
New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis

 , ,
, ,  and
and
Abstract

1. Introduction
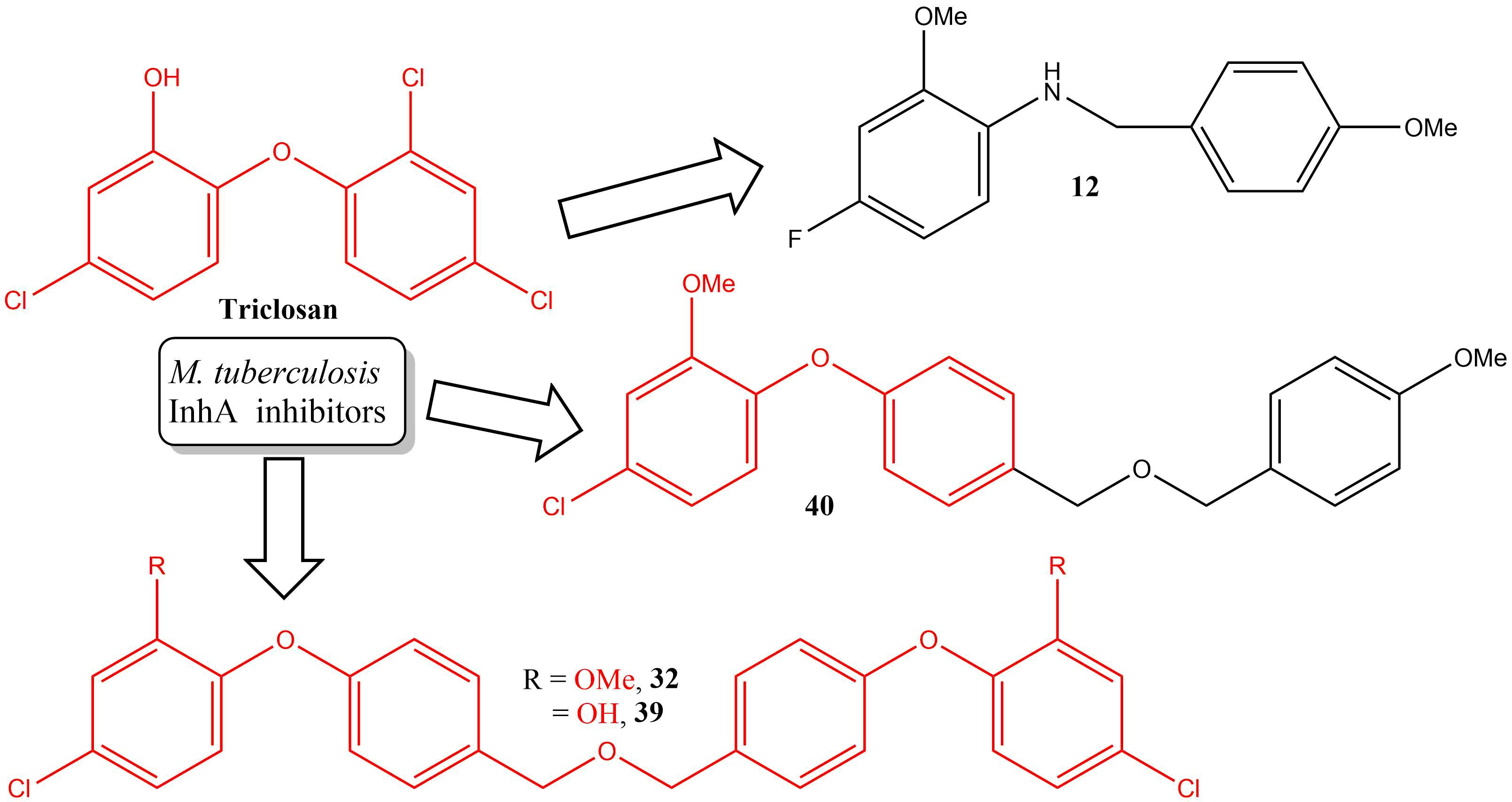
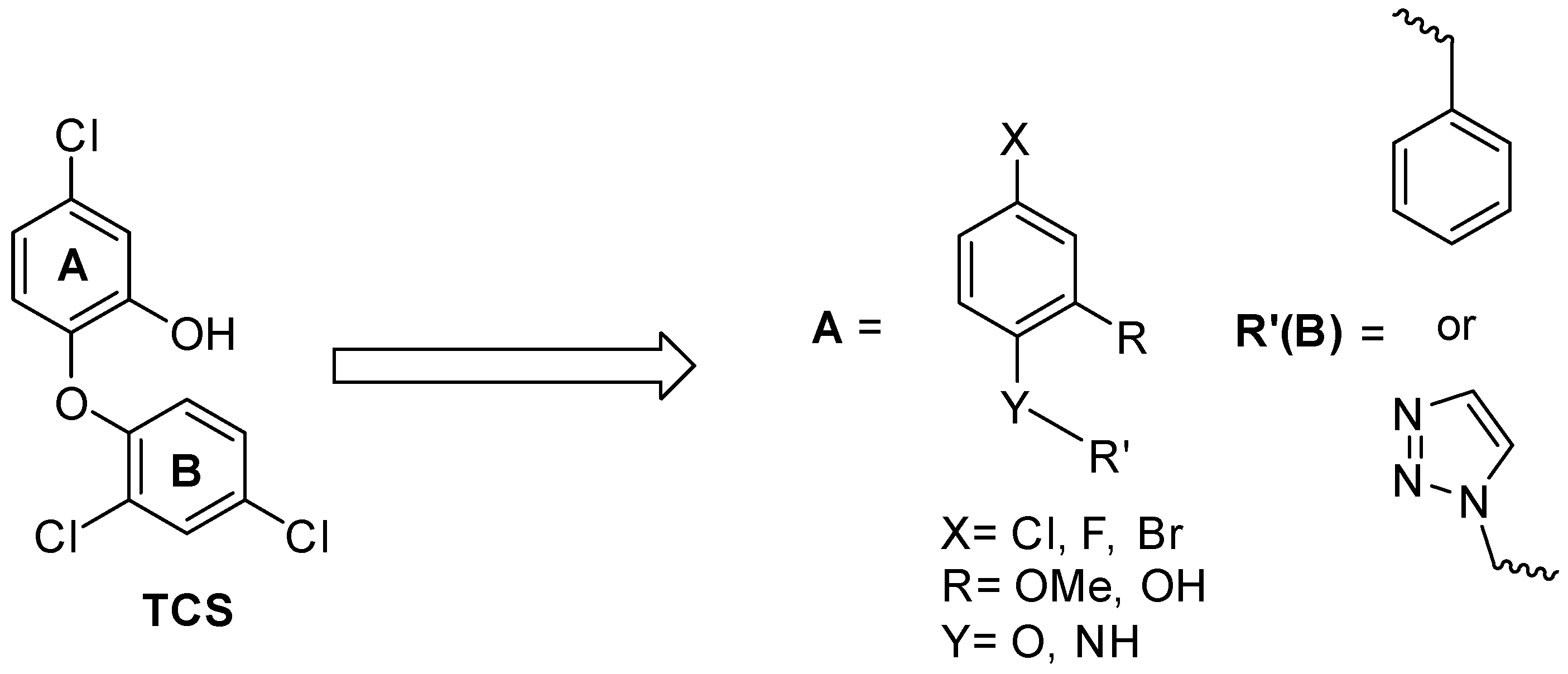
SAR and Design of Compounds
2. Results and Discussion
2.1. Drug-Like Properties
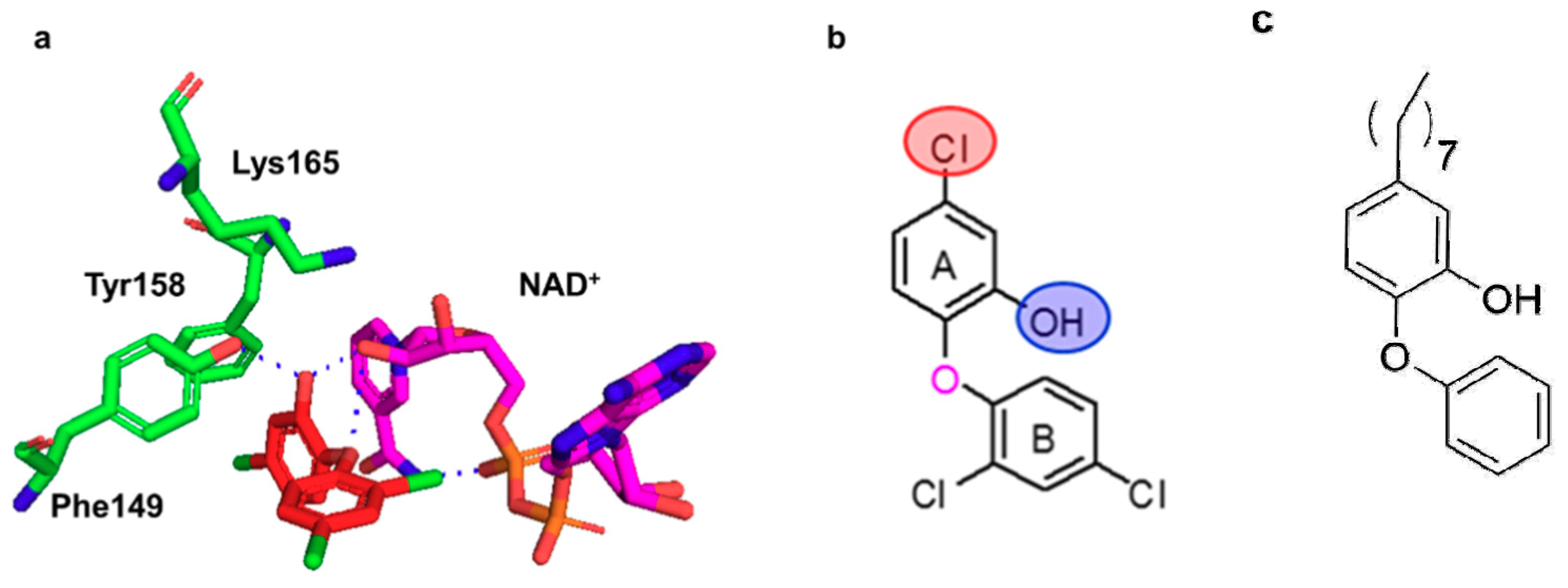
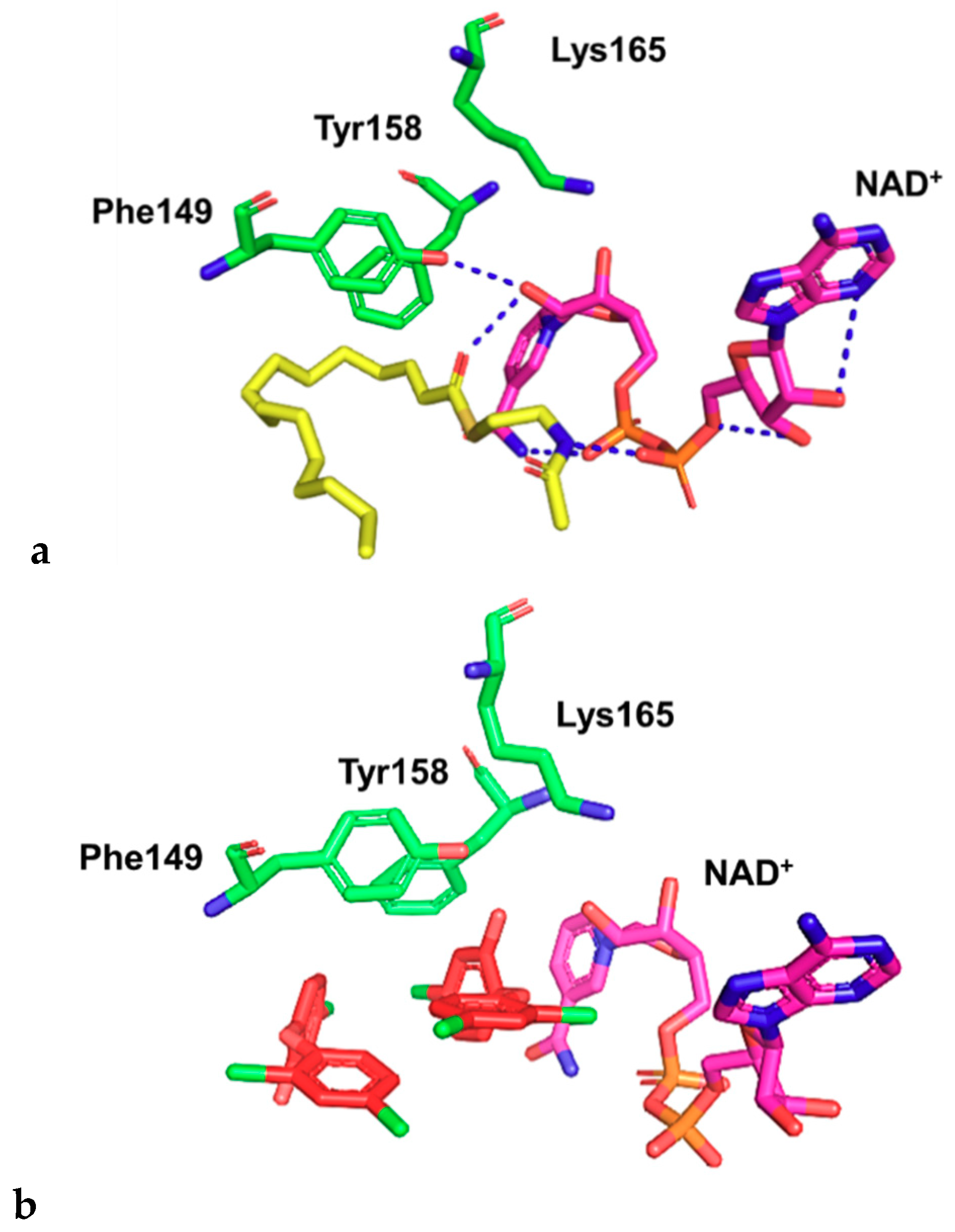
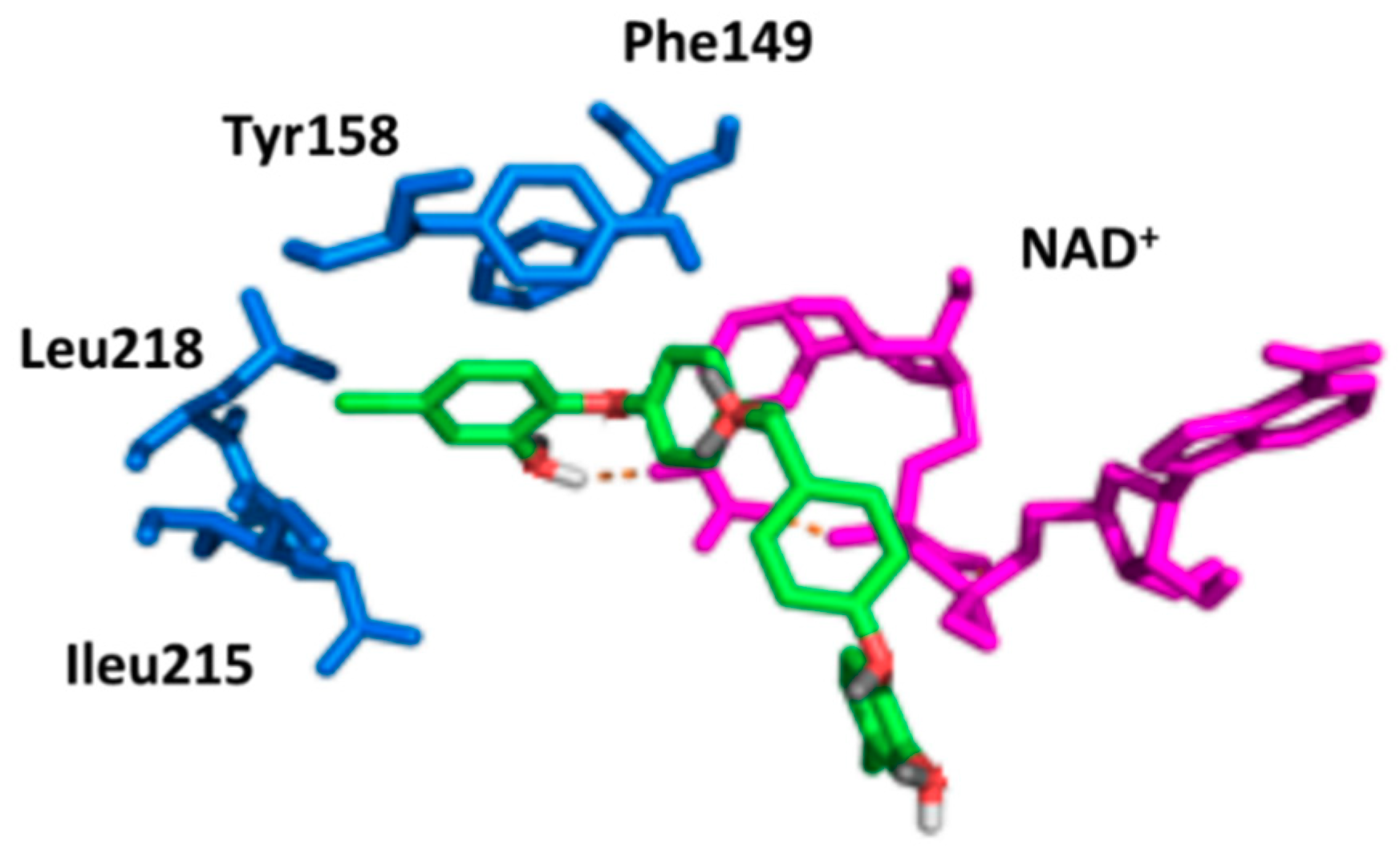
2.2. Docking Studies
