
Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance


Abstract
1. Background
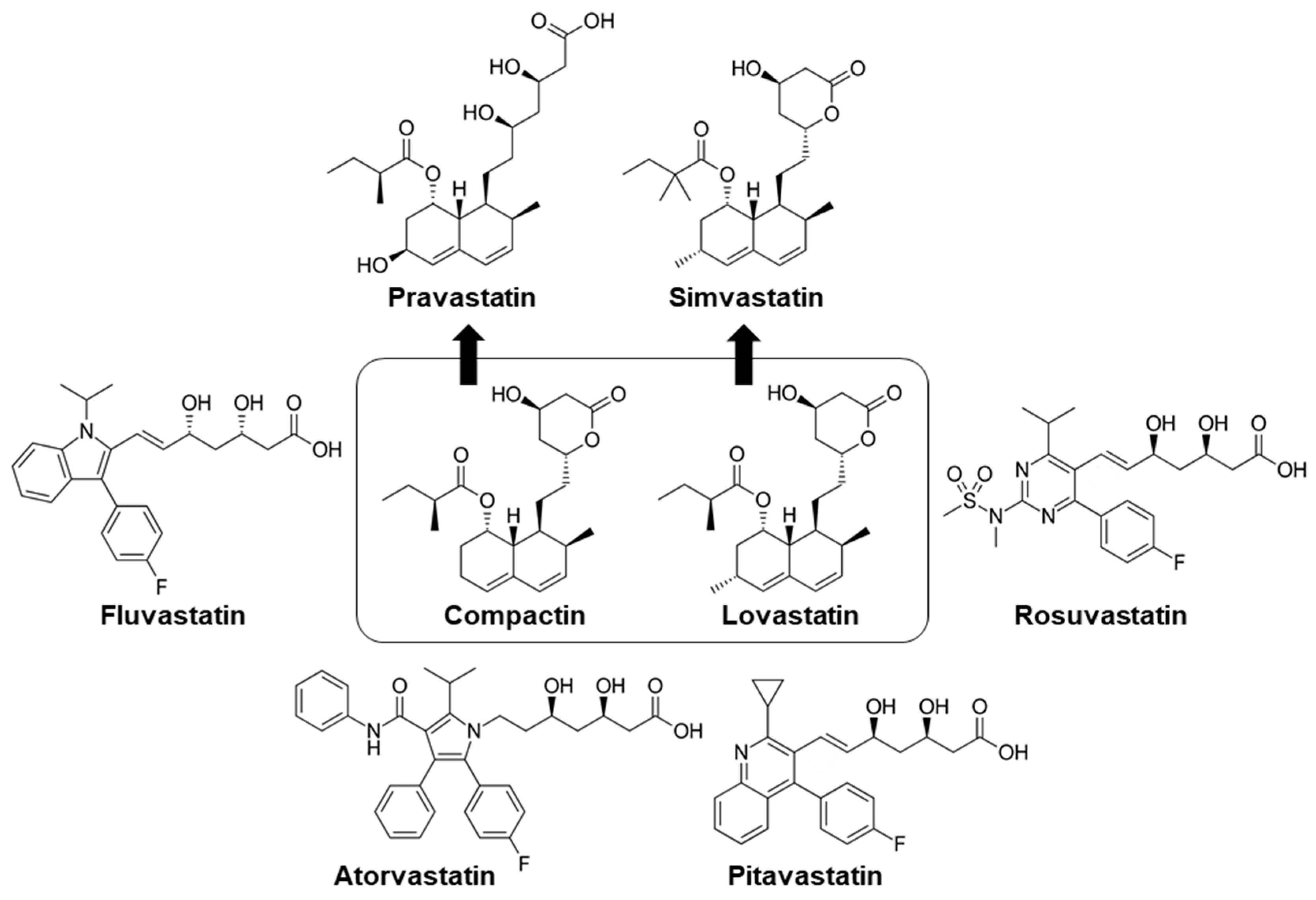
2. The Emergence of Statin as a Lipid-Lowering Drug
3. Statin Repurposed in Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N. | Statin | Clinical Indication | Doses | Human Plasma Concentration | Toxicity | Ref |
|---|---|---|---|---|---|---|
| 1 | Lovastatin | Multiple myeloma | 2 mg/kg days 1–5, 8–12 and 0.5 mg/kg days 15–28 of each cycle | - | Somnolence, fatigue and constipation, deep vein thrombosis, pulmonary embolism | [44,45] |
| 2 | Simvastatin | Refractory multiple myeloma, pancreatic cancer, colorectal cancer, | 30 mg, 80 mg daily | - | Hematoxicity, bone pain, gastrointestinal side effects, infections, muscle pain, fatigue, anemia, depression | [46,47,48] |
| 3 | Pravastatin | Gastric cancer, hepatocellular carcinoma (HCC) | 20–40 mg/kg | - | Diarrhea, stomatitis | [49] |
| 4 | Fluvastaatin | Prostate cancer | 80 mg | 63.4 ng/mL or 0.2 μM (0.0–437.0 ng/mL or 0.0–1.1 μM) | - | [50] |
| 5 | Atorvastatin | Prostate cancer | 80 mg | 3.6 ng/mL | - | [51] |
| 6 | Rosuvastatin | Advanced solid malignancies | 20 mg, 80 mg daily | - | Fatigue, myalgia, muscle weakness | [52,53] |
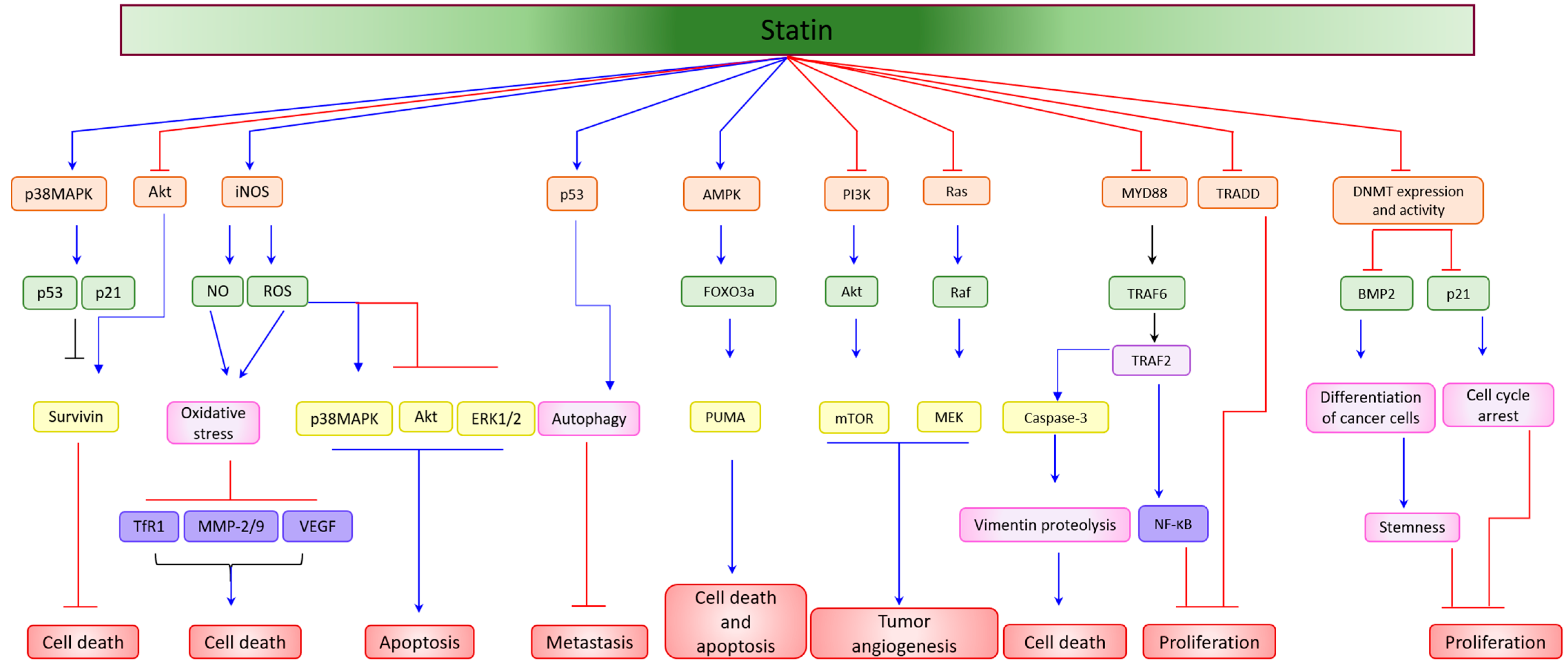
4. Statin as a Single Agent in the Suppression of Cancer Cell Proliferation, and the Induction of Apoptosis
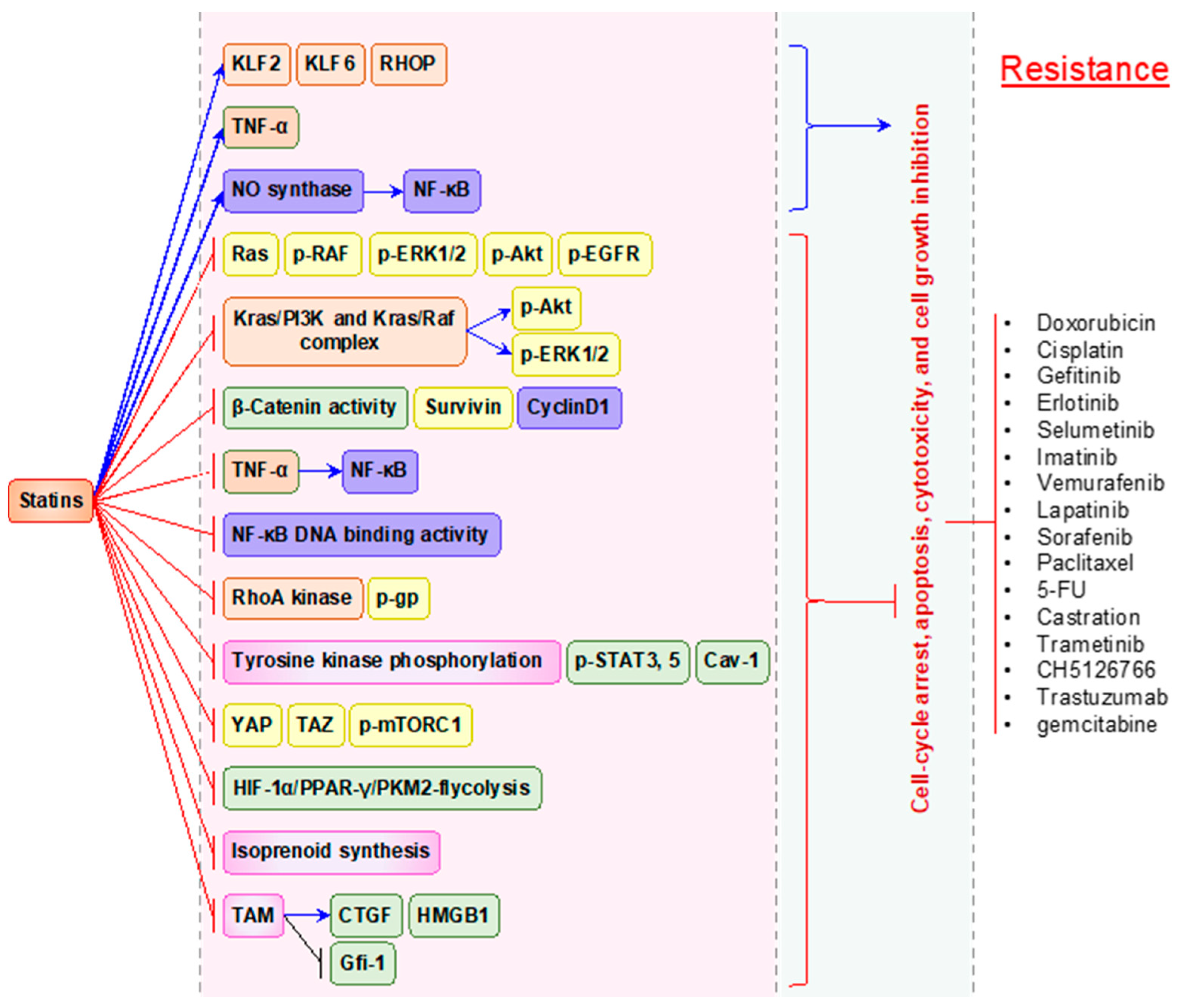
5. Statins Possess Synergistic Action to Overcome the Resistance to Anti-cancer Therapies
5.1. Doxorubicin
5.2. 5-Fluorouracil (5-FU) and Capecitabine
5.3. Sorafenib
5.4. Gefitinib, Erlotinib, and Imatinib
5.5. Cisplatin
5.6. Gemcitabine
5.7. Vemurafenib
5.8. TRAIL
5.9. Prednisolone
| Cancer Types/Cells | Statin | Concurrent Therapy | Statin Dose | Pathway | Ref |
|---|---|---|---|---|---|
| Colorectal cancer | Lovastatin | - | 2 μM | Inhibits DNMT and demethylates the BMP2, TIMP3, and HIC1 promoters | [61] |
| Breast cancer | - | 4,8,16 μM | Cell cycle arrest at G(0)/G (1) phase | [62] | |
| MDA-MB-231 breast cancer | - | 1–10 μM | Upregulates Raf1, amyloid β, MEK6, STAT1, myelin-oligodendrocyte glycoprotein, Vitamin D3 receptor, downregulates CREB, and γ glutamyl transferase | [63] | |
| Glioblastoma | Gefitinib | 10 μM | Decreases Akt | [126] | |
| Human cholangiosarcoma | Gefitinib | 5–10 μM | Increases cell cycle arrest, TNF-alpha, and decreases LKB1 activation | [127] | |
| Human non-small cell lung carcinoma | Gefitinib | 1–5 μM2 | Increases PARP, caspase-3, decreases Bcl-2, RAS, p-RAF, p-ERK1/2, p-AKT, and p-EGFR | [129] | |
| Chronic myeloid leukemia | Imatinib | 5–20 μM | Decreases ABCB1 and ABCG2 | [134] | |
| Gall bladder cancer | Cisplatin | 10–50 μM | Impairs DNA damage response | [137] | |
| Prostate cancer | TRAIL | 5 μM | Increases PRRA replication, CAR, and integrin | [149] | |
| Glioblastoma | Temozolomide | 0.625–20 μM | Impairs autophagy flux | [152] | |
| Anaplastic thyroid cancer | Troglitazone | 1–100 μM | Increases cell cycle arrest, p21, and p27 | [153] | |
| Multiple myeloma | Pravastatin | - | 0.3, 0.6, and 0.9 μM | Increases cells in G0/G1 phase of the cell cycle and reduces the factors VEGF, and bFGF | [70] |
| Human hepatoma | - | Decreases p38 activity and expressions of p-p38, RhoC, and MMP-2, while elevates MKP-1 expression | [71] | ||
| Esophageal cancer | Simvastatin | - | 0.625, 1.25, 2.5, 5, and 10 μM | Inhibits PTEN-PI3K/AKT pathway | [69] |
| Cholangiocarcinoma | - | 1–100 μM, 25–50 μM | Reduces Rac1 activity, lowers expression of ABCA1 and ABCG1 | [65,66] | |
| Prostate cancer | Docetaxel | 25 μM | Increases Bad, reduces Bcl-2, Bcl-xL and cleaved caspases 9/3, increases TNF, Fas-L, Traf1, and cleaved caspase 8 | [64] | |
| Prostate cancer | Doxorubicin | 2.5–20 μM | Decreases ABGC4 protein | [98] | |
| Malignant mesothelioma | Doxorubicin | 10 μM | Increases NF-kB and NO production | [102] | |
| Colon carcinoma | 5-FU | 5 mg/kg | Decreases tumor angiogenesis, Bcl-2 and increases Bax | [116] | |
| Human salivary adenoid cystic carcinoma | MiR-21 inhibitor (miR-21i) | 1–100 μM | Decreases N-Cadherin and increases E-Cadherin, decreases in Bcl-2 and survivin, while increase in p53, Bax, and caspase-9 | [67] | |
| Pancreatic ductal carcinoma | Simvastatin | Gemcitabine | 5–40 μM | Increases Gfi-1, decreases CTGF | [144] |
| Chronic myeloid leukemia | Imatinib | 10–50 μM | Increases cell cycle arrest, decreases STAT5 and STAT3 | [135] | |
| Metastatic melanoma | Dacarbazine | 0.5–1 μM | Decreases RhoA/RhoC/LIM, increases p53, p21, p27, casp-3, and PARP | [154] | |
| Breast cancer | Pentoxifylline | 0.1–50 μM | Increases apoptosis, autophagy, and cell cycle arrest | [155] | |
| Prostate cancer | Castration | 0.1–20 μM | Increases cell cycle arrest, apoptosis, and decreases Akt | [156] | |
| Blood cancer | Ventoclax | 5–20 μM | Increases p53, PUMA | [157] | |
| Non-small cell lung cancer | Gefitinib, Erlotinib | 5 μM | Decreases Akt, b-catenin, survivin, cyclin D1 | [130] | |
| Gastric cancer xenograft | Capecitbine | 10–50 μM | Decreases NF-kB | [117] | |
| Melanoma cells | 5,6-dimethylsanthenone-4-acetic acid | 1.5–14 μM | Decreases HIF-alpha | [158] | |
| Breast cancer | Anti-HER2 | 1–5 μM | Decreases YAP/TAZ signaling | [159] | |
| Colon cancer | Simvastatin + phenothiazines | Doxorubicin | 2.5 μM | Decreases ABCB1, COX-2 enzymes, Bcl-2 and increases Bax | [103] |
| Human myeloid leukemia | Simvastatin, Mevastatin, Lovastatin, Pravastatin | Doxorubicin, Paclitaxel, 5-FU | 5–50 μM | Decreases NF-kB | [109] |
| Pancreatic cancer | Simvastatin + bisphosphonates | Gemcitabine | 0.1–100 μM | Decreases cell viability | [141] |
| Colon cancer | Simvastatin + Oxicam derivatives | Doxorubicin | 5 μM | Increases caspase-3, Bax, decreases Bcl-2 and COX-2 | [107] |
| Prostate cancer | Simvastatin + Valproic acid | Docetaxel | 1–100 μM | Decreases YAP | [160] |
| Acute myeloid leukemia (AML), | Fluvastatin | Tyrosine kinase inhibitor (lestaurtinib) | 0.2–2 μM | Inhibits FLT3 glycosylation | [73] |
| C6 glioma cell line | - | 1 to 10 μM | Decreases p-ERK1/2 expression, upregulates p-JNK1/2, and reduces MMP-9 and VEGF concentrations | [76] | |
| Breast cancer | - | 5–20 μM | Downregulates vimentin, | [79] | |
| Breast cancer | - | 10 μM | Increases p53 and induces autophagy | [81] | |
