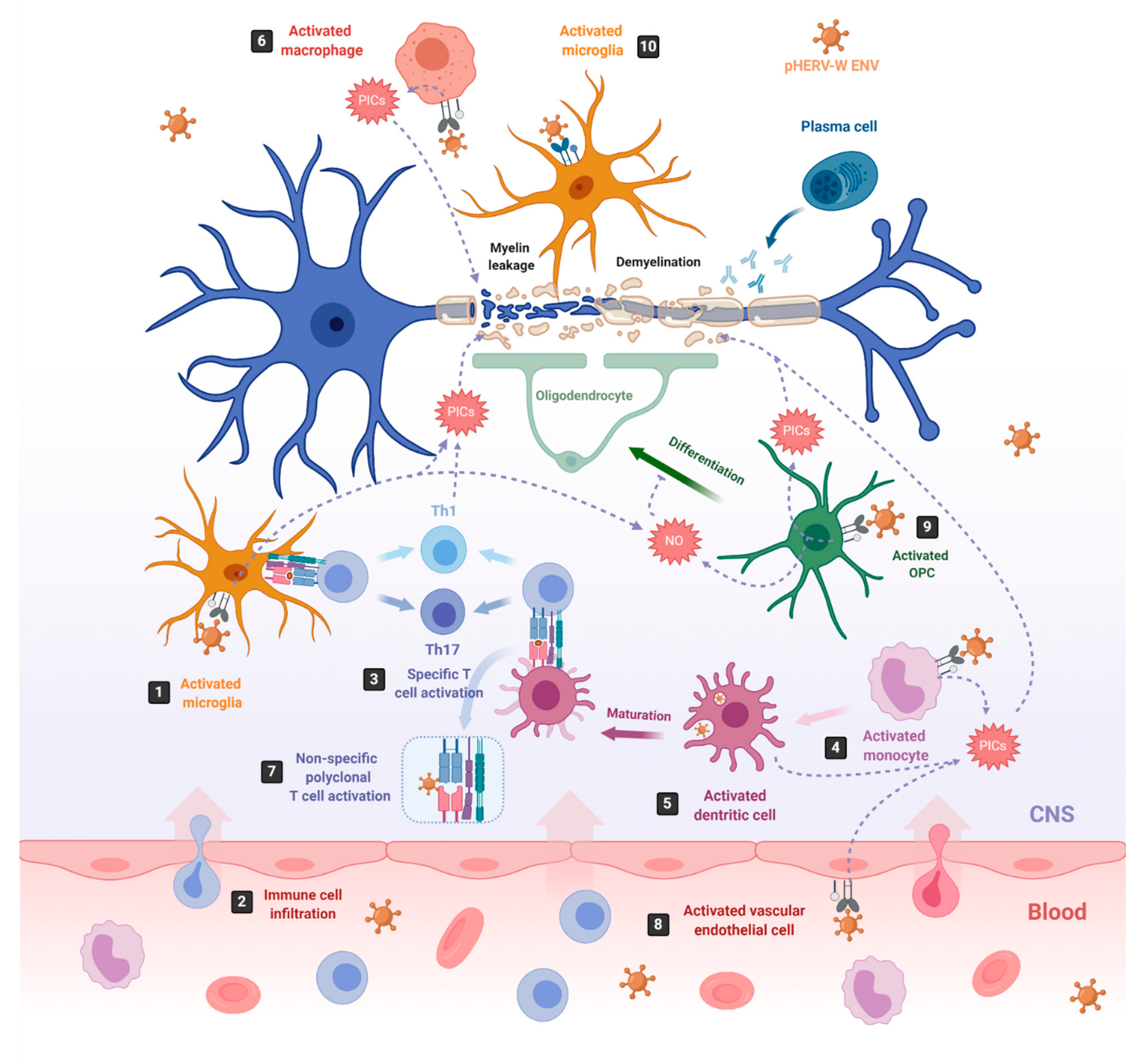
4. HERV-Associated Pathogenesis
4.1. HERV-W Related Diseases
4.2. HERV-K Related Diseases
5. HERV-Targeted Treatments
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, 1–5. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable elements, inflammation, and neurological disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef]
- Löwer, R.; Löwer, J.; Kurth, R. The viruses in all of us: Characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 5177–5184. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Retroviruses and primate evolution. Bioessays 2000, 22, 161–171. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genom. Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Belshaw, R.; Watson, J.; Katzourakis, A.; Howe, A.; Woolven-Allen, J.; Burt, A.; Tristem, M. Rate of recombinational deletion among human endogenous retroviruses. J. Virol. 2007, 81, 9437–9442. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Perron, H.; Feschotte, C. Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA 2018, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, W.; Frank, O.; Zeilfelder, U.; Spiess, B.; Greenwood, A.D.; Hehlmann, R.; Leib-Mösch, C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 2005, 79, 341–352. [Google Scholar] [CrossRef]
- Stauffer, Y.; Theiler, G.; Sperisen, P.; Lebedev, Y.; Jongeneel, C.V. Digital expression profiles of human endogenous retroviral families in normal and cancerous tissues. Cancer Immun. Arch. 2004, 4, 2. [Google Scholar]
- Oja, M.; Peltonen, J.; Blomberg, J.; Kaski, S. Methods for estimating human endogenous retrovirus activities from EST databases. BMC Bioinform. 2007, 8, S2–S11. [Google Scholar] [CrossRef]
- Shin, W.; Lee, J.; Son, S.Y.; Ahn, K.; Kim, H.S.; Han, K. Human-specific HERV-K insertion causes genomic variations in the human genome. PLoS ONE 2013, 8, e60605. [Google Scholar] [CrossRef] [PubMed]
- Medstrand, P.; Landry, J.R.; Mager, D.L. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J. Biol. Chem. 2001, 276, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef]
- Suntsova, M.; Garazha, A.; Ivanova, A.; Kaminsky, D.; Zhavoronkov, A.; Buzdin, A. Molecular functions of human endogenous retroviruses in health and disease. Cell Mol. Life Sci. 2015, 72, 3653–3675. [Google Scholar] [CrossRef]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef]
- Hu, T.; Pi, W.; Zhu, X.; Yu, M.; Ha, H.; Shi, H.; Choi, J.-H.; Tuan, D. Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucleic Acids Res. 2017, 45, 4479–4492. [Google Scholar] [CrossRef]
- Wilson, K.D.; Ameen, M.; Guo, H.; Abilez, O.J.; Tian, L.; Mumbach, M.R.; Diecke, S.; Qin, X.; Liu, Y.; Yang, H.; et al. Endogenous Retrovirus-Derived lncRNA BANCR Promotes Cardiomyocyte Migration in Humans and Non-human Primates. Dev. Cell 2020, 54, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Mallet, F.; Bouton, O.; Prudhomme, S.; Cheynet, V.; Oriol, G.; Bonnaud, B.; Lucotte, G.; Duret, L.; Mandrand, B. The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc. Natl. Acad. Sci. USA 2004, 101, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Larsson, E.; Andersson, G. Beneficial role of human endogenous retroviruses: Facts and hypotheses. Scand. J. Immunol. 1998, 48, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; Renard, M.; Schlecht-Louf, G.; Bouallaga, I.; Heidmann, O.; Letzelter, C.; Richaud, A.; Ducos, B.; Heidmann, T. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20534–20539. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Jouvin-Marche, E.; Michel, M.; Ounanian-Paraz, A.; Camelo, S.; Dumon, A.; Jolivet-Reynaud, C.; Marcel, F.; Souillet, Y.; Borel, E.; et al. Multiple sclerosis retrovirus particles and recombinant envelope trigger an abnormal immune response in vitro, by inducing polyclonal Vbeta16 T-lymphocyte activation. Virology 2001, 287, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Song, F.; Fernandez-Escobar, A.; Luo, G.; Wang, J.H.; Sun, Y. The Properties of Cytokines in Multiple Sclerosis: Pros and Cons. Am. J. Med. Sci. 2018, 356, 552–560. [Google Scholar] [CrossRef]
- Mameli, G.; Astone, V.; Arru, G.; Marconi, S.; Lovato, L.; Serra, C.; Sotgiu, S.; Bonetti, B.; Dolei, A. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not Human herpesvirus 6. J. Gen. Virol. 2007, 88, 264–274. [Google Scholar] [CrossRef]
- Nowak, J.; Januszkiewicz, D.; Pernak, M.; Liweń, I.; Zawada, M.; Rembowska, J.; Nowicka, K.; Lewandowski, K.; Hertmanoswka, H.; Wender, M. Multiple sclerosis-associated virus-related pol sequences found both in multiple sclerosis and healthy donors are more frequently expressed in multiple sclerosis patients. J. Neurovirol. 2003, 9, 112–117. [Google Scholar] [CrossRef]
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology 2009, 6, 104. [Google Scholar] [CrossRef]
- Reale, M.; Costantini, E.; Greig, N.H. Cytokine Imbalance in Schizophrenia. From Research to Clinic: Potential Implications for Treatment. Front. Psychiatry 2021, 12, 536257. [Google Scholar] [CrossRef]
- Karlsson, H.; Schröder, J.; Bachmann, S.; Bottmer, C.; Yolken, R.H. HERV-W-related RNA detected in plasma from individuals with recent-onset schizophrenia or schizoaffective disorder. Mol. Psychiatry 2004, 9, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Mekaoui, L.; Bernard, C.; Veas, F.; Stefas, I.; Leboyer, M. Endogenous retrovirus type W GAG and envelope protein antigenemia in serum of szhizophrenia patients. Biol. Psychiatry 2008, 64, 1019–1023. [Google Scholar] [CrossRef]