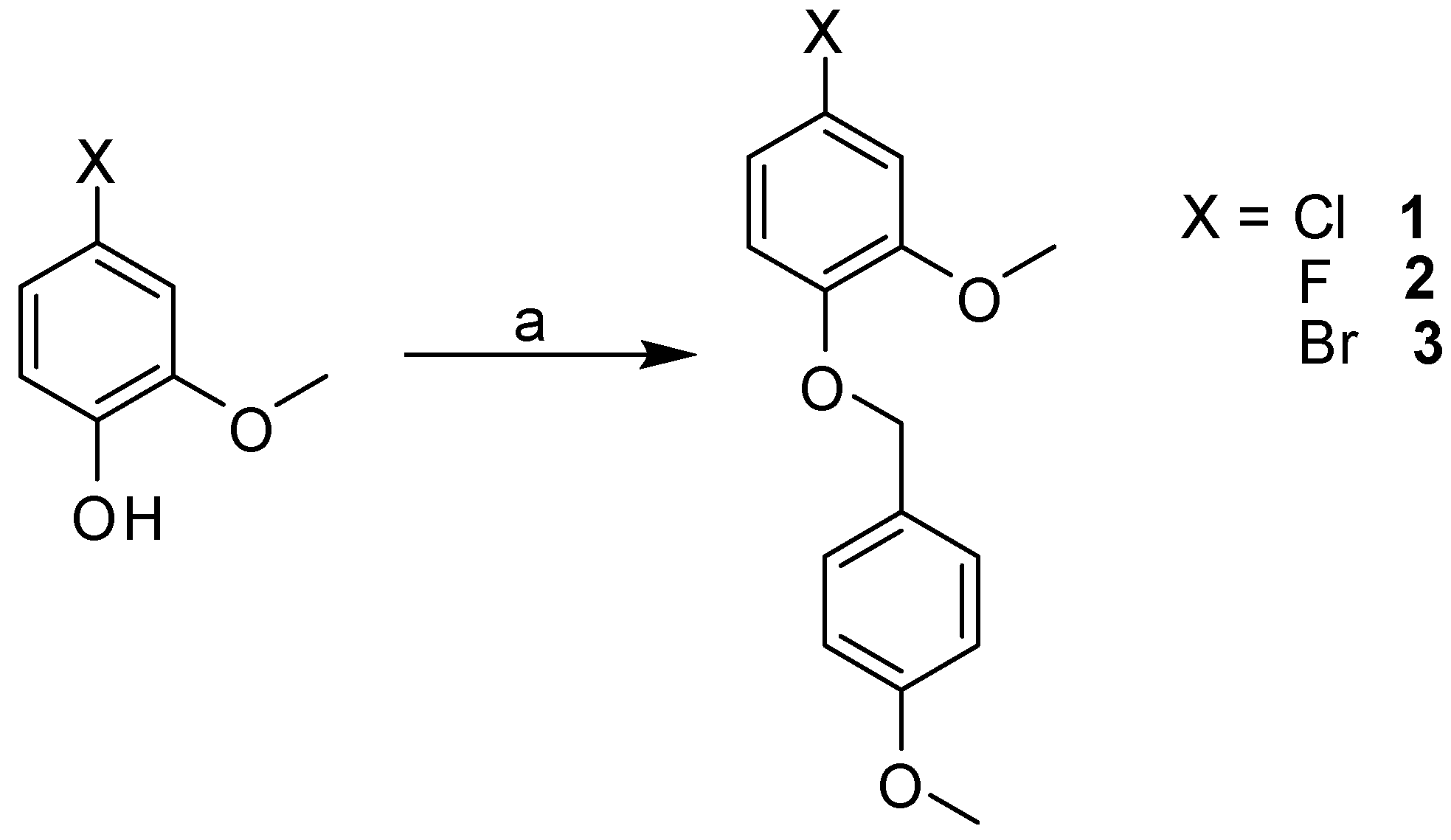
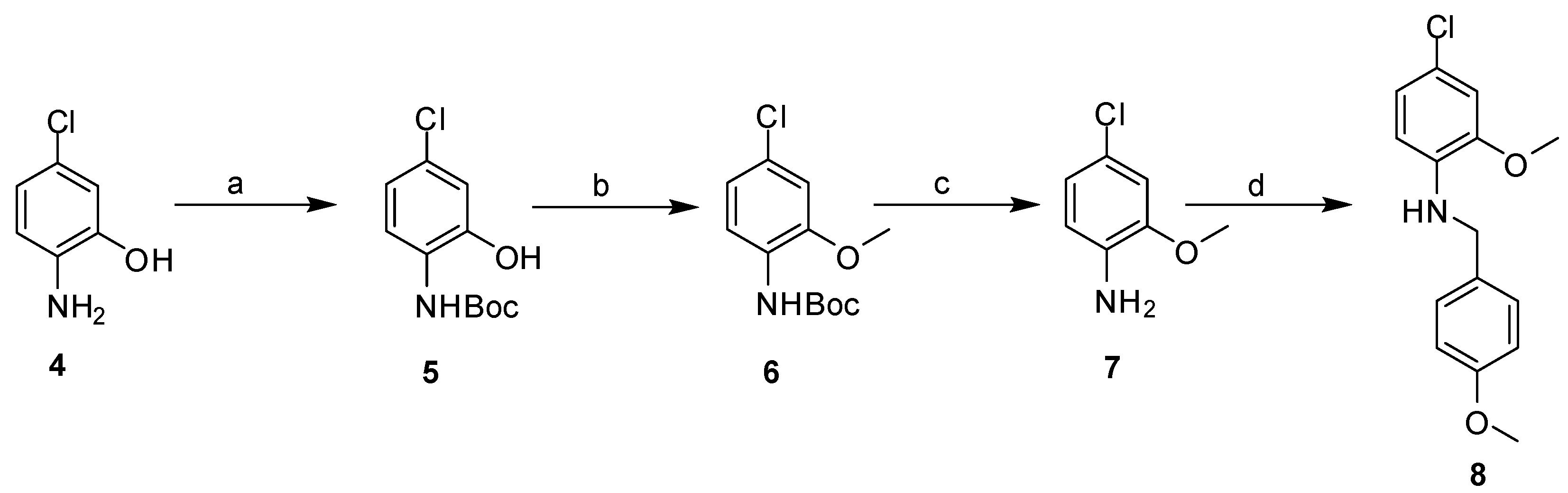
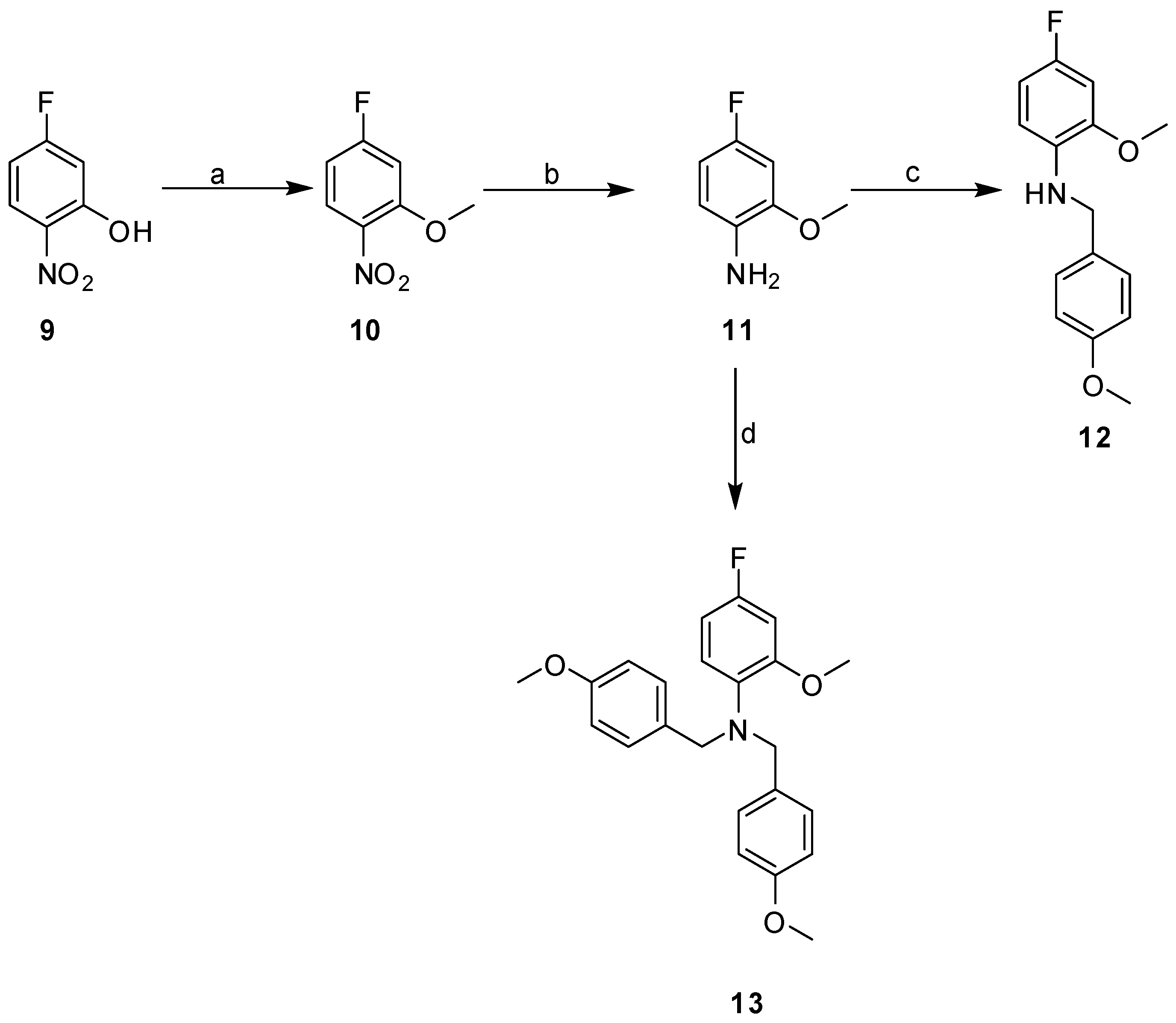
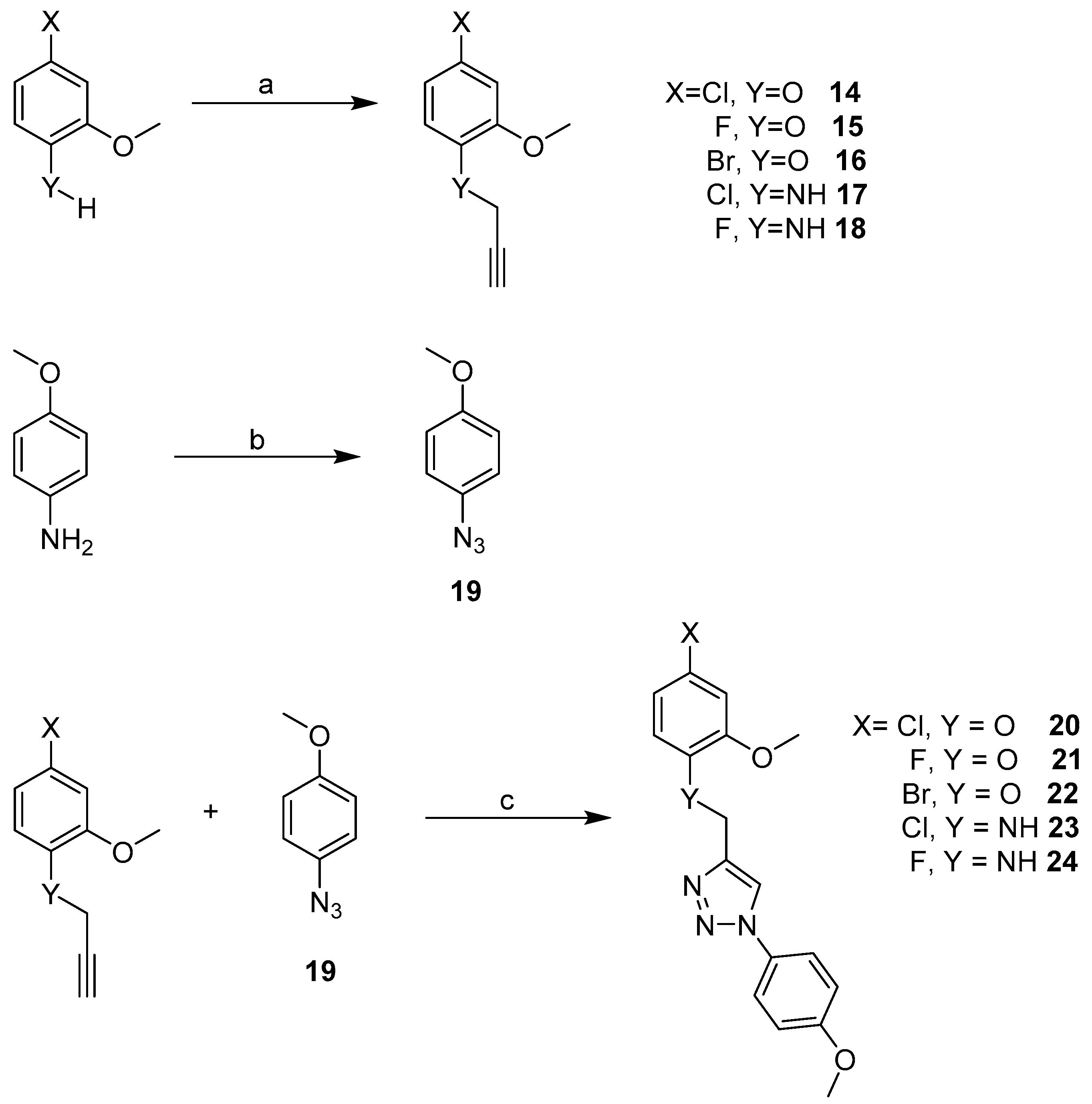
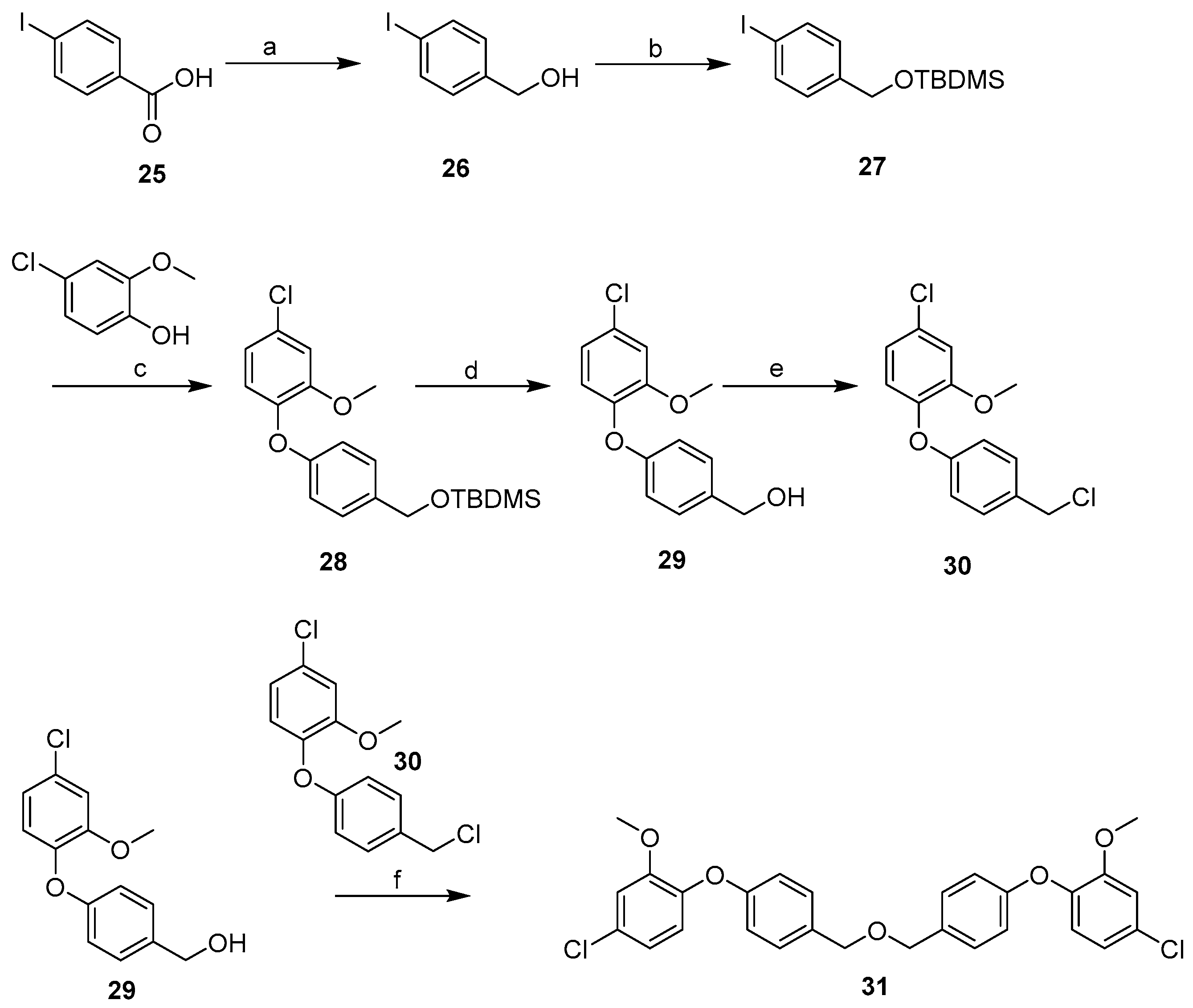
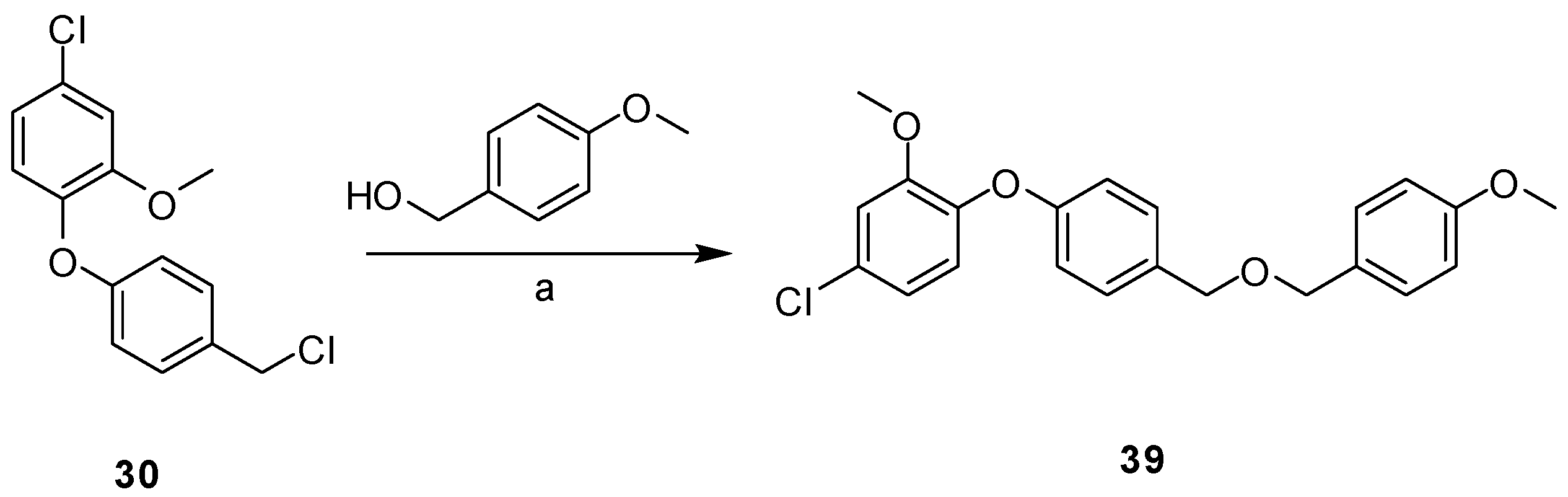
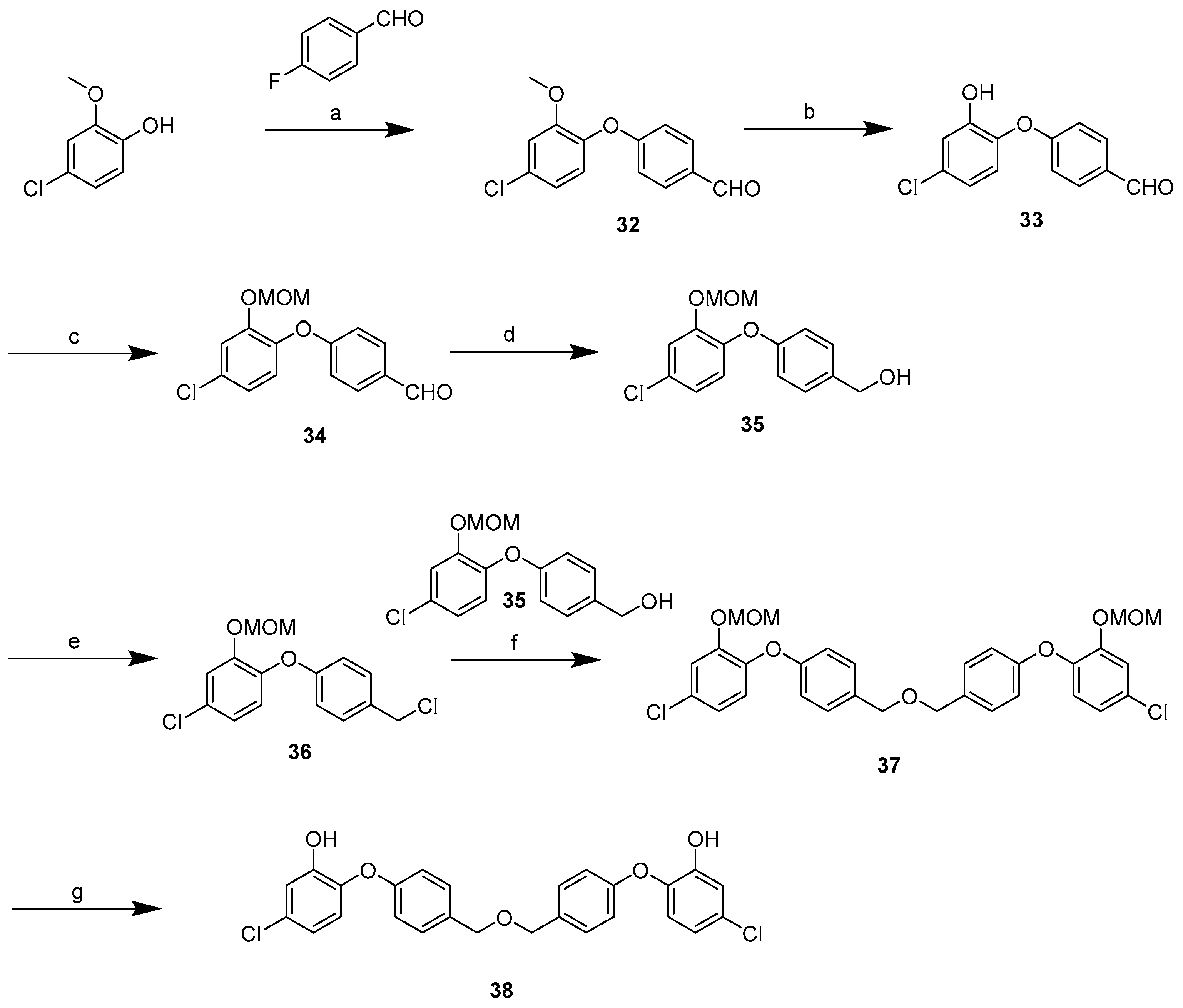
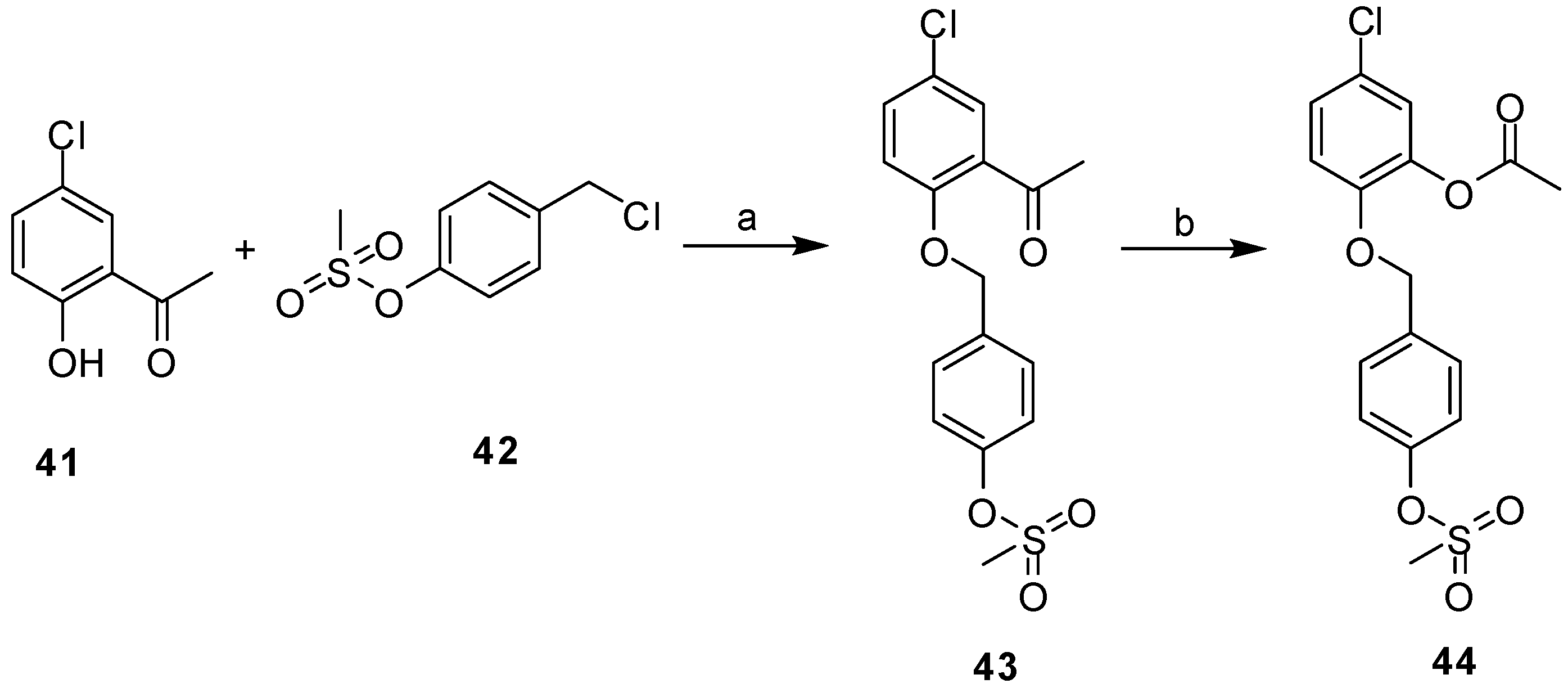
2.3. Synthesis
2.4. Inhibition Studies with Purified InhA
2.5. Minimum Inhibitory Concentration (MIC)