| Human hepatoma cells (HepG2) | Trans-activator transcription peptide (TAT) | 1–1000 μM | Accumulates cells in the pre-G phase | [82] | |
| Melanoma cells | Sorafenib | 1 μM | Increases PARP, and JNK | [119] | |
| Hepatocellular carcinoma | Sorafenib | 10 mg/kg | Inactivates MAPK and NF-kB | [121] | |
| Melanoma cells | Vemurafenib | 1–10 μM | Decreases Akt | [148] | |
| Cervical cancer | Fluvastatin, Atorvastatin, and simvastatin | - | 10–160 μM | Increases ROS and nitrite production | [75] |
| Lymphoma cells | Fluvastatin, atorvastatin, and simvastatin | - | 0–5 μM | Enhances the DNA fragmentation and the activation of proapoptotic members such as caspase-3, PARP and Bax, increases reactive oxygen species (ROS), p38 MAPK activation but suppresses activation of anti-apoptotic molecule Bcl-2, decrease mitochondrial membrane potential and activation of Akt and Erk pathways | [78] |
| Human breast cancer | Fluvastatin and atorvastatin | Estradiol | - | Deregulates Bcl-2 rather than up-regulation of Fas-L or p53 | [74] |
| Breast cancer | Fluvastatin and simvastatin | - | 10 to 20 μM | Increases nitric oxide levels via iNOS expression, increases MnSOD, catalase and GSH which in turn, diminished H2O2 levels, down regulates transferrin receptor (TfR1), TfR1, MMP-2, 9 | [79] |
| glioblastoma cell lines | Fluvastatin, cerivastatin, and pitavastatin | - | IC50 value: Ceri:0.0010 μM Pita:0.0023 μM Flu:0.109 μM | Increase autophagy | [80] |
| Breast cancer | Fluvastatin and simvastatin | CH51126766 or trametinib | 0.3 μM | Decreases Akt and increases PARP | [150] |
| Non-small cell lung cancer | Fluvastatin and pitavastatin | Erlotinib | 100 μM | Increases casp-3 and PARP | [133] |
| Cervical cancer | Fluvastatin, atorvastatin, and simvastatin | - | 10–160 μM | Increases ROS and nitrite production | [75] |
| Lymphoma cells | - | 0–5 μM | Enhances DNA fragmentation, caspase-3, PARP and Bax, but suppresses Bcl-2, increases reactive oxygen species (ROS) and activation of p38 MAPK, decreases mitochondrial membrane potential and activation of Akt and Erk pathways | [78] | |
| NCI-H332M, DU-145, PC-3 and HOP-92 cell lines | Atorvastatin | - | 0–30 μM | Inhibits protein prenylation | [83] |
| Human osteosarcoma | Doxorubicin and cisplatin | 10 μM | Decreases MMP2 | [100] | |
| Hepatocellular carcinoma | Hypoxia | 1–10 μM | Inactivates YAP | [120] | |
| Human cholangiocarcinoma | Gemcitabine | 5–100 μM | Decreases Yes-associated protein | [142] | |
| Non-small cell lung cancer | Gefitinib | 1–5 μM | Decreases Akt and ERK | [132] | |
| Melanoma cancer | Tamoxifen | 1–100 μM | Increases Bax and cytochrome C | [161] | |
| Colon cancer | Celecoxib | 15–45 μM | Increases cell cycle arrest and apoptosis | [162] | |
| Prostate cancer | Rosuvastatin | - | 5–50 μM | Decreases Vimentin and Zeb-1, and inhibits spheroid formation | [84] |
| Hepatic cancer | NA | The IC50 values ranged from 12 to 112 μg/mL | Enhances apoptosis and induces cell cycle arrest at G2/M phase | [87] | |
| Murine mammary adenocarcinoma | Nilotinib | 7.5 mg/kg | Increases caspase 3, decreases ER alpha, and tumor nitric oxide level | [163] | |
| Hepatocellular carcinoma | Dasatinib | 10, 25, 50 μM | Decreases p-FAK/p-Src, p-Ras/p-Raf, p-STAT3, p-Akt, HGF, VEGF, MMP-9, and Ki67 | [164] | |
| Adrenocortical carcinoma | Mitotane | 100 μM | Decreases cell viability, ABCA1 and induces apoptosis | [165] | |
| Ovarian cancer | Pitavastatin | - | 1 μM | Increases caspase activity and apoptotic cell death | [89] |
| Oral squamous cell carcinoma | - | 0.05–0.25 μM | Increases p-AMPK, FOXO3a, and PUMA while decreases p-Akt | [90] | |
| Breast and melanoma model | Radiation | 1.25, 2.5 or 5 μM | Increases senescence and delays DNA repair | [91] | |
| Pancreatic ductal carcinoma | Gemcitabine | 0.5 μM | Increases caspase-3, PARP, RIP1-RIP3-MLKL complex, decreases cyclineA2/CDK2, increases p21 | [143] | |
| Melanoma | Dacarbazine | 1 μM | Increases apoptosis and autophagy cell death | [166] | |
| Breast cancer | Cerivastatin | - | 25 ng/mL | Down-regulates cyclin D1, PCNA, c-myc, and up-regulates p21, p19INK4d, integrin h8, (decrease in u-PA, MMP-9, u-PAR, PAI-1 and increase in anti-oncogenes Wnt-5a and H-cadherin | [92] |
| Human glioblastoma | - | 10–100 μM | Down-regulates tyrosine phosphorylation of FAK | [93] | |
| Breast cancer | NA | 25 ng/mL | Induces cell cycle arrest at G1/S, inactivates Rho, NF-kB, and decreases MMP-9 | [94] | |
| Breast cancer | Doxorubicin and cisplatin | 0.0195–0.624 μM | Increases p21 | [101] | |
| Colorectal cancer | 5-FU | 0.01–10 μM | Decreases nuclear factor kB binding activity | [115] | |
| Malignant mesothelioma | Mevastatin | Doxorubicin | 100 μM | Increases NF-kB and NO production | [102] |
6. The Dark Side of Statin Therapy (Resistance and Intolerance to Statins)
7. Statin-Mediated Resistance
8. A Clinical Trial of Statin in Cancer
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Stauss, H.J.; Tran, M.G.B. TCR Gene Therapy: Challenges, Opportunities, and Future Directions. Cells 2020, 9, 2567. [Google Scholar] [CrossRef]
- Bondhopadhyay, B.; Sisodiya, S.; Chikara, A.; Khan, A.; Tanwar, P.; Afroze, D.; Singh, N.; Agrawal, U.; Mehrotra, R.; Hussain, S. Cancer immunotherapy: A promising dawn in cancer research. Am. J. Blood Res. 2020, 10, 375–385. [Google Scholar]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic screening in cancer drug discovery—Past, present and future. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef]
- Petsko, G.A. When failure should be the option. BMC Biol. 2010, 8, 61. [Google Scholar] [CrossRef]
- Umscheid, C.A.; Margolis, D.J.; Grossman, C.E. Key concepts of clinical trials: A narrative review. Postgrad. Med. 2011, 123, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J. Trial watch: Phase II failures: 2008–2010. Nat. Rev. Drug Discov. 2011, 10, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014, 8, 442. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Kelderman, S.; Schumacher, T.N.; Haanen, J.B. Acquired and intrinsic resistance in cancer immunotherapy. Mol. Oncol. 2014, 8, 1132–1139. [Google Scholar] [CrossRef]