- Huang, W.; Li, S.; Hu, Y.; Yu, H.; Luo, F.; Zhang, Q.; Zhu, F. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Hamdani, N.; Faucard, R.; Lajnef, M.; Jamain, S.; Daban-Huard, C.; Sarrazin, S.; Leguen, E.; Houenou, J.; Delavest, M.; et al. Molecular characteristics of Human Endogenous Retrovirus type-W in schizophrenia and bipolar disorder. Transl. Psychiatry 2012, 2, e201. [Google Scholar] [CrossRef]
- Fries, G.R.; Walss-Bass, C.; Bauer, M.E.; Teixeira, A.L. Revisiting inflammation in bipolar disorder. Pharmacol. Biochem. Behav. 2019, 177, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiat-ric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Balestrieri, E.; Cipriani, C.; Matteucci, C.; Benvenuto, A.; Coniglio, A.; Argaw-Denboba, A.; Toschi, N.; Bucci, I.; Miele, M.T.; Grelli, S.; et al. Children With Autism Spectrum Disorder and Their Mothers Share Abnormal Expression of Selected Endogenous Retroviruses Families and Cytokines. Front. Immunol. 2019, 10, 2244. [Google Scholar] [CrossRef]
- Anand, D.; Colpo, G.D.; Zeni, G.; Zeni, C.P.; Teixeira, A.L. Attention-Deficit/Hyperactivity Disorder And Inflammation: What Does Current Knowledge Tell Us? A Systematic Review. Front. Psychiatry 2017, 8, 228. [Google Scholar] [CrossRef]
- D’Agati, E.; Pitzianti, M.; Balestrieri, E.; Matteucci, C.; Sinibaldi Vallebona, P.; Pasini, A. First evidence of HERV-H transcriptional activity reduction after methylphenidate treatment in a young boy with ADHD. New Microbiol. 2016, 39, 237–239. [Google Scholar]
- Chiara, C.; Bernanda, P.M.; Claudia, M.; Elisa, D.A.; Tony, M.M.; Valentina, R.; Sandro, G.; Paolo, C.; Paola, S.V.; Augusto, P.; et al. The Decrease in Human Endogenous Retrovirus-H Activity Runs in Parallel with Improvement in ADHD Symptoms in Patients Undergoing Methylphenidate Therapy. Int. J. Mol. Sci. 2018, 19, 3286. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Geldern, G.V.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef] [PubMed]
- Polverino, A.; Rucco, R.; Stillitano, I.; Bonavita, S.; Grimaldi, M.; Minino, R.; Pesoli, M.; Trojsi, F.; D’Ursi, A.M.; Sorrentino, G.; et al. In Amyotrophic Lateral Sclerosis Blood Cytokines Are Altered, but Do Not Correlate with Changes in Brain Topology. Brain Connect. 2020, 10, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pintó, I.; Agmon-Levin, N.; Howard, A.; Shoenfeld, Y. Fibromyalgia and cytokines. Immunol Lett. 2014, 161, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, T.; Sadones, O.; Sánchez-Fito, T.; Almenar-Pérez, E.; Espejo, J.A.; Martín-Martínez, E.; Nathanson, L.; Oltra, E. Activation of transposable elements in immune cells of fibromyalgia patients. Int. J. Mol. Sci. 2020, 21, 1366. [Google Scholar] [CrossRef]
- Yang, T.; Yang, Y.; Wang, D.; Li, C.; Qu, Y.; Guo, J.; Shi, T.; Bo, W.; Sun, Z.; Asakawa, T. The clinical value of cytokines in chronic fatigue syndrome. J. Transl. Med. 2019, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.S.; da Silva Nali, L.H.; Leal, C.O.D.; Sabino, E.C.; Lacerda, E.M.; Kingdon, C.C.; Nacul, L.; Romano, C.M.; Kingdon, C. HERV-K and HERV-W transcriptional activity in myalgic encephalomyelitis/chronic fatigue syndrome. Auto Immun. Highlights 2019, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Hurst, T.P.; Magiorkinis, G. Epigenetic control of human endogenous retrovirus expression: Focus on regulation of long-terminal repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Turelli, P.; Playfoot, C.; Grun, D.; Raclot, C.; Pontis, J.; Coudray, A.; Thorball, C.; Duc, J.; Pankevich, E.V.; Deplancke, B.; et al. Primate-restricted KRAB zinc finger proteins and target retrotransposons control gene expression in human neurons. Sci. Adv. 2020, 6, eaba3200. [Google Scholar] [CrossRef]
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef]
- Ecco, G.; Imbeault, M.; Trono, D. A tale of domestication: The endovirome, its polydactyl controllers and the species-specificity of human biology. Development 2020, 144, 2719–2729. [Google Scholar] [CrossRef]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.A.; Oda, S.-I.; Chong, Z.-S.; Yu, M.; McLaughlin, S.H.; Modis, Y. Structure of KAP1 tripartite motif identifies molecular interfaces required for retroelement silencing. Proc. Natl. Acad. Sci. USA 2019, 116, 15042–15051. [Google Scholar] [CrossRef]
- Grassi, D.A.; Jonsson, M.E.; Brattas, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19, e45000. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J., III. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box–zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 system contributes to the early embryonic establishment of site-specific DNA methylation patterns maintained during development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef]
- Turelli, P.; Castro-Diaz, N.; Marzetta, F.; Kapopoulou, A.; Raclot, C.; Duc, J.; Tieng, V.; Quenneville, S.; Trono, D. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res. 2014, 24, 1260–1270. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef]