2.6. Correlation of the Docking and Isolated Enzyme Results
2.7. Structure Activity Relationships for the Compounds Evaluated
3. Materials and Methods
3.1. Molecular Docking and Design studies
3.2. Synthesis
3.2.1. Materials and General Methods
3.2.2. General Procedure for Library Synthesis
- General Procedure for Synthesis of the Benzylphenyl ether Compounds (A)
- General Procedure for the Synthesis of the Propargyl Analogues (B)
- General Procedure for the Synthesis of the Azide (C)
- General Procedure for the Synthesis of the Triazole-Linked Biaryl Anilines and Ethers via Click Chemistry (D)
- Purification by Precipitation (E)
3.3. InhA Enzyme Assay
3.4. Minimum Inhibitory Concentration Determination
3.5. MIC Determination for Di-Triclosan Derivative 39
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CuAAC | Copper(I) Azide-Alkyne Cycloaddition |
| ENR | Enoyl reductase |
| XDR | Extensively drug-resistant |
| INH | Isoniazid |
| GOLD | Genetic Optimization for Ligand Docking |
| HBA | Hydrogen bond acceptors |
| HBD | Hydrogen bond donors |
| InhA | M. tuberculosis enoyl reductase |
| MDR | Multidrug-resistance |
| Mtb | Mycobacterium tuberculosis |
| NAD+ | Nicotinamide adenosine dinucleotide |
| r.t. | Room temperature |
| SAR | Structure-activity relationship |
| TPSA | Topological polar surface area |
| TCS | Triclosan |
| TB | Tuberculosis |
References
- Global Tuberculosis Report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 12 June 2019).
- Tuberculosis. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 10 February 2020).
- HIV/AIDS. Available online: www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 10 February 2020).