- Dinić, J.; Efferth, T.; García-Sosa, A.T.; Grahovac, J.; Padrón, J.M.; Pajeva, I.; Rizzolio, F.; Saponara, S.; Spengler, G.; Tsakovska, I. Repurposing old drugs to fight multidrug resistant cancers. Drug Resist. Updates 2020, 52, 100713. [Google Scholar] [CrossRef]
- Tobert, J.A. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Manu, P.; Rogozea, L. The Discovery of Statins. Am. J. Ther. 2016, 23, e980–e981. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P. The discovery of statins. Cell 2008, 134, 903–905. [Google Scholar] [CrossRef]
- Roth, B.D. The discovery and development of atorvastatin, a potent novel hypolipidemic agent. Prog. Med. Chem. 2002, 40, 1–22. [Google Scholar] [CrossRef]
- van Leuven, S.I.; Kastelein, J.J. Atorvastatin. Expert Opin. Pharmacother. 2005, 6, 1191–1203. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Moreno-Bueno, G. Updating the role of obesity and cholesterol in breast cancer. Breast Cancer Res. 2019, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Agnihotri, N. Role of cholesterol homeostasis and its efflux pathways in cancer progression. J. Steroid Biochem. Mol. Biol. 2019, 191, 105377. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef]
- Göbel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond—The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188351. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, H.; Zhou, X.; Luo, H.; Tang, F.; Yang, J.; Alterovitz, G.; Cheng, L.; Ren, B. Lovastatin synergizes with itraconazole against planktonic cells and biofilms of Candida albicans through the regulation on ergosterol biosynthesis pathway. Appl. Microbiol. Biotechnol. 2018, 102, 5255–5264. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.M.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J. Investig. Med. 2018, 66, 1083–1087. [Google Scholar] [CrossRef]
- Braun, L.R.; Feldpausch, M.N.; Czerwonka, N.; Weiss, J.; Branch, K.; Lee, H.; Martinez-Salazar, E.L.; Torriani, M.; Sponseller, C.A.; Grinspoon, S.K.; et al. Effects of Pitavastatin on Insulin Sensitivity and Liver Fat: A Randomized Clinical Trial. J. Clin. Endocrinol. Metab. 2018, 103, 4176–4186. [Google Scholar] [CrossRef]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927.e6–3938.e6. [Google Scholar] [CrossRef]
- Göbel, A.; Zinna, V.M.; Dell’Endice, S.; Jaschke, N.; Kuhlmann, J.D.; Wimberger, P.; Rachner, T.D. Anti-tumor effects of mevalonate pathway inhibition in ovarian cancer. BMC Cancer 2020, 20, 703. [Google Scholar] [CrossRef]
- Calvillo-Argüelles, O.; Abdel-Qadir, H.; Michalowska, M.; Billia, F.; Suntheralingam, S.; Amir, E.; Thavendiranathan, P. Cardioprotective Effect of Statins in Patients With HER2-Positive Breast Cancer Receiving Trastuzumab Therapy. Can. J. Cardiol. 2019, 35, 153–159. [Google Scholar] [CrossRef]
- Ricco, N.; Flor, A.; Wolfgeher, D.; Efimova, E.V.; Ramamurthy, A.; Appelbe, O.K.; Brinkman, J.; Truman, A.W.; Spiotto, M.T.; Kron, S.J. Mevalonate pathway activity as a determinant of radiation sensitivity in head and neck cancer. Mol. Oncol. 2019, 13, 1927–1943. [Google Scholar] [CrossRef]
- Tan, X.L.; E., J.Y.; Lin, Y.; Rebbeck, T.R.; Lu, S.E.; Shang, M.; Kelly, W.K.; D’Amico, A.; Stein, M.N.; Zhang, L.; et al. Individual and joint effects of metformin and statins on mortality among patients with high-risk prostate cancer. Cancer Med. 2020, 9, 2379–2389. [Google Scholar] [CrossRef]
- Hus, M.; Grzasko, N.; Szostek, M.; Pluta, A.; Helbig, G.; Woszczyk, D.; Adamczyk-Cioch, M.; Jawniak, D.; Legiec, W.; Morawska, M.; et al. Thalidomide, dexamethasone and lovastatin with autologous stem cell transplantation as a salvage immunomodulatory therapy in patients with relapsed and refractory multiple myeloma. Ann. Hematol. 2011, 90, 1161–1166. [Google Scholar] [CrossRef]
- Kawata, S.; Yamasaki, E.; Nagase, T.; Inui, Y.; Ito, N.; Matsuda, Y.; Inada, M.; Tamura, S.; Noda, S.; Imai, Y.; et al. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br. J. Cancer 2001, 84, 886–891. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Baumann, P.; Bumeder, I.; Meinhardt, G.; Straka, C.; Emmerich, B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur. J. Haematol. 2007, 79, 240–243. [Google Scholar] [CrossRef]
- Moon, D.C.; Lee, H.S.; Lee, Y.I.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; Chung, J.B.; Bang, S. Concomitant Statin Use Has a Favorable Effect on Gemcitabine-Erlotinib Combination Chemotherapy for Advanced Pancreatic Cancer. Yonsei Med. J. 2016, 57, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Baas, J.M.; Krens, L.L.; Tije, A.J.T.; Erdkamp, F.; van Wezel, T.; Morreau, H.; Gelderblom, H.; Guchelaar, H.J. Safety and efficacy of the addition of simvastatin to cetuximab in previously treated KRAS mutant metastatic colorectal cancer patients. Investig. New Drugs 2015, 33, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Konings, I.R.; van der Gaast, A.; van der Wijk, L.J.; de Jongh, F.E.; Eskens, F.A.; Sleijfer, S. The addition of pravastatin to chemotherapy in advanced gastric carcinoma: A randomised phase II trial. Eur. J. Cancer 2010, 46, 3200–3204. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; Hamilton, R.J.; Masoomian, M.; Khurram, N.; Branchard, E.; Mullen, P.J.; Elbaz, M.; Hersey, K.; Chadwick, D.; Ghai, S.; et al. A pilot window-of-opportunity study of preoperative fluvastatin in localized prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Knuuttila, E.; Riikonen, J.; Syvälä, H.; Auriola, S.; Murtola, T.J. Access and concentrations of atorvastatin in the prostate in men with prostate cancer. Prostate 2019, 79, 1412–1419. [Google Scholar] [CrossRef]
- Goss, G.D.; Jonker, D.J.; Laurie, S.A.; Weberpals, J.I.; Oza, A.M.; Spaans, J.N.; La Porte, C.; Dimitroulakos, J. A phase I study of high-dose rosuvastatin with standard dose erlotinib in patients with advanced solid malignancies. J. Transl. Med. 2016, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Nabati, M.; Janbabai, G.; Esmailian, J.; Yazdani, J. Effect of Rosuvastatin in Preventing Chemotherapy-Induced Cardiotoxicity in Women With Breast Cancer: A Randomized, Single-Blind, Placebo-Controlled Trial. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 233–241. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.L.; Chen, C.Y.; Hsu, Y.F.; Kuo, W.S.; Ou, G.; Chiu, P.T.; Huang, Y.H.; Hsu, M.J. Simvastatin induced HCT116 colorectal cancer cell apoptosis through p38MAPK-p53-survivin signaling cascade. Biochim. Biophys. Acta 2013, 1830, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.E.; Na, K.S.; Park, D.S.; Choi, K.H.; Kim, B.R.; Shim, H.; Jeong, E.T.; Kim, H.R. Apoptotic induction by simvastatin in human lung cancer A549 cells via Akt signaling dependent down-regulation of survivin. Investig. New Drugs 2011, 29, 945–952. [Google Scholar] [CrossRef]