- Ishimoto, K.; Kawamata, N.; Uchihara, Y.; Okubo, M.; Fujimoto, R.; Gotoh, E.; Kakinouchi, K.; Mizohata, E.; Hino, N.; Okada, Y.; et al. Ubiquitination of lysine 867 of the human SETDB1 protein upregulates its histone H3 lysine 9 (H3K9) methyltransferase activity. PLoS ONE 2016, 11, e0165766. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, M.E.; Garza, R.; Sharma, Y.; Petri, R.; Södersten, E.; Johansson, J.G.; Johansson, P.A.; Atacho, D.A.; Pircs, K.; Madsen, S.; et al. Activation of endogenous retroviruses during brain development causes an inflammatory response. EMBO J. 2021, 40, e106423. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Collins, P.L.; Kyle, K.E.; Egawa, T.; Shinkai, Y.; Oltz, E.M. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 8367–8372. [Google Scholar] [CrossRef]
- Tan, S.L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.-I.; Wolf, M.; Kurumizaka, H. Structural basis of heterochromatin formation by human HP1. Mol. Cell. 2018, 69, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Azebi, S.; England, P.; Christensen, T.; Møller-Larsen, A.; Petersen, T.; Batsché, E.; Muchardt, C. Citrullination of histone H3 interferes with HP1-mediated transcriptional repression. PLoS Genet. 2012, 8, e1002934. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J., III. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef]
- Stewart, M.D.; Li, J.; Wong, J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell Biol. 2005, 25, 2525–2538. [Google Scholar] [CrossRef]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jönsson, M.E.; Männe, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef]
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157. [Google Scholar] [CrossRef]
- Sharif, J.; Endo, T.A.; Nakayama, M.; Karimi, M.M.; Shimada, M.; Katsuyama, K.; Goyal, P.; Brind’Amour, J.; Sun, M.-A.; Sun, Z.; et al. Activation of endogenous retroviruses in dnmt1(−/−) ESCs involves disruption of SETDB1-mediated repression by NP95 binding to hemimethylated DNA. Cell Stem Cell. 2016, 19, 81–94. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-H.; Liao, H.-F.; Wolf, D.; Tai, K.-Y.; Chuang, C.-Y.; Lee, H.-S.; Kuo, H.-C.; Hata, K.; Zhang, X.; Cheng, X.; et al. Ectopic DNMT3L triggers assembly of a repressive complex for retroviral silencing in somatic cells. J. Virol. 2014, 88, 10680–10695. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, G.; Tao, T.; Zhang, D.; Liu, X.; Qiu, H.; Han, L.; Xu, Z.; Xiao, Y.; Cheng, C.; Shen, A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology 2016, 26, 820–833. [Google Scholar] [CrossRef]
- Gao, J.; Yang, Y.; Qiu, R.; Zhang, K.; Teng, X.; Liu, R.; Wang, Y. Proteomic analysis of the OGT interactome: Novel links to epithelial-mesenchymal transition and metastasis of cervical cancer. Carcinogenesis 2018, 39, 1222–1234. [Google Scholar] [CrossRef]
- Boulard, M.; Rucli, S.; Edwards, J.R.; Bestor, T.H. Methylation-directed glycosylation of chromatin factors represses retrotransposon promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 14292–14298. [Google Scholar] [CrossRef] [PubMed]
- Foroushani, A.K.; Chim, B.; Wong, M.; Rastegar, A.; Barbian, K.; Martens, C.; Hafner, M.; Muljo, S.A. Post-transcriptional regulation of human endogenous retroviruses by RNA-Binding Motif Protein 4, RBM4. Proc. Natl. Acad. Sci. USA 2020, 117, 26520–26530. [Google Scholar] [CrossRef]
- Chelmicki, T.; Roger, E.; Teissandier, A.; Dura, M.; Bonneville, L.; Rucli, S.; Dossin, F.; Fouassier, C.; Lameiras, S.; Bourc’his, D. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 2021, 591, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Garson, J.A.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef]
- Karlsson, H.; Bachmann, S.; Schröder, J.; McArthur, J.; Torrey, E.F.; Yolken, R.H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4634–4639. [Google Scholar] [CrossRef] [PubMed]
- Slokar, G.; Hasler, G. Human endogenous retroviruses as pathogenic factors in the development of schizophrenia. Front. Psychiatry 2015, 6, 183. [Google Scholar] [CrossRef]
- Johansson, E.M.; Bouchet, D.; Tamouza, R.; Ellul, P.; Morr, A.S.; Avignone, E.; Germi, R.; Leboyer, M.; Perron, H.; Groc, L. Human endogenous retroviral protein triggers deficit in glutamate synapse maturation and behaviors associated with psychosis. Sci. Adv. 2020, 6, eabc0708. [Google Scholar] [CrossRef]
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Blond, J.L.; Besème, F.; Duret, L.; Bouton, O.; Bedin, F.; Perron, H.; Mandrand, B.; Mallet, F. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J. Virol. 1999, 73, 1175–1185. [Google Scholar] [CrossRef]
- Pavlícek, A.; Paces, J.; Elleder, D.; Hejnar, J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: Their integration, stability, and distribution. Genome Res. 2002, 12, 391–399. [Google Scholar] [CrossRef]
- Costas, J. Characterization of the intragenomic spread of the human endogenous retrovirus family HERV-W. Mol. Biol. Evol. 2002, 19, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Bouton, O.; Bedin, F.; Duret, L.; Mandrand, B.; Mallet, F.; Paranhos-Baccala, G. Chromosomal distribution and coding capacity of the human endogenous retrovirus HERV-W family. AIDS Res. Hum. Retroviruses 2000, 16, 731–740. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, N.; Lazar, V.; Casella, J.-F.; Benit, L.; Heidmann, T. Survey of human genes of retroviral origin: Identification and transcriptome of the genes with coding capacity for complete envelope proteins. J. Virol. 2003, 77, 10414–10422. [Google Scholar] [CrossRef] [PubMed]