- Grange, J.M. Mycobacteria and Human Disease, 2nd ed.; Arnold-Hodder Headline Group: London, UK, 1996; pp. 159–187. [Google Scholar]
- Finch, R.G.; Greenwood, D.; Ragnar Norby, S.; Whitley, R.J. Antibiotics and Chemotherapy, Anti-Infective Agents and Their Use in Treatment, 8th ed.; Elsevier Science Ltd.: Edinburgh, UK, 2003; 427p. [Google Scholar]
- Bhatt, A.; Molle, V.; Besra, G.S.; Jacobs, W.R.; Kremer, L. The Mycobacterium tuberculosis FAS-II condensing enzymes: Their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007, 64, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; White, S.W.; Rock, C.O. Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid Res. 2001, 40, 467–497. [Google Scholar] [CrossRef]
- Janin, Y.L. Antituberculosis drugs: Ten years of research. Bioorg. Med. Chem. 2007, 15, 2479–2513. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R., Jr.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, H.; Yu, S.; Wang, F.; Sacchettini, J.C.; Magliozzo, R.S. Hydrogen peroxide-mediated isoniazid activation catalyzed by Mycobacterium tuberculosis catalase-peroxidase (KatG) and its S315T mutant. Biochemistry 2006, 45, 4131–4140. [Google Scholar] [CrossRef]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Quemard, A.; Sacchettini, J.C.; Dessen, A.; Vilcheze, C.; Bittman, R.; Jacobs, W.R., Jr.; Blanchard, J.S. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995, 34, 8235–8241. [Google Scholar] [CrossRef]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; De Lisle, G.; Jacobs, W.R. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Sciecnce 1994, 263, 227–230. [Google Scholar] [CrossRef]
- Vilchèze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef]
- Cole, S.T.; Davies, K.E.; Mcmurray, D.N.; Jacobs, W.R. Tuberculosis and the Tubercle Bacillus, 1st ed.; ASM Press, American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Zhang, Y.; Yew, W.-W. Mechanisms of drug resistance in Mycobacterium tuberculosis: Update 2015. Int. J. Tuberc. Lung Dis. 2015, 19, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.V.B.; Vasconcelos, I.B.; Prado, A.M.X.; Fadel, V.; Basso, L.A.; de Azevedo, W.F., Jr.; Santos, D.S. Crystallographic studies on the binding of isonicotinyl-NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. J. Struct. Biol. 2007, 159, 369–380. [Google Scholar] [CrossRef]
- Rožman, K.; Sosič, I.; Fernandez, R.; Young, R.J.; Mendoza, A.; Gobec, S.; Encinas, L. A new ‘golden age’ for the antitubercular target InhA. Drug Discov. Today 2017, 22, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Rubin, J.R.; Holland, D.R.; Zhang, E.; Snow, M.E.; Rock, C.O. Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 1999, 274, 11110–11114. [Google Scholar] [CrossRef] [PubMed]
- Vosatka, R.; Kratky, M.; Vinsova, J. Triclosan and its derivatives as antimycobacterial active agents. Eur. J. Pharm. Sci. 2018, 114, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Rajkhowa, S.; Jha, A.N.; Deka, R.C. Anti-tubercular drug development: Computational strategies to identify potential compounds. J. Mol. Graph. Model. 2015, 62, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Falany, C.N.; James, M.O. Triclosan as a substrate and inhibitor of 3’-phosphoadenosine 5’-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug Metab. Dispos. 2004, 32, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.R.; Morbidoni, H.R.; Alland, D.; Sneddon, S.F.; Gourlie, B.B.; Staveski, M.M. Targeting tuberculosis and malaria through inhibition of Enoyl reductase: Compound activity and structural data. J. Biol. Chem. 2003, 278, 20851–20859. [Google Scholar] [CrossRef]
- Sivaraman, S.; Sullivan, T.J.; Johnson, F.; Novichenok, P.; Cui, G.; Simmerling, A.C.; Tonge, P.J. Inhibition of the Bacterial Enoyl Reductase FabI by Triclosan: A Structure−Reactivity Analysis of FabI Inhibition by Triclosan Analogues. J. Med. Chem. 2004, 47, 509–518. [Google Scholar] [CrossRef]
- Sivaraman, S.; Zwahlen, J.; Bell, A.F.; Hedstrom, L.; Tonge, P.J. Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: Kinetic analysis of mutant FabIs. Biochemistry 2003, 42, 4406–4413. [Google Scholar] [CrossRef]
- Sullivan, T.J.; Truglio, J.J.; Boyne, M.E.; Novichenok, P.; Zhang, X.; Stratton, C.F.; Li, H.-J.; Kaur, T.; Amin, A.; Johnson, F.; et al. High Affinity InhA Inhibitors with Activity against Drug-Resistant Strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal Structure of the Mycobacterium tuberculosis Enoyl-ACP Reductase, InhA, in Complex with NAD+ and a C16 Fatty Acyl Substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef] [PubMed]
- Ende, C.W.A.; Knudson, S.E.; Liu, N.; Childs, J.; Sullivan, T.J.; Boyne, M.; Xu, H.; Gegina, Y.; Knudson, D.L.; Johnson, F.; et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 3029–3033. [Google Scholar] [CrossRef] [PubMed]
- Freundlich, J.S.; Wang, F.; Vilcheze, C.; Gulten, G.; Langley, R.; Schiehser, G.A. Triclosan derivatives: Towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem 2009, 4, 241–248. [Google Scholar] [CrossRef]
- Luckner, S.R.; Liu, N.; Am Ende, C.W.; Tonge, P.J.; Kisker, C. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 2010, 285, 14330–14337. [Google Scholar] [CrossRef]
- He, X.; Alian, A.; Stroud, R.; Ortiz de Montellano, P.R. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 6308–6323. [Google Scholar] [CrossRef] [PubMed]
- Cinu, T.A.; Sidhartha, S.K.; Indira, B.; Varadaraj, B.G.; Vishnu, P.S.; Shenoy, G.G. Design, synthesis and evaluation of antitubercular activity of Triclosan analogues. Arab. J. Chem. 2019, 12, 3316–3323. [Google Scholar] [CrossRef]
- Kar, S.S.; Bhat, G.V.; Rao, P.P.; Shenoy, V.P.; Bairy, I.; Shenoy, G.G. Rational design and synthesis of novel diphenyl ether derivatives as antitubercular agents. Drug Des. Dev. Ther. 2016, 10, 2299–2310. [Google Scholar]
- Khade, A.B.; Boshoff, H.I.M.; Arora, K.; Vandana, K.E.; Verma, R.; Shenoy, G.G. Design, synthesis, evaluation, and molecular dynamic simulation of triclosan mimic diphenyl ether derivatives as antitubercular and antibacterial agents. Struct. Chem. 2020, 31, 983–998. [Google Scholar] [CrossRef]
- Verma, R.; Boshoff, H.I.; Arora, K.; Bairy, I.; Tiwari, M.; Bhat, V.G.; Shenoy, G.G.; Bhat, G.V. Synthesis, antitubercular evaluation, molecular docking and molecular dynamics studies of 4,6-disubstituted-2-oxo-dihydropyridine-3-carbonitriles. J. Mol. Struct. 2019, 1197, 117–133. [Google Scholar] [CrossRef]
- Verma, R.; Boshoff, H.I.; Arora, K.; Bairy, I.; Tiwari, M.; Varadaraj, B.G.; Shenoy, G.G. Synthesis, evaluation, molecular docking, and molecular dynamics studies of novel N-(4-[pyridin-2-yloxy]benzyl)arylamine derivatives as potential antitubercular agents. Drug Dev. Res. 2020, 81, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Boyne, M.E.; Sullivan, T.J.; amEnde, C.W.; Lu, H.; Gruppo, V.; Heaslip, D. Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: Evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors. Antimicrob. Agents Chemother. 2007, 51, 3562–3567. [Google Scholar] [CrossRef] [PubMed]
- Kini, S.G.; Bhat, A.; Pan, Z.; Dayan, F.E. Synthesis and antitubercular activity of heterocycle substituted diphenyl ether derivatives. J. Enzyme Inhib. Med. Chem. 2010, 25, 730–736. [Google Scholar] [CrossRef]
- Kini, S.G.; Bhat, A.R.; Bryant, B.; Williamson, J.S.; Dayan, F.E. Synthesis, antitubercular activity and docking study of novel cyclic azole substituted diphenyl ether derivatives. Eur. J. Med. Chem. 2009, 44, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Z.; Yang, J.; Yang, T.; Pi, W.; Ang, W.; Lin, Y.; Liu, Y.; Li, Z.; Luo, Y.; et al. Design, synthesis and evaluation of novel molecules with a diphenyl ether nucleus as potential antitubercular agents. Bioorganic Med. Chem. Lett. 2012, 22, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Knudson, S.E.; Bommineni, G.R.; Li, H.J.; Lai, C.T.; Liu, N. Time-dependent diaryl ether inhibitors of InhA: Structure-activity relationship studies of enzyme inhibition, antibacterial activity, and in vivo efficacy. ChemMedChem 2014, 9, 776–791. [Google Scholar] [CrossRef]
- Pan, P.; Tonge, P.J. Targeting InhA, the FASII Enoyl-ACP Reductase: SAR Studies on Novel Inhibitor Scaffolds. Curr. Top. Med. Chem. 2012, 12, 672–693. [Google Scholar] [CrossRef] [PubMed]
- Roberto Chiarelli, L.; Mori, G.; Esposito, M.; Silvia Orena, B.; Rosalia Pasca, M. Biological evaluation of potent triclosan-derived inhibitors of the enoyl-acyl carrier protein reductase InhA in drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. ChemMedChem 2014, 9, 2528–2537. [Google Scholar]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorganic Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef]
- Armstrong, T.; Lamont, M.; Lanne, A.; Alderwick, L.J.; Thomas, N.R. Inhibition of Mycobacterium tuberculosis InhA: Design, synthesis and evaluation of new di-triclosan derivatives. Bioorganic Med. Chem. 2020, 28, 115744. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Saffon, N.; Sammartino, J.C.; Degiacomi, G.; Pasca, M.R.; Lherbet, C. First triclosan-based macrocyclic inhibitors of InhA enzyme. Bioorganic Chem. 2020, 95, 103498. [Google Scholar] [CrossRef] [PubMed]
- Owono, L.C.O.; Ntie-Kang, F.; Keita, M.; Megnassan, E.; Frecer, V.; Miertuš, S. Virtually Designed Triclosan-Based Inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis and of Plasmodium falciparum. Mol. Inform. 2015, 34, 292–307. [Google Scholar] [CrossRef]
- Available online: www.acdlabs.com (accessed on 10 August 2011).