- Relja, B.; Meder, F.; Wilhelm, K.; Henrich, D.; Marzi, I.; Lehnert, M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int. J. Mol. Med. 2010, 26, 735–741. [Google Scholar] [CrossRef]
- Gallelli, L.; Falcone, D.; Scaramuzzino, M.; Pelaia, G.; D’Agostino, B.; Mesuraca, M.; Terracciano, R.; Spaziano, G.; Maselli, R.; Navarra, M.; et al. Effects of simvastatin on cell viability and proinflammatory pathways in lung adenocarcinoma cells exposed to hydrogen peroxide. BMC Pharmacol. Toxicol. 2014, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Tsai, Y.C.; Tseng, J.H.; Liou, J.J.; Horng, S.; Wen, H.C.; Fan, Y.C.; Zhong, W.B.; Hsu, S.P. Simvastatin Inhibits Cell Proliferation and Migration in Human Anaplastic Thyroid Cancer. Int. J. Mol. Sci. 2017, 18, 2690. [Google Scholar] [CrossRef]
- Dongoran, R.A.; Wang, K.H.; Lin, T.J.; Yuan, T.C.; Liu, C.H. Anti-Proliferative Effect of Statins Is Mediated by DNMT1 Inhibition and p21 Expression in OSCC Cells. Cancers 2020, 12, 2084. [Google Scholar] [CrossRef]
- Kodach, L.L.; Jacobs, R.J.; Voorneveld, P.W.; Wildenberg, M.E.; Verspaget, H.W.; van Wezel, T.; Morreau, H.; Hommes, D.W.; Peppelenbosch, M.P.; van den Brink, G.R.; et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 2011, 60, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mi, M.T.; Zhu, J.D.; Zhang, Q.Y. Effects of lovastatin on proliferation and gap junctional intercellular communication of human breast cancer cell MCF-7. Ai Zheng Aizheng Chin. J. Cancer 2003, 22, 257–261. [Google Scholar]
- Yang, T.; Yao, H.; He, G.; Song, L.; Liu, N.; Wang, Y.; Yang, Y.; Keller, E.T.; Deng, X. Effects of Lovastatin on MDA-MB-231 Breast Cancer Cells: An Antibody Microarray Analysis. J. Cancer 2016, 7, 192–199. [Google Scholar] [CrossRef]
- Goc, A.; Kochuparambil, S.T.; Al-Husein, B.; Al-Azayzih, A.; Mohammad, S.; Somanath, P.R. Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis. BMC Cancer 2012, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Yang, F.; Wise, C.E.; Meng, F.; Priester, S.; Munshi, M.K.; Guerrier, M.; Dostal, D.E.; Glaser, S.S. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig. Liver Dis. 2011, 43, 395–403. [Google Scholar] [CrossRef]
- Seeree, P.; Janvilisri, T.; Kangsamaksin, T.; Tohtong, R.; Kumkate, S. Downregulation of ABCA1 and ABCG1 transporters by simvastatin in cholangiocarcinoma cells. Oncol. Lett. 2019, 18, 5173–5184. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, T.; Yan, F.; Cai, W.; Zheng, J.; Jiang, X.; Sun, J. Effect of simvastatin and microRNA-21 inhibitor on metastasis and progression of human salivary adenoid cystic carcinoma. Biomed. Pharmacother. 2018, 105, 1054–1061. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lin, Y.J.; Lin, C.L.; Lin, H.J.; Wu, H.S.; Hsu, H.Y.; Sun, Y.C.; Wu, H.Y.; Lai, C.H.; Kao, C.H. Simvastatin Therapy for Drug Repositioning to Reduce the Risk of Prostate Cancer Mortality in Patients with Hyperlipidemia. Front. Pharmacol. 2018, 9, 225. [Google Scholar] [CrossRef]
- Jin, Y.; Xu, K.; Chen, Q.; Wang, B.; Pan, J.; Huang, S.; Wei, Y.; Ma, H. Simvastatin inhibits the development of radioresistant esophageal cancer cells by increasing the radiosensitivity and reversing EMT process via the PTEN-PI3K/AKT pathway. Exp. Cell Res. 2018, 362, 362–369. [Google Scholar] [CrossRef]
- Trojan, P.J.; Bohatch-Junior, M.S.; Otuki, M.F.; Souza-Fonseca-Guimarães, F.; Svidnicki, P.V.; Nogaroto, V.; Fernandes, D.; Krum, E.A.; Favero, G.M. Pravastatin induces cell cycle arrest and decreased production of VEGF and bFGF in multiple myeloma cell line. Braz. J. Biol. 2016, 76, 59–65. [Google Scholar] [CrossRef]
- Zhang, W.J.; Yang, S.H.; Tian, S.J.; Li, Z.X.; Gong, Y.X.; Qu, Y.; Mao, W.W. Effects of pravastatin on the proliferation and invasion of human hepatocarcinoma HepG2 cell line. Zhonghua Gan Zang Bing Za Zhi Zhonghua Ganzangbing Zazhi Chin. J. Hepatol. 2010, 18, 280–283. [Google Scholar] [CrossRef]
- Bourgier, C.; Auperin, A.; Rivera, S.; Boisselier, P.; Petit, B.; Lang, P.; Lassau, N.; Taourel, P.; Tetreau, R.; Azria, D.; et al. Pravastatin Reverses Established Radiation-Induced Cutaneous and Subcutaneous Fibrosis in Patients with Head and Neck Cancer: Results of the Biology-Driven Phase 2 Clinical Trial Pravacur. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Li, L.; Nguyen, B.; Brown, P.; Levis, M.; Small, D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 2012, 120, 3069–3079. [Google Scholar] [CrossRef] [PubMed]
- Mück, A.O.; Seeger, H.; Wallwiener, D. Inhibitory effect of statins on the proliferation of human breast cancer cells. Int. J. Clin. Pharmacol. Ther. 2004, 42, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Crescencio, M.E.; Rodríguez, E.; Páez, A.; Masso, F.A.; Montaño, L.F.; López-Marure, R. Statins inhibit the proliferation and induce cell death of human papilloma virus positive and negative cervical cancer cells. Int. J. Biomed. Sci. 2009, 5, 411–420. [Google Scholar]
- Sławińska-Brych, A.; Zdzisińska, B.; Kandefer-Szerszeń, M. Fluvastatin inhibits growth and alters the malignant phenotype of the C6 glioma cell line. Pharmacol. Rep. 2014, 66, 121–129. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Gollavilli, P.N.; Vasamsetti, S.B.; Karnewar, S.; Gopoju, R.; Ummanni, R.; Kotamraju, S. Statin-induced inhibition of breast cancer proliferation and invasion involves attenuation of iron transport: Intermediacy of nitric oxide and antioxidant defence mechanisms. FEBS J. 2014, 281, 3719–3738. [Google Scholar] [CrossRef]