- Roebke, C.; Wahl, S.; Laufer, G.; Stadelmann, C.; Sauter, M.; Mueller-Lantzsch, N.; Mayer, J.; Ruprecht, K. An N-terminally truncated envelope protein encoded by a human endogenous retrovirus W locus on chromosome Xq22.3. Retrovirology 2010, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Kim, D.-S.; Huh, J.-W.; Ahn, K.; Yi, J.-M.; Lee, J.-R.; Hirai, H. Molecular characterization of the HERV-W env gene in humans and primates: Expression, FISH, phylogeny, and evolution. Mol. Cells 2008, 26, 53–60. [Google Scholar]
- Lavillette, D.; Marin, M.; Ruggieri, A.; Mallet, F.; Cosset, F.-L.; Kabat, D. The envelope glycoprotein of human endogenous retrovirus type W uses a divergent family of amino acid transporters/cell surface receptors. J. Virol. 2002, 76, 6442–6452. [Google Scholar] [CrossRef]
- Gong, R.; Peng, X.; Kang, S.; Feng, H.; Huang, J.; Zhang, W.; Lin, D.; Tien, P.; Xiao, G. Structural characterization of the fusion core in syncytin, envelope protein of human endogenous retrovirus family W. Biochem. Biophys. Res. Commun. 2005, 331, 1193–1200. [Google Scholar] [CrossRef]
- Komurian-Pradel, F.; Paranhos-Baccala, G.; Bedin, F.; Ounanian-Paraz, A.; Sodoyer, M.; Ott, C.; Rajoharison, A.; Garcia, E.; Mallet, F.; Mandrand, B.; et al. Molecular cloning and characterization of MSRV-related sequences associated with retrovirus-like particles. Virology 1999, 260, 1–9. [Google Scholar] [CrossRef]
- Ruprecht, K.; Mayer, J. On the origin of a pathogenic HERV-W envelope protein present in multiple sclerosis lesions. Proc. Natl. Acad. Sci. USA 2019, 116, 19791–19792. [Google Scholar] [CrossRef]
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed endogenous retroviral insertions in the human population. J. Virol. 2014, 88, 9529–9537. [Google Scholar] [CrossRef]
- Serra, C.; Mameli, G.; Arru, G.; Sotgiu, S.; Rosati, G.; Dolei, A. In vitro modulation of the multiple sclerosis (MS)-associated retrovirus by cytokines: Implications for MS pathogenesis. J. Neurovirol. 2003, 9, 637–643. [Google Scholar] [CrossRef]
- Kremer, D.; Gruchot, J.; Weyers, V.; Oldemeier, L.; Göttle, P.; Healy, L.; Jang, J.H.; Xu, Y.K.T.; Volsko, C.; Dutta, R.; et al. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15216–15225. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Reynaud, J.M.; Gourru-Lesimple, G.; Perron, H.; Marche, P.N.; Horvat, B. Induction of Proinflammatory Multiple Sclerosis-Associated Retrovirus Envelope Protein by Human Herpesvirus-6A and CD46 Receptor Engagement. Front. Immunol. 2018, 9, 2803. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Pierquin, J.; Brunel, J.; Gorter, R.; Quétard, C.; Horvat, B.; Amor, S.; Portoukalian, J.; Perron, H. Human Endogenous Retrovirus Type W Envelope from Multiple Sclerosis Demyelinating Lesions Shows Unique Solubility and Antigenic Characteristics. Virol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Z.; Wang, P.; Li, S.; Zeng, J.; Tu, X.; Yan, Q.; Xiao, Z.; Pan, M.; Zhu, F. Syncytin-1, an endogenous retroviral protein, triggers the activation of CRP via TLR3 signal cascade in glial cells. Brain Behav. Immun. 2018, 67, 324–334. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef]
- Boller, K.; König, H.; Sauter, M.; Mueller-Lantzsch, N.; Löwer, R.; Löwer, J.; Kurth, R. Evidence that HERV-K is the endogenous retrovirus sequence that codes for the human teratocarcinoma-derived retrovirus HTDV. Virology 1993, 96, 349–353. [Google Scholar] [CrossRef]
- Bieda, K.; Hoffmann, A.; Boller, K. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J. Gen. Virol. 2001, 82, 591–596. [Google Scholar] [CrossRef]
- Dewannieux, M.; Blaise, S.; Heidmann, T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J. Virol. 2005, 79, 15573–15577. [Google Scholar] [CrossRef]
- Boller, K.; Schönfeld, K.; Lischer, S.; Fischer, N.; Hoffmann, A.; Kurth, R.; Tönjes, R.R. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J. Gen. Virol. 2008, 89, 567–572. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, M.; Brocks, D.; Sheng, Y.-H.; Islam, M.S.; Ressnerova, A.; Assenov, Y.; Milde, T.; Oehme, I.; Witt, O.; Goyal, A.; et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 2018, 17, 811–822. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Lang, A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin. Rev. Allergy Immunol. 2010, 39, 51–61. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Multiple sclerosis: Autoimmunity and viruses. Curr. Opin. Rheumatol. 2013, 25, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Limosin, F.; Rouillon, F.; Payan, C.; Cohen, J.-M.; Strub, N. Prenatal exposure to influenza as a risk factor for adult schizophrenia: Influenza and schizophrenia. Acta Psychiatry Scand. 2003, 107, 331–335. [Google Scholar] [CrossRef]
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 2003, 23, 297–302. [Google Scholar] [CrossRef]
- Ebert, T.; Kotler, M. Prenatal exposure to influenza and the risk of subsequent development of schizophrenia. Isr. Med. Assoc. J. 2005, 7, 35–38. [Google Scholar] [CrossRef]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Nellåker, C.; Yao, Y.; Jones-Brando, L.; Mallet, F.; Yolken, R.H.; Karlsson, H. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology 2006, 3, 44. [Google Scholar] [CrossRef]
- Li, F.; Nellåker, C.; Sabunciyan, S.; Yolken, R.H.; Jones-Brando, L.; Johansson, A.-S.; Owe-Larsson, B.; Karlsson, H. Transcriptional derepression of the ERVWE1 locus following influenza A virus infection. J. Virol. 2014, 88, 4328–4337. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Domingues, P.; Golebiowski, F.; Patzina, C.; Tatham, M.H.; Hay, R.T.; Hale, B.G. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl. Acad. Sci. USA 2019, 16, 17399–17408. [Google Scholar] [CrossRef]
