
pH Dependence of T2 for Hyperpolarizable 13C-Labelled Small Molecules Enables Spatially Resolved pH Measurement by Magnetic Resonance Imaging


Abstract