- Qi, X.F.; Zheng, L.; Lee, K.J.; Kim, D.H.; Kim, C.S.; Cai, D.Q.; Wu, Z.; Qin, J.W.; Yu, Y.H.; Kim, S.K. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 2013, 4, e518. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Dhople, V.M.; Völker, U.; Ummanni, R.; Kotamraju, S. Fluvastatin mediated breast cancer cell death: A proteomic approach to identify differentially regulated proteins in MDA-MB-231 cells. PLoS ONE 2014, 9, e108890. [Google Scholar] [CrossRef]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S.C.; et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Al-Saleem, M.S.M.; Ahmed, O.A.A.; Fahmy, U.A.; Alhakamy, N.A.; Eid, B.G.; Abdel-Naim, A.B.; Abdel-Mageed, W.M.; AlRasheed, M.M.; Shazly, G.A. Optimized Conjugation of Fluvastatin to HIV-1 TAT Displays Enhanced Pro-Apoptotic Activity in HepG2 Cells. Int. J. Mol. Sci. 2020, 21, 4138. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Hosaka, Y.Z.; Beckwitt, C.; Wells, A.; Oltvai, Z.N.; Warita, K. Concomitant attenuation of HMG-CoA reductase expression potentiates the cancer cell growth-inhibitory effect of statins and expands their efficacy in tumor cells with epithelial characteristics. Oncotarget 2018, 9, 29304–29315. [Google Scholar] [CrossRef] [PubMed]
- Deezagi, A.; Safari, N. Rosuvastatin inhibit spheroid formation and epithelial-mesenchymal transition (EMT) in prostate cancer PC-3 cell line. Mol. Biol. Rep. 2020, 47, 8727–8737. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.S.; Thirupataiah, B.; Medishetti, R.; Ray, A.; Bele, S.D.; Hossain, K.A.; Reddy, G.S.; Edwin, R.K.; Joseph, A.; Kumar, N.; et al. Rosuvastatin based novel 3-substituted isocoumarins/3-alkylidenephthalides: Ultrasound assisted synthesis and identification of new anticancer agents. Eur. J. Med. Chem. 2020, 201, 112335. [Google Scholar] [CrossRef] [PubMed]
- Hosny, K.M.; Rizg, W.Y.; Khallaf, R.A. Preparation and Optimization of In Situ Gel Loaded with Rosuvastatin-Ellagic Acid Nanotransfersomes to Enhance the Anti-Proliferative Activity. Pharmaceutics 2020, 12, 263. [Google Scholar] [CrossRef]
- Aldalaen, S.; El-Gogary, R.I.; Nasr, M. Fabrication of rosuvastatin-loaded polymeric nanocapsules: A promising modality for treating hepatic cancer delineated by apoptotic and cell cycle arrest assessment. Drug Dev. Ind. Pharm. 2019, 45, 55–62. [Google Scholar] [CrossRef]
- Hamidreza Kheiri, M.; Alimohammadi, N.; Danafar, H. Preparation of biocompatible copolymeric micelles as a carrier of atorvastatin and rosuvastatin for potential anticancer activity study. Pharm. Dev. Technol. 2019, 24, 303–313. [Google Scholar] [CrossRef]
- de Wolf, E.; Abdullah, M.I.; Jones, S.M.; Menezes, K.; Moss, D.M.; Drijfhout, F.P.; Hart, S.R.; Hoskins, C.; Stronach, E.A.; Richardson, A. Dietary geranylgeraniol can limit the activity of pitavastatin as a potential treatment for drug-resistant ovarian cancer. Sci. Rep. 2017, 7, 5410. [Google Scholar] [CrossRef]
- Lee, N.; Tilija Pun, N.; Jang, W.J.; Bae, J.W.; Jeong, C.H. Pitavastatin induces apoptosis in oral squamous cell carcinoma through activation of FOXO3a. J. Cell Mol. Med. 2020, 24, 7055–7066. [Google Scholar] [CrossRef]
- Efimova, E.V.; Ricco, N.; Labay, E.; Mauceri, H.J.; Flor, A.C.; Ramamurthy, A.; Sutton, H.G.; Weichselbaum, R.R.; Kron, S.J. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence after Tumor Irradiation. Mol. Cancer Ther. 2018, 17, 407–418. [Google Scholar] [CrossRef]
- Denoyelle, C.; Albanese, P.; Uzan, G.; Hong, L.; Vannier, J.P.; Soria, J.; Soria, C. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell. Signal. 2003, 15, 327–338. [Google Scholar] [CrossRef]
- Obara, S.; Nakata, M.; Takeshima, H.; Kuratsu, J.; Maruyama, I.; Kitajima, I. Inhibition of migration of human glioblastoma cells by cerivastatin in association with focal adhesion kinase (FAK). Cancer Lett. 2002, 185, 153–161. [Google Scholar] [CrossRef]
- Denoyelle, C.; Vasse, M.; Körner, M.; Mishal, Z.; Ganné, F.; Vannier, J.P.; Soria, J.; Soria, C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: An in vitro study. Carcinogenesis 2001, 22, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Tan, M.M.; Xia, Z.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Cerivastatin triggers tumor-specific apoptosis with higher efficacy than lovastatin. Clin. Cancer Res. 2001, 7, 2067–2075. [Google Scholar] [PubMed]
- Vincent, L.; Chen, W.; Hong, L.; Mirshahi, F.; Mishal, Z.; Mirshahi-Khorassani, T.; Vannier, J.P.; Soria, J.; Soria, C. Inhibition of endothelial cell migration by cerivastatin, an HMG-CoA reductase inhibitor: Contribution to its anti-angiogenic effect. FEBS Lett. 2001, 495, 159–166. [Google Scholar] [CrossRef]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Mallappa, S.; Neeli, P.K.; Karnewar, S.; Kotamraju, S. Doxorubicin induces prostate cancer drug resistance by upregulation of ABCG4 through GSH depletion and CREB activation: Relevance of statins in chemosensitization. Mol. Carcinog. 2019, 58, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Shin, D.H.; Kim, J.S. Repurposing of Fluvastatin as an Anticancer Agent against Breast Cancer Stem Cells via Encapsulation in a Hyaluronan-Conjugated Liposome. Pharmaceutics 2020, 12, 1133. [Google Scholar] [CrossRef] [PubMed]
- Fromigué, O.; Hamidouche, Z.; Marie, P.J. Statin-induced inhibition of 3-hydroxy-3-methyl glutaryl coenzyme a reductase sensitizes human osteosarcoma cells to anticancer drugs. J. Pharmacol. Exp. Ther. 2008, 325, 595–600. [Google Scholar] [CrossRef]
- Kozar, K.; Kaminski, R.; Legat, M.; Kopec, M.; Nowis, D.; Skierski, J.S.; Koronkiewicz, M.; Jakóbisiak, M.; Golab, J. Cerivastatin demonstrates enhanced antitumor activity against human breast cancer cell lines when used in combination with doxorubicin or cisplatin. Int. J. Oncol. 2004, 24, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Orecchia, S.; Pescarmona, G.; Betta, P.G.; Ghigo, D.; Bosia, A. Statins revert doxorubicin resistance via nitric oxide in malignant mesothelioma. Int. J. Cancer 2006, 119, 17–27. [Google Scholar] [CrossRef]
- Środa-Pomianek, K.; Michalak, K.; Palko-Łabuz, A.; Uryga, A.; Świątek, P.; Majkowski, M.; Wesołowska, O. The Combined Use of Phenothiazines and Statins Strongly Affects Doxorubicin-Resistance, Apoptosis, and Cox-2 Activity in Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 955. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhang, R.; Zhu, Z.L.; Xu, S.N.; Li, Y.F. Effects of Simvastatin on Expression of Multidrug Resistance Gene and Protein of SHI-1 Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2016, 24, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, M.; Riganti, C.; Vitale, C.; Griggio, V.; Campia, I.; Robino, M.; Foglietta, M.; Castella, B.; Sciancalepore, P.; Buondonno, I.; et al. Simvastatin and downstream inhibitors circumvent constitutive and stromal cell-induced resistance to doxorubicin in IGHV unmutated CLL cells. Oncotarget 2015, 6, 29833–29846. [Google Scholar] [CrossRef] [PubMed]