- Bhende, P.M.; Seaman, W.T.; Delecluse, H.-J.; Kenney, S.C. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 2004, 36, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kwun, H.J.; Kim, H.S.; Jang, K.L. Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol. Cells 2003, 15, 75–80. [Google Scholar]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Moore, F.G.A.; Wolfson, C. Human herpes virus 6 and multiple sclerosis: Human herpes virus 6 and multiple sclerosis. Acta Neurol. Scand. 2002, 106, 63–83. [Google Scholar] [CrossRef]
- Mameli, G.; Madeddu, G.; Mei, A.; Uleri, E.; Poddighe, L.; Delogu, L.G.; Maida, I.; Babudieri, S.; Serra, C.; Manetti, R.; et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: The missing link with multiple sclerosis? PLoS ONE 2013, 8, e78474. [Google Scholar] [CrossRef]
- Sutkowski, N.; Conrad, B.; Thorley-Lawson, D.A.; Huber, B.T. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity 2001, 15, 579–589. [Google Scholar] [CrossRef]
- Sutkowski, N.; Chen, G.; Calderon, G.; Huber, B.T. Epstein-Barr virus latent membrane protein LMP-2A is sufficient for transactivation of the human endogenous retrovirus HERV-K18 superantigen. J. Virol. 2004, 78, 7852–7860. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, F.C.; Lin, M.; Tai, A.; Chen, G.; Huber, B.T. Cutting edge: Epstein-Barr virus transactivates the HERV-K18 superantigen by docking to the human complement receptor 2 (CD21) on primary B cells. J. Immunol. 2006, 177, 2056–2060. [Google Scholar] [CrossRef]
- Tai, A.K.; Luka, J.; Ablashi, D.; Huber, B.T. HHV-6A infection induces expression of HERV-K18-encoded superantigen. J. Clin. Virol. 2009, 46, 47–48. [Google Scholar] [CrossRef]
- Turcanova, V.L.; Bundgaard, B.; Höllsberg, P. Human herpesvirus-6B induces expression of the human endogenous retrovirus K18-encoded superantigen. J. Clin. Virol. 2009, 46, 15–19. [Google Scholar] [CrossRef]
- Mak, M.; Samochowiec, J.; Frydecka, D.; Pełka-Wysiecka, J.; Szmida, E.; Karpiński, P.; Sasiadek, M.M.; Piotrowski, P.; Samochowiec, A.; Misiak, B. First-episode schizophrenia is associated with a reduction of HERV-K methylation in peripheral blood. Psychiatry Res. 2019, 271, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 induce endogenous retrovirus K expression via interferon-stimulated response elements in its 5′ long terminal repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Miyagawa, K.; Chen, S.Y.; Tamosiuniene, R.; Wang, L.; Sharp, O.; Samayoa, E.; Harada, D.; Moonen, J.R.A.; Cao, A.; et al. Upregulation of HERV-K is Linked to Immunity and Inflammation in Pulmonary Arterial Hypertension. Circulation 2017, 136, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. The environment and susceptibility to schizophrenia. Prog. Neurobiol. 2011, 93, 23–58. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef]
- Lafon, M.; Jouvin-Marche, E.; Marche, P.N.; Perron, H. Human viral superantigens: To be or not to be transactivated? Trends Immunol. 2002, 23, 238–239. [Google Scholar] [CrossRef]
- Firouzi, R.; Rolland, A.; Michel, M.; Jouvin-Marche, E.; Hauw, J.J.; Malcus-Vocanson, C.; Lazarini, F.; Gebuhrer, L.; Seigneurin, J.M.; Touraine, J.L.; et al. Multiple sclerosis-associated retrovirus particles cause T lymphocyte-dependent death with brain hemorrhage in humanized SCID mice model. J. Neurovirol. 2003, 9, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montojo, M.; Dominguez-Mozo, M.; Arias-Leal, A.; Garcia-Martinez, Á.; De las Heras, V.; Casanova, I.; Faucard, R.; Gehin, N.; Madeira, A.; Arroyo, R.; et al. The DNA copy number of human endogenous retrovirus-W (MSRV-type) is increased in multiple sclerosis patients and is influenced by gender and disease severity. PLoS ONE 2013, 8, e53623. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Prinz, M. Role of microglia in CNS autoimmunity. Clin. Dev. Immunol. 2013, 2013, 209093. [Google Scholar] [CrossRef]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Monoghan, K.L.; Zheng, W.; Hu, G.; Wan, E.C.K. Monocytes and Monocyte-Derived Antigen-Presenting Cells Have Distinct Gene Signatures in Experimental Model of Multiple Sclerosis. Front. Immunol. 2019, 10, 2779. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends. Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta 2016, 1862, 352–367. [Google Scholar] [CrossRef]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef]
- Martino, G.; Adorini, L.; Rieckmann, P.; Hillert, J.; Kallmann, B.; Comi, G.; Filippi, M. Inflammation in multiple sclerosis: The good, the bad, and the complex. Lancet Neurol. 2002, 1, 499–509. [Google Scholar] [CrossRef]
- Hemmer, B.; Nessler, S.; Zhou, D.; Kieseier, B.; Hartung, H.-P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.G.; Lucchinetti, C.F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012, 7, 185–217. [Google Scholar] [CrossRef]
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P.-O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553. [Google Scholar] [CrossRef]
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; van der Valk, P.; Horssen, J.V.; Hartung, H.-P.; Perron, H.; Küry, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation: Retroviruses and Myelin Repair. Ann. Neurol. 2013, 74, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.K.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef]