- Wang, J.; Li, P.; Shen, Q.; Song, G. Concise synthesis of aromatic tertiary amines via a double Petasis–borono Mannich reaction of aromatic amines, formaldehyde, and organoboronic acids. Tetrahedron Lett. 2014, 55, 3888–3891. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Liu, M.S.; Zhang, Y.; Niu, Y.; Huang, F.; Ka, J.-W.; Yip, H.-L.; Tian, Y.; Jen, A.K.-Y. Thermally Cross-Linkable Hole-Transporting Materials on Conducting Polymer: Synthesis, Characterization, and Applications for Polymer Light-Emitting Devices. Chem. Mater. 2008, 20, 413–422. [Google Scholar] [CrossRef]
- SYBYL 8.1, SYBYL X. Available online: http://www.tripos.co.uk (accessed on 4 April 2021).
- GOLD 1.1 ed. Available online: http://www.ccducam.ac.uk/ (accessed on 4 April 2021).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics Server. Available online: http://www.molinspiration.com (accessed on 20 February 2020).
- Thomas, N.R.; Yang, Y.; Drew, W.C.; Lees, K.J. Biomolecular Labelling Using Multifunctional Biotin Analogues. United Kingdom Patent WO 2010/106347 A2, 22 March 2010. [Google Scholar]
- Pal, M.; Parasuraman, K.; Yeleswarapu, K.R. Palladium-Catalyzed Cleavage of O/N-Propargyl Protecting Groups in Aqueous Media under a Copper-Free Condition1. Org. Lett. 2003, 5, 349–352. [Google Scholar] [CrossRef]
- Spletstoser, J.T.; Flaherty, P.T.; Himes, R.H.; Georg, G.I. Synthesis and Anti-Tubulin Activity of a 3-(4-Azidophenyl)-3-dephenylpaclitaxel Photoaffinity Probe. J. Med. Chem. 2004, 47, 6459–6465. [Google Scholar] [CrossRef]
- Drewe, W.C.; Neidle, S. Click chemistry assembly of G-quadruplex ligands incorporating a diarylurea scaffold and triazole linkers. Chem. Commun. 2008, 5295–5297. [Google Scholar] [CrossRef]
- Parikh, S.; Moynihan, D.P.; Xiao, G.; Tonge, P.J. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry 1999, 38, 13623–13634. [Google Scholar] [CrossRef]
- Danquah, C.A.; Maitra, A.; Gibbons, S.; Faull, J.; Bhakta, S. HT-SPOTi: A rapid, gold standard drug susceptibility test (DST), to detect antibiotic resistance profile as well as to evaluate novel chemical entities for new anti-infective drug discovery. Curr. Protoc. Microbiol. 2015, 2015, 17–18. [Google Scholar]
- Evangelopoulos, D.; Bhakta, S. Rapid methods for testing inhibitors of mycobacterial growth. Methods Mol. Biol. 2010, 642, 193–201. [Google Scholar] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | MW | ClogP | TPSA | HBD | HBA | Fitness Scores | % Inhibition | MIC (µg mL−1) |
|---|---|---|---|---|---|---|---|---|
| TCS | 289.54 | 5.53 | 29.46 | 1 | 2 | - | 92 | 20 [48] |
| INH | 137.14 | −0.67 | 68.01 | 2 | 4 | N/A | N/A | 0.05 |
| 8PP | 298.42 | 7.53 | 29.46 | 1 | 2 | - | - | 1.9 [28] |
| 1 | 278.73 | 4.44 ± 0.27 | 27.70 | 0 | 3 | 57.22 | NI | >50 |
| 2 | 262.28 | 3.70 ± 0.62 | 27.70 | 0 | 4 | 53.86 | 18 | 50 |
| 3 | 323.18 | 4.68 ± 0.52 | 27.70 | 0 | 3 | 58.66 | 25 | >50 |
| 8 | 277.75 | 4.27 ± 0.56 | 30.50 | 1 | 3 | 60.28 | 63 | >50 |
| 12 | 261.29 | 3.76 ± 0.60 | 30.50 | 1 | 4 | 55.62 | NI | 7.8 |
| 13 | 381.44 | 5.85 ± 0.80 | 21.71 | 0 | 5 | 69.25 | 43 | >125 |
| 20 | 345.78 | 4.03 ± 0.82 | 58.42 | 0 | 6 | 75.59 | 36 | >50 |
| 21 | 329.32 | 3.29 ± 1.05 | 58.42 | 0 | 7 | 71.55 | 56 | >50 |
| 22 | 390.23 | 4.26 ± 0.94 | 58.42 | 0 | 6 | 75.80 | NI | >50 |
| 23 | 344.80 | 3.86 ± 0.96 | 61.21 | 1 | 6 | 76.22 | 45 | >50 |
| 24 | 328.34 | 3.34 ± 1.03 | 61.21 | 1 | 7 | 72.63 | 23 | >125 |
| 31 | 511.39 | 8.21 ± 0.50 | 46.17 | 0 | 5 | 90.79 | 42 | >125 |
| 38 | 483.34 | 7.88 ± 0.54 | 68.16 | 2 | 5 | 86.50 | 100 | 125 |
| 39 | 384.85 | 5.40 ± 0.48 | 27.70 | 0 | 4 | 72.92 | 27 | 7.8 |
| 40 | 450.05 | 6.01 ± 0.45 | 9.23 | 0 | 1 | 59.03 | 30 | >50 |
| 43 | 354.80 | 3.50 ± 0.28 | 69.68 | 0 | 5 | 68.54 | 35 | 7.8 |
| 44 | 370.81 | 2.92 ± 0.26 | 78.92 | 0 | 6 | 62.44 | 14 | 15.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chetty, S.; Armstrong, T.; Sharma Kharkwal, S.; Drewe, W.C.; De Matteis, C.I.; Evangelopoulos, D.; Bhakta, S.; Thomas, N.R. New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis. Pharmaceuticals 2021, 14, 361. https://doi.org/10.3390/ph14040361
Chetty S, Armstrong T, Sharma Kharkwal S, Drewe WC, De Matteis CI, Evangelopoulos D, Bhakta S, Thomas NR. New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis. Pharmaceuticals. 2021; 14(4):361. https://doi.org/10.3390/ph14040361
Chicago/Turabian StyleChetty, Sarentha, Tom Armstrong, Shalu Sharma Kharkwal, William C. Drewe, Cristina I. De Matteis, Dimitrios Evangelopoulos, Sanjib Bhakta, and Neil R. Thomas. 2021. "New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis" Pharmaceuticals 14, no. 4: 361. https://doi.org/10.3390/ph14040361
APA StyleChetty, S., Armstrong, T., Sharma Kharkwal, S., Drewe, W. C., De Matteis, C. I., Evangelopoulos, D., Bhakta, S., & Thomas, N. R. (2021). New InhA Inhibitors Based on Expanded Triclosan and Di-Triclosan Analogues to Develop a New Treatment for Tuberculosis. Pharmaceuticals, 14(4), 361. https://doi.org/10.3390/ph14040361