- Greife, A.; Tukova, J.; Steinhoff, C.; Scott, S.D.; Schulz, W.A.; Hatina, J. Establishment and characterization of a bladder cancer cell line with enhanced doxorubicin resistance by mevalonate pathway activation. Tumour Biol. 2015, 36, 3293–3300. [Google Scholar] [CrossRef]
- Środa-Pomianek, K.; Michalak, K.; Palko-Łabuz, A.; Uryga, A.; Szczęśniak-Sięga, B.; Wesołowska, O. Simvastatin Strongly Augments Proapoptotic, Anti-inflammatory and Cytotoxic Activity of Oxicam Derivatives in Doxorubicin-resistant Colon Cancer Cells. Anticancer Res. 2019, 39, 727–734. [Google Scholar] [CrossRef]
- Palko-Łabuz, A.; Środa-Pomianek, K.; Wesołowska, O.; Kostrzewa-Susłow, E.; Uryga, A.; Michalak, K. MDR reversal and pro-apoptotic effects of statins and statins combined with flavonoids in colon cancer cells. Biomed. Pharmacother. 2019, 109, 1511–1522. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem. Pharmacol. 2008, 75, 907–913. [Google Scholar] [CrossRef]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Henslee, A.B.; Steele, T.A. Combination statin and chemotherapy inhibits proliferation and cytotoxicity of an aggressive natural killer cell leukemia. Biomark. Res. 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, P.; Marchal, J.A.; Boulaiz, H.; Carrillo, E.; Vélez, C.; Rodríguez-Serrano, F.; Melguizo, C.; Prados, J.; Madeddu, R.; Aranega, A. 5-Fluorouracil derivatives: A patent review. Expert Opin. Ther. Pat. 2012, 22, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, C.H.; Yee, L.K.; Venzon, D.J.; Schuler, B.; Grollman, F.; Chabuk, C.; Hamilton, J.M.; Chen, A.P.; Allegra, C.J.; Grem, J.L. High inter- and intrapatient variation in 5-fluorouracil plasma concentrations during a prolonged drug infusion. Clin. Cancer Res. 1999, 5, 1347–1352. [Google Scholar] [PubMed]
- Peters, G.J.; Lankelma, J.; Kok, R.M.; Noordhuis, P.; van Groeningen, C.J.; van der Wilt, C.L.; Meyer, S.; Pinedo, H.M. Prolonged retention of high concentrations of 5-fluorouracil in human and murine tumors as compared with plasma. Cancer Chemother. Pharmacol. 1993, 31, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Collie-Duguid, E.; Cassidy, J. Cerivastatin enhances the cytotoxicity of 5-fluorouracil on chemosensitive and resistant colorectal cancer cell lines. FEBS Lett. 2002, 531, 415–420. [Google Scholar] [CrossRef]
- Luput, L.; Sesarman, A.; Porfire, A.; Achim, M.; Muntean, D.; Casian, T.; Patras, L.; Rauca, V.F.; Drotar, D.M.; Stejerean, I.; et al. Liposomal simvastatin sensitizes C26 murine colon carcinoma to the antitumor effects of liposomal 5-fluorouracil in vivo. Cancer Sci. 2020, 111, 1344–1356. [Google Scholar] [CrossRef]
- Manu, K.A.; Shanmugam, M.K.; Li, F.; Chen, L.; Siveen, K.S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J. Mol. Med. 2014, 92, 267–276. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Zhang, S.; Doudican, N.A.; Quay, E.; Orlow, S.J. Fluvastatin enhances sorafenib cytotoxicity in melanoma cells via modulation of AKT and JNK signaling pathways. Anticancer Res. 2011, 31, 3259–3265. [Google Scholar]
- Zhou, T.Y.; Zhuang, L.H.; Hu, Y.; Zhou, Y.L.; Lin, W.K.; Wang, D.D.; Wan, Z.Q.; Chang, L.L.; Chen, Y.; Ying, M.D.; et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells. Sci. Rep. 2016, 6, 30483. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.; Zheng, H.; Wang, B.; Liu, Y.; Liu, D.; Chen, J.; Xu, W.; Li, A.; Zhu, Y. Synergistic anti-tumor efficacy of sorafenib and fluvastatin in hepatocellular carcinoma. Oncotarget 2017, 8, 23265–23276. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Dai, W.; Mao, Y.; Wu, L.; Li, J.; Chen, K.; Yu, Q.; Kong, R.; Li, S.; Zhang, J.; et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis. J. Exp. Clin. Cancer Res. 2020, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Riaño, I.; Martín, L.; Varela, M.; Serrano, T.; Núñez, O.; Mínguez, B.; Rodrigues, P.M.; Perugorria, M.J.; Banales, J.M.; Arenas, J.I. Efficacy and Safety of the Combination of Pravastatin and Sorafenib for the Treatment of Advanced Hepatocellular Carcinoma (ESTAHEP Clinical Trial). Cancers 2020, 12, 1900. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Barber, T.D.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N. Engl. J. Med. 2004, 351, 2883. [Google Scholar] [CrossRef] [PubMed]
- Cemeus, C.; Zhao, T.T.; Barrett, G.M.; Lorimer, I.A.; Dimitroulakos, J. Lovastatin enhances gefitinib activity in glioblastoma cells irrespective of EGFRvIII and PTEN status. J. Neurooncol. 2008, 90, 9–17. [Google Scholar] [CrossRef]
- Yang, S.H.; Lin, H.Y.; Chang, V.H.; Chen, C.C.; Liu, Y.R.; Wang, J.; Zhang, K.; Jiang, X.; Yen, Y. Lovastatin overcomes gefitinib resistance through TNF-α signaling in human cholangiocarcinomas with different LKB1 statuses in vitro and in vivo. Oncotarget 2015, 6, 23857–23873. [Google Scholar] [CrossRef]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Johnson, D.H.; et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef]
- Park, I.H.; Kim, J.Y.; Jung, J.I.; Han, J.Y. Lovastatin overcomes gefitinib resistance in human non-small cell lung cancer cells with K-Ras mutations. Investig. New Drugs 2010, 28, 791–799. [Google Scholar] [CrossRef]
- Hwang, K.E.; Kwon, S.J.; Kim, Y.S.; Park, D.S.; Kim, B.R.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp. Cell Res. 2014, 323, 288–296. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, I.K.; Lee, H.I.; Mo, J.Y.; Yeo, C.D.; Kang, H.H.; Moon, H.S.; Lee, S.H. The apoptotic effect of simvastatin via the upregulation of BIM in nonsmall cell lung cancer cells. Exp. Lung Res. 2016, 42, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bi, H.; Hou, J.; Zhang, X.; Zhang, C.; Yue, L.; Wen, X.; Liu, D.; Shi, H.; Yuan, J.; et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis. 2013, 4, e814. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Otahal, A.; Aydemir, D.; Tomasich, E.; Minichsdorfer, C. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci. Rep. 2020, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Glodkowska-Mrowka, E.; Mrowka, P.; Basak, G.W.; Niesiobedzka-Krezel, J.; Seferynska, I.; Wlodarski, P.K.; Jakobisiak, M.; Stoklosa, T. Statins inhibit ABCB1 and ABCG2 drug transporter activity in chronic myeloid leukemia cells and potentiate antileukemic effects of imatinib. Exp. Hematol. 2014, 42, 439–447. [Google Scholar] [CrossRef]