- Göttle, P.; Förster, M.; Gruchot, J.; Kremer, D.; Hartung, H.P.; Perron, H.; Küry, P. Rescuing the negative impact of human endogenous retrovirus envelope protein on oligodendroglial differentiation and myelination. Glia 2019, 67, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Dougier-Reynaud, H.-L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.-B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human endogenous retrovirus protein activates innate immunity and promotes experimental allergic encephalomyelitis in mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef]
- Perron, H.; Germi, R.; Bernard, C.; García-Montojo, M.; Deluen, C.; Farinelli, L.; Faucard, R.; Veas, F.; Stefas, I.; Fabriek, B.O.; et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. 2012, 18, 1721–1736. [Google Scholar] [CrossRef]
- Perron, H.; Lazarini, F.; Ruprecht, K.; Péchoux-Longin, C.; Seilhean, D.; Sazdovitch, V.; Créange, A.; Battail-Poirot, N.; Sibaï, G.; Santoro, L.; et al. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: Physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J. Neurovirol. 2005, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Garson, J.A.; Tuke, P.W.; Giraud, P.; Paranhos-Baccala, G.; Perron, H. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet 1998, 351, 33. [Google Scholar] [CrossRef]
- Dolei, A.; Serra, C.; Mameli, G.; Pugliatti, M.; Sechi, G.; Cirotto, M.C.; Rosati, G.; Sotgiu, S. Multiple sclerosis-associated retrovirus (MSRV) in Sardinian MS patients. Neurology 2002, 58, 471–473. [Google Scholar] [CrossRef]
- Sotgiu, S.; Arru, G.; Mameli, G.; Serra, C.; Pugliatti, M.; Rosati, G.; Dolei, A. Multiple sclerosis-associated retrovirus in early multiple sclerosis: A six-year follow-up of a Sardinian cohort. Mult. Scler. 2006, 12, 698–703. [Google Scholar] [CrossRef]
- Mameli, G.; Serra, C.; Astone, V.; Castellazzi, M.; Poddighe, L.; Fainardi, E.; Neri, W.; Granieri, W.; Dolei, A. Inhibition of multiple-sclerosis-associated retrovirus as biomarker of interferon therapy. J. Neurovirol. 2008, 14, 73–77. [Google Scholar] [CrossRef]
- Sotgiu, S.; Pugliatti, M.; Sanna, A.; Sotgiu, A.; Castiglia, P.; Solinas, G.; Dolei, A.; Serra, C.; Bonetti, B.; Rosati, G. Multiple sclerosis complexity in selected populations: The challenge of Sardinia, insular Italy. Eur. J. Neurol. 2002, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Mameli, G.; Serra, C.; Zarbo, I.R.; Arru, G.; Dolei, A. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult. Scler. 2010, 16, 1248–1251. [Google Scholar] [CrossRef]
- Gerondakis, S.; Grumont, R.J.; Banerjee, A. Regulating B-cell activation and survival in response to TLR signals. Immunol. Cell Biol. 2007, 85, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Akira, S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006, 117, 979–987. [Google Scholar] [CrossRef]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.C.; Rasley, A.; Tranguch, S.L.; Marriott, I. Cultured astrocytes express toll-like receptors for bacterial products. Glia 2003, 43, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Hemmi, H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 2003, 85, 85–95. [Google Scholar] [CrossRef]
- Planas, R.; Metz, I.; Martin, R.; Sospedra, M. Detailed Characterization of T Cell Receptor Repertoires in Multiple Sclerosis Brain Lesions. Front. Immunol. 2018, 9, 509. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin-from mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Touil, T.; Deloire-Grassin, M.S.; Vital, C.; Petry, K.G.; Brochet, B. In vivo damage of CNS myelin and axons induced by peroxynitrite. Neuroreport 2001, 12, 3637–3644. [Google Scholar] [CrossRef]
- Karamita, M.; Barnum, C.; Möbius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017, 2. [Google Scholar] [CrossRef]
- Madeira, A.; Burgelin, I.; Perron, H.; Curtin, F.; Lang, A.B.; Faucard, R. MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll-like receptor 4: Relevance of GNbAC1 in multiple sclerosis treatment. J. Neuroimmunol. 2016, 291, 29–38. [Google Scholar] [CrossRef]
- Faucard, R.; Madeira, A.; Gehin, N.; Authier, F.-J.; Panaite, P.-A.; Lesage, C.; Burgelin, I.; Bertel, M.; Bernard, C.; Curtin, F.; et al. Human endogenous retrovirus and neuroinflammation in chronic inflammatory demyelinating polyradiculoneuropathy. EBioMedicine 2016, 6, 190–198. [Google Scholar] [CrossRef]
- Christensen, T. HERVs in neuropathogenesis. J. Neuroimmune Pharmacol. 2010, 5, 326–335. [Google Scholar] [CrossRef]
- Leboyer, M.; Tamouza, R.; Charron, D.; Faucard, R.; Perron, H. Human endogenous retrovirus type W (HERV-W) in schizophrenia: A new avenue of research at the gene-environment interface. World J. Biol. Psychiatry 2011, 14, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 2017, 7, e1024. [Google Scholar] [CrossRef]
- Müller, N.; Wagner, J.K.; Krause, D.; Weidinger, E.; Wildenauer, A.; Obermeier, M.; Dehning, S.; Gruber, R.; Shwarz, M.J. Impaired monocyte activation in schizophrenia. Psychiatry Res. 2012, 198, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory cytokine alterations in schizophrenia: A systematic quantitative review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef]