- Oh, B.; Kim, T.Y.; Min, H.J.; Kim, M.; Kang, M.S.; Huh, J.Y.; Kim, Y.; Lee, D.S. Synergistic killing effect of imatinib and simvastatin on imatinib-resistant chronic myelogenous leukemia cells. Anticancer Drugs 2013, 24, 20–31. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Duan, J.; Wang, H.; Zhang, Y.; Qiao, K.; Wang, J. Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle 2019, 18, 3337–3350. [Google Scholar] [CrossRef]
- Koi, C.; Izumi, H.; Kurita, T.; Nguyen, T.T.; Murakami, M.; Yoshiura, Y.; Hachisuga, T.; Morimoto, Y. Lovastatin induced Kruppel like factor 2 (KLF2), Kruppel like factor 6 (KLF6) and Ras homolog family member B (RHOB) genes and preferentially led to viability reduction of Cisplatin-resistant cells. Oncotarget 2017, 8, 106429–106442. [Google Scholar] [CrossRef]
- Hu, T.; Shen, H.; Huang, H.; Yang, Z.; Zhou, Y.; Zhao, G. Cholesterol-lowering drug pitavastatin targets lung cancer and angiogenesis via suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling. Anticancer Drugs 2020, 31, 377–384. [Google Scholar] [CrossRef]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22, 3–10. [Google Scholar]
- Kawashiri, T.; Tokunaga, A.; Kobayashi, D.; Shimazoe, T. Anti-tumor Activities of 3-Hydroxy-3-methylglutaryl Coenzyme A (HMG-CoA) Reductase Inhibitors and Bisphosphonates in Pancreatic Cell Lines Which Show Poor Responses to Gemcitabine. Biol. Pharm. Bull. 2020, 43, 49–52. [Google Scholar] [CrossRef]
- Kitagawa, K.; Moriya, K.; Kaji, K.; Saikawa, S.; Sato, S.; Nishimura, N.; Namisaki, T.; Akahane, T.; Mitoro, A.; Yoshiji, H. Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2020, 21, 7588. [Google Scholar] [CrossRef]
- Chen, Y.H.; Chen, Y.C.; Lin, C.C.; Hsieh, Y.P.; Hsu, C.S.; Hsieh, M.C. Synergistic Anticancer Effects of Gemcitabine with Pitavastatin on Pancreatic Cancer Cell Line MIA PaCa-2 in vitro and in vivo. Cancer Manag. Res. 2020, 12, 4645–4665. [Google Scholar] [CrossRef] [PubMed]
- Xian, G.; Zhao, J.; Qin, C.; Zhang, Z.; Lin, Y.; Su, Z. Simvastatin attenuates macrophage-mediated gemcitabine resistance of pancreatic ductal adenocarcinoma by regulating the TGF-β1/Gfi-1 axis. Cancer Lett. 2017, 385, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shah, S.R.; Illum, H.; Dowell, J. Vemurafenib: Targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 2012, 72, 2207–2222. [Google Scholar] [CrossRef]
- Held, M.A.; Langdon, C.G.; Platt, J.T.; Graham-Steed, T.; Liu, Z.; Chakraborty, A.; Bacchiocchi, A.; Koo, A.; Haskins, J.W.; Bosenberg, M.W.; et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 2013, 3, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Theodosakis, N.; Langdon, C.G.; Micevic, G.; Krykbaeva, I.; Means, R.E.; Stern, D.F.; Bosenberg, M.W. Inhibition of isoprenylation synergizes with MAPK blockade to prevent growth in treatment-resistant melanoma, colorectal, and lung cancer. Pigment. Cell Melanoma Res. 2019, 32, 292–302. [Google Scholar] [CrossRef]
- Nishiya, M.; Yasuhira, S.; Shibazaki, M.; Oikawa, H.; Masuda, T.; Maesawa, C. Fluvastatin exerts an antitumor effect in vemurafenib-resistant melanoma cells. Anticancer Drugs 2019, 30, 451–457. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Gong, Z.; Shen, L.; Kao, C.; Hock, J.M.; Sun, L.; Li, X. Lovastatin enhances adenovirus-mediated TRAIL induced apoptosis by depleting cholesterol of lipid rafts and affecting CAR and death receptor expression of prostate cancer cells. Oncotarget 2015, 6, 3055–3070. [Google Scholar] [CrossRef] [PubMed]
- Iizuka-Ohashi, M.; Watanabe, M.; Sukeno, M.; Morita, M.; Hoang, N.T.H.; Kuchimaru, T.; Kizaka-Kondoh, S.; Sowa, Y.; Sakaguchi, K.; Taguchi, T.; et al. Blockage of the mevalonate pathway overcomes the apoptotic resistance to MEK inhibitors with suppressing the activation of Akt in cancer cells. Oncotarget 2018, 9, 19597–19612. [Google Scholar] [CrossRef]
- Abdullah, M.I.; Abed, M.N.; Khanim, F.; Richardson, A. Screening a library of approved drugs reveals that prednisolone synergizes with pitavastatin to induce ovarian cancer cell death. Sci. Rep. 2019, 9, 9632. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, P.; Li, N.; Kiang, K.M.Y.; Cheng, S.Y.; Wong, V.K.; Leung, G.K. Lovastatin Enhances Cytotoxicity of Temozolomide via Impairing Autophagic Flux in Glioblastoma Cells. Biomed. Res. Int. 2019, 2019, 2710693. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.B.; Tsai, Y.C.; Chin, L.H.; Tseng, J.H.; Tang, L.W.; Horng, S.; Fan, Y.C.; Hsu, S.P. A Synergistic Anti-Cancer Effect of Troglitazone and Lovastatin in a Human Anaplastic Thyroid Cancer Cell Line and in a Mouse Xenograft Model. Int. J. Mol. Sci. 2018, 19, 1834. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Obata, N.; Kawashima, K.; Tabata, M.; Imano, M.; Satou, T.; Nishida, S. Combination therapy with dacarbazine and statins improved the survival rate in mice with metastatic melanoma. J. Cell. Physiol. 2019, 234, 17975–17989. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Esparza, Y.C.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; García Latorre, E.A.; Boyer, O.; Varin, R.; Jiménez-Zamudio, L.A.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar] [CrossRef]
- Ingersoll, M.A.; Miller, D.R.; Martinez, O.; Wakefield, C.B.; Hsieh, K.C.; Simha, M.V.; Kao, C.L.; Chen, H.T.; Batra, S.K.; Lin, M.F. Statin derivatives as therapeutic agents for castration-resistant prostate cancer. Cancer Lett. 2016, 383, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Roberts, A.; Juarez, D.; Vo, T.T.; Bhatt, S.; Herzog, L.O.; Mallya, S.; Bellin, R.J.; Agarwal, S.K.; Salem, A.H.; et al. Statins enhance efficacy of venetoclax in blood cancers. Sci. Transl. Med. 2018, 10, eaaq1240. [Google Scholar] [CrossRef]
- Rauca, V.F.; Licarete, E.; Luput, L.; Sesarman, A.; Patras, L.; Bulzu, P.; Rakosy-Tican, E.; Banciu, M. Combination therapy of simvastatin and 5, 6-dimethylxanthenone-4-acetic acid synergistically suppresses the aggressiveness of B16.F10 melanoma cells. PLoS ONE 2018, 13, e0202827. [Google Scholar] [CrossRef]