- Misiak, B.; Stańczykiewicz, B.; Kotowicz, K.; Rybakowski, J.K.; Samochowiec, J.; Frydecka, D. Cytokines and C-reactive protein alterations with respect to cognitive impairment in schizophrenia and bipolar disorder: A systematic review. Schizophr. Res. 2018, 192, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.; Stallings, C.; Origoni, A.; Boronow, J.; Yolken, R. C-reactive protein is associated with the severity of cognitive impairment but not of psychiatric symptoms in individuals with schizophrenia. Schizophr. Res. 2007, 93, 261–265. [Google Scholar] [CrossRef]
- Melbourne, J.K.; Chase, K.A.; Feiner, B.; Rosen, C.; Sharma, R.P. Long non-coding and endogenous retroviral RNA levels are associated with proinflammatory cytokine mRNA expression in peripheral blood cells: Implications for schizophrenia. Psychiatry Res. 2018, 262, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Deb-Rinker, P.; Klempan, T.A.; O’Reilly, R.L.; Torrey, E.F.; Singh, S.M. Molecular characterization of a MSRV-like sequence identified by RDA from monozygotic twin pairs discordant for schizophrenia. Genomics 1999, 61, 133–144. [Google Scholar] [CrossRef]
- Yao, Y.; Schröder, J.; Nellåker, C.; Bottmer, C.; Bachmann, S.; Yolken, R.H.; Karlsson, H. Elevated levels of human endogenous retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 2008, 7, 103–112. [Google Scholar] [CrossRef]
- Yolken, R.H.; Karlsson, H.; Yee, F.; Johnston-Wilson, N.L.; Torrey, E.F. Endogenous retroviruses and schizophrenia. Brain Res. Rev. 2000, 31, 193–199. [Google Scholar] [CrossRef]
- Gourion, D.; Goldberger, C.; Leroy, S.; Bourdel, M.-C.; Olié, J.-P.; Krebs, M.-O. Age at onset of schizophrenia: Interaction between brain-derived neurotrophic factor and dopamine D3 receptor gene variants. Neuroreport 2005, 16, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Gros-Louis, F.; Gaspar, C.; Rouleau, G.A. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Truax, A.C.; Vanmassenhove, B.; Kretzschmar, H.A.; Van Deerlin, V.M.; Clarck, C.M.; Grossman, M.; Miller, B.L.; Trojanowski, J.Q.; et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 2007, 66, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Maldarelli, F.; Mellors, J.; Coffin, J.M. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J. Virol. 2014, 88, 11108–11120. [Google Scholar] [CrossRef]
- Manghera, M.; Ferguson-Parry, J.; Douville, R.N. TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol. Dis. 2016, 94, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.-J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Wang, T.; Medynets, M.; Johnson, K.R.; Doucet-O’Hare, T.T.; DiSanza, B.; Li, W.; Xu, Y.; Bagnell, A.; Tyagi, R.; Sampson, K.; et al. Regulation of stem cell function and neuronal differentiation by HERV-K via mTOR pathway. Proc. Natl. Acad. Sci. USA 2020, 117, 17842–17853. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, N.V.; Liu, H.; Ciceri, G.; Jungverdorben, J.; Frishman, G.; Tchieu, J.; Cederquist, G.Y.; Rothenaigner, I.; Schorpp, K.; Klepper, L.; et al. Activation of HERV-K(HML-2) disrupts cortical patterning and neuronal differentiation by increasing NTRK3. Cell Stem Cell 2021, S1934-5909, 00166–1. [Google Scholar] [CrossRef]
- Bartkowska, K.; Paquin, A.; Gauthier, A.S.; Kaplan, D.R.; Miller, F.D. Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 2007, 134, 4369–4380. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Dembny, P.; Newman, A.G.; Singh, M.; Hinz, M.; Szczepek, M.; Krüger, C.; Adalbert, R.; Dzaye, O.; Trimbuch, T.; Wallach, T.; et al. Human endogenous retrovirus HERV-K(HML-2) RNA causes neurodegeneration through Toll-like receptors. JCI Insight 2020, 5, e131093. [Google Scholar] [CrossRef]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. The immune system in health and disease. In Immunobiology, 6th ed.; Churchill Livingstone: London, UK, 2004. [Google Scholar]
- Curtin, F.; Lang, A.B.; Perron, H.; Laumonier, M.; Vidal, V.; Porchet, H.C.; Hartung, H.-P. GNbAC1, a humanized monoclonal antibody against the envelope protein of multiple sclerosis-associated endogenous retrovirus: A first-in-humans randomized clinical study. Clin. Ther. 2012, 34, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Curtin, F.; Perron, H.; Kromminga, A.; Porchet, H.; Lang, A.B. Preclinical and early clinical development of GNbAC1, a humanized IgG4 monoclonal antibody targeting endogenous retroviral MSRV-Env protein. MAbs 2015, 7, 265–275. [Google Scholar] [CrossRef]
- Curtin, F.; Perron, H.; Faucard, R.; Porchet, H.; Lang, A.B. Treatment against human endogenous retrovirus: A possible personalized medicine approach for multiple sclerosis. Mol. Diagn. Ther. 2015, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Curtin, F.; Vidal, V.; Bernard, C.; Kromminga, A.; Lang, A.B.; Porchet, H. Serum pharmacokinetics and cerebrospinal fluid concentration analysis of the new IgG4 monoclonal antibody GNbAC1 to treat multiple sclerosis: A Phase 1 study. MAbs 2016, 8, 854–860. [Google Scholar] [CrossRef]
- Derfuss, T.; Curtin, F.; Guebelin, C.; Bridel, C.; Rasenack, M.; Matthey, A.; Pasquier, R.D.; Schluep, M.; Desmeules, J.; Lang, A.B.; et al. A phase IIa randomized clinical study testing GNbAC1, a humanized monoclonal antibody against the envelope protein of multiple sclerosis associated endogenous retrovirus in multiple sclerosis patients-a twelve month follow-up. J. Neuroimmunol. 2015, 285, 68–70. [Google Scholar] [CrossRef][Green Version]
- Arru, G.; Leoni, S.; Pugliatti, M.; Mei, A.; Serra, C.; Delogu, L.G.; Manetti, R.; Dolei, A.; Sotgiu, S.; Mameli, G. Natalizumab inhibits the expression of human endogenous retroviruses of the W family in multiple sclerosis patients: A longitudinal cohort study. Mult. Scler. 2014, 20, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Sanderson, N.S.R.; Rasenack, M.; Lalive, P.H.; Lang, A.B.; Curtin, F.; Lindberg, R.L.P.; Kappos, L.; Derfuss, T. Immunologic monitoring during a phase 2a trial of the GNbAC1 antibody in patients with MS. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e144. [Google Scholar] [CrossRef] [PubMed]
- Curtin, F.; Porchet, H.; Glanzman, R.; Schneble, H.M.; Vidal, V.; Audoli-Inthavong, M.L.; Lambert, E.; Hartung, H.P. A placebo randomized controlled study to test the efficacy and safety of GNbAC1, a monoclonal antibody for the treatment of multiple sclerosis-Rationale and design. Mult. Scler. Relat. Disord. 2016, 9, 95–100. [Google Scholar] [CrossRef]
- Kremer, D.; Weyers, V.; Gruchot, J.; Göttle, P.; Hartung, H.P.; Perron, H.; Küry, P. Meeting report:“Human endogenous retroviruses: HERVs or transposable elements in autoimmune, chronic inflammatory and degenerative diseases or cancer”, Lyon, France, november 5th and 6th 2019–an MS scientist’s digest. Mult. Scler. Relat. Disord. 2020, 42, 102068. [Google Scholar] [CrossRef] [PubMed]
- Levet, S.; Medina, J.; Joanou, J.; Demolder, A.; Queruel, N.; Réant, K.; Normand, M.; Sefals, M.; Dimier, J.; Germi, R.; et al. An ancestral retroviral protein identified as a therapeutic target in type-1 diabetes. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Ruiz, P.L.D.; Tapia, G.; Bakken, I.J.; Håberg, S.E.; Hungnes, O.; Gulseth, H.L.; Stene, L.C. Pandemic influenza and subsequent risk of type 1 diabetes: A nationwide cohort study. Diabetologia 2018, 61, 1996–2004. [Google Scholar] [CrossRef]
- Tauriainen, S.; Oikarinen, S.; Oikarinen, M.; Hyöty, H. Enteroviruses in the pathogenesis of type 1 diabetes. Semin. Immunopathol. 2011, 33, 45–55. [Google Scholar] [CrossRef]
- Garay-Malpartida, H.M.; Mourão, R.F.; Mantovani, M.; Santos, I.A.; Sogayar, M.C.; Goldberg, A.C. Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol. 2011, 12, 18. [Google Scholar] [CrossRef]
- Qiao, Y.-C.; Chen, Y.-L.; Pan, Y.-H.; Tian, F.; Xu, Y.; Zhang, X.-X.; Zhao, H.-L. The change of serum tumor necrosis factor alpha in patients with type 1 diabetes mellitus: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0176157. [Google Scholar] [CrossRef]
- Curtin, F.; Bernard, C.; Levet, S.; Perron, H.; Porchet, H.; Médina, J.; Malpass, S.; Lloyd, D.; Simpson, R.; RAINBOW-T1D Investigators. A new therapeutic approach for type 1 diabetes: Rationale for GNbAC1, an anti-HERV-W-Env monoclonal antibody. Diabetes Obes. Metab. 2018, 20, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Curtin, F.; Champion, B.; Davoren, P.; Duke, S.; Ekinci, E.I.; Gilfillan, C.; Morbey, C.; Nathow, T.; O’Moore-Sullivan, T.; O’Neal, D.; et al. A safety and pharmacodynamics study of temelimab, an antipathogenic human endogenous retrovirus type W envelope monoclonal antibody, in patients with type 1 diabetes. Diabetes Obes. Metab. 2020, 22, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis-success from bench to bedside. Nat. Rev. Neurol. 2019, 15, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Medina, J.; Nath, A.; Steiner, J.P.; Wenxue, L.I.; Myoung-Hwa, L.E.E. Anti-HERV-K Envelope Antibody and Uses Thereof. US20200308258, 17 June 2020. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969352&tab=NATIONALBIBLIO&_cid=P22-KJQZUJ-45299-1 (accessed on 21 May 2021).


{kind=link}
{kind=link}
| Disease | Retrotransposon | Elevated Cytokines | Ref. |
|---|---|---|---|
| Multiple Sclerosis | HERV-W ENV, POL HERV-H ENV | IFN-γ, IL-6, TNF-α, IL-17, IL-22, IL-12, IL-1 | [26,27,28,29] |
| Schizophrenia | HERV-W ENV, GAG | IL-6, IL-1β, IL-2, sIL-2R, IL-8, IL-1, IL-1RA, TNF-α, IFN-γ, IL-4, TGF-β, IL-18, IL-10 | [30,31,32,33,34] |
| Bipolar disorder (BD) | HERV-W ENV | IL-6, TNF-α, IL-10, sIL-2R, IL-1β, IL-1RA | [34,35,36] |
| Autism spectrum disorder (ASD) | HERV-H ENV HERV-K ENV HERV-W ENV | IFN-γ, IL-1β, IL-6, TNF-α, IL-10 | [37] |
| Attention deficit hyperactivity disorder (ADHD) | HERV-H ENV | IL-6, TNF-β, IFN-γ, IL-2, IL-10, IL-13, IL-16 | [38,39,40] |
| Sporadic amyotrophic lateral sclerosis (sALS) | HERV-K GAG, POL, ENV | TNF-α, IFN-γ, IL-6, IL-8, IL-1β, IL1RA, IL-2, IL-4, IL-5, IL-7, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17, IL-18, IL-21 | [41,42] |
| Fibromyalgia (FM) | HERV-W POL HERV-K ENV HERV-H GAG | INF-β, INF-γ, IL-1RA, IL-6, IL-17A, | [43,44] |
| Chronic fatigue syndrome/ myalgic encephalomyelitis (CFS/MS) | HERV-K POL | IFN-γ, IFN-α, IL-4, IL-5, IL-7, IL-13, IL-6, IL-1β, IL-2, IL-12 | [45,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giménez-Orenga, K.; Oltra, E. Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease. Pharmaceuticals 2021, 14, 495. https://doi.org/10.3390/ph14060495
Giménez-Orenga K, Oltra E. Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease. Pharmaceuticals. 2021; 14(6):495. https://doi.org/10.3390/ph14060495
Chicago/Turabian StyleGiménez-Orenga, Karen, and Elisa Oltra. 2021. "Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease" Pharmaceuticals 14, no. 6: 495. https://doi.org/10.3390/ph14060495
APA StyleGiménez-Orenga, K., & Oltra, E. (2021). Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease. Pharmaceuticals, 14(6), 495. https://doi.org/10.3390/ph14060495