- Sethunath, V.; Hu, H.; de Angelis, C.; Veeraraghavan, J.; Qin, L.; Wang, N.; Simon, L.M.; Wang, T.; Fu, X.; Nardone, A.; et al. Targeting the Mevalonate Pathway to Overcome Acquired Anti-HER2 Treatment Resistance in Breast Cancer. Mol. Cancer Res. 2019, 17, 2318–2330. [Google Scholar] [CrossRef]
- Iannelli, F.; Roca, M.S.; Lombardi, R.; Ciardiello, C.; Grumetti, L.; de Rienzo, S.; Moccia, T.; Vitagliano, C.; Sorice, A.; Costantini, S.; et al. Synergistic antitumor interaction of valproic acid and simvastatin sensitizes prostate cancer to docetaxel by targeting CSCs compartment via YAP inhibition. J. Exp. Clin. Cancer Res. 2020, 39, 213. [Google Scholar] [CrossRef]
- Ghasemi, M.; Malek, M.; Javanmard, S.H.; Ghasemi, A.; Esfahani, H.N.; Vaseghi, G. Atorvastatin enhances apoptotic effects of tamoxifen on melanoma cancer cells. Bratisl. Lek. Listy 2019, 120, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, Q.; Lin, Y.; Reddy, B.S.; Yang, C.S. Combination of atorvastatin and celecoxib synergistically induces cell cycle arrest and apoptosis in colon cancer cells. Int. J. Cancer 2008, 122, 2115–2124. [Google Scholar] [CrossRef]
- Goda, A.E.; Elsisi, A.E.; Sokkar, S.S.; Abdelrazik, N.M. Enhanced in vivo targeting of estrogen receptor alpha signaling in murine mammary adenocarcinoma by nilotinib/rosuvastatin novel combination. Toxicol. Appl. Pharmacol. 2020, 404, 115185. [Google Scholar] [CrossRef]
- El Sayed, I.; Helmy, M.W.; El-Abhar, H.S. Inhibition of SRC/FAK cue: A novel pathway for the synergistic effect of rosuvastatin on the anti-cancer effect of dasatinib in hepatocellular carcinoma. Life Sci. 2018, 213, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Boulate, G.; Amazit, L.; Naman, A.; Seck, A.; Paci, A.; Lombes, A.; Pussard, E.; Baudin, E.; Lombes, M.; Hescot, S. Potentiation of mitotane action by rosuvastatin: New insights for adrenocortical carcinoma management. Int. J. Oncol. 2019, 54, 2149–2156. [Google Scholar] [CrossRef]
- Al-Qatati, A.; Aliwaini, S. Combined pitavastatin and dacarbazine treatment activates apoptosis and autophagy resulting in synergistic cytotoxicity in melanoma cells. Oncol. Lett. 2017, 14, 7993–7999. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Gouda, K.; Chisaki, I.; Asada, K.; Ogura, J.; Takahashi, N.; Konishi, T.; Koshida, Y.; Sasaki, S.; Yamaguchi, H.; et al. Regulation of multidrug resistance protein 2 (MRP2, ABCC2) expression by statins: Involvement of SREBP-mediated gene regulation. Int. J. Pharm. 2013, 452, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Scheers, E.; Grabner, M.; Verpaalen, B.; Helsen, N.; Vanstreels, E.; Daelemans, D.; Verfaillie, C.; Neyts, J. Understanding the molecular mechanism of host-based statin resistance in hepatitis C virus replicon containing cells. Biochem. Pharmacol. 2015, 96, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Masko, E.M.; Alfaqih, M.A.; Solomon, K.R.; Barry, W.T.; Newgard, C.B.; Muehlbauer, M.J.; Valilis, N.A.; Phillips, T.E.; Poulton, S.H.; Freedland, A.R.; et al. Evidence for Feedback Regulation Following Cholesterol Lowering Therapy in a Prostate Cancer Xenograft Model. Prostate 2017, 77, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Lettiero, B.; Inasu, M.; Kimbung, S.; Borgquist, S. Insensitivity to atorvastatin is associated with increased accumulation of intracellular lipid droplets and fatty acid metabolism in breast cancer cells. Sci. Rep. 2018, 8, 5462. [Google Scholar] [CrossRef]
- Göbel, A.; Breining, D.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Induction of 3-hydroxy-3-methylglutaryl-CoA reductase mediates statin resistance in breast cancer cells. Cell Death Dis. 2019, 10, 91. [Google Scholar] [CrossRef]
- El-Ashmawy, N.E.; Al-Ashmawy, G.M.; Amr, E.A.; Khedr, E.G. Inhibition of lovastatin- and docosahexaenoic acid-initiated autophagy in triple negative breast cancer reverted resistance and enhanced cytotoxicity. Life Sci. 2020, 259, 118212. [Google Scholar] [CrossRef]
- Reiner, Z. Resistance and intolerance to statins. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Cauley, J.A.; McTiernan, A.; Rodabough, R.J.; LaCroix, A.; Bauer, D.C.; Margolis, K.L.; Paskett, E.D.; Vitolins, M.Z.; Furberg, C.D.; Chlebowski, R.T. Statin use and breast cancer: Prospective results from the Women’s Health Initiative. J. Natl. Cancer Inst. 2006, 98, 700–707. [Google Scholar] [CrossRef]
- Simon, T.G.; Bonilla, H.; Yan, P.; Chung, R.T.; Butt, A.A. Atorvastatin and fluvastatin are associated with dose-dependent reductions in cirrhosis and hepatocellular carcinoma, among patients with hepatitis C virus: Results from ERCHIVES. Hepatology 2016, 64, 47–57. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Lin, X.; Albiges, L.; Fay, A.P.; Kaymakcalan, M.D.; Mickey, S.S.; Ghoroghchian, P.P.; Bhatt, R.S.; Kaffenberger, S.D.; Simantov, R.; et al. Statins and survival outcomes in patients with metastatic renal cell carcinoma. Eur. J. Cancer 2016, 52, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef]
- Chen, T.L.; Estey, E.H.; Othus, M.; Gardner, K.M.; Markle, L.J.; Walter, R.B. Cyclosporine modulation of multidrug resistance in combination with pravastatin, mitoxantrone and etoposide for adult patients with relapsed/refractory acute myeloid leukemia: A phase 1/2 study. Leuk. Lymphoma 2013, 54, 2534–2536. [Google Scholar] [CrossRef]
- Farooqi, M.A.M.; Malhotra, N.; Mukherjee, S.D.; Sanger, S.; Dhesy-Thind, S.K.; Ellis, P.; Leong, D.P. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2018, 13, e0209486. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tilija Pun, N.; Jeong, C.-H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. https://doi.org/10.3390/ph14050470
Tilija Pun N, Jeong C-H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals. 2021; 14(5):470. https://doi.org/10.3390/ph14050470
Chicago/Turabian StyleTilija Pun, Nirmala, and Chul-Ho Jeong. 2021. "Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance" Pharmaceuticals 14, no. 5: 470. https://doi.org/10.3390/ph14050470
APA StyleTilija Pun, N., & Jeong, C.-H. (2021). Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals, 14(5), 470. https://doi.org/10.3390/ph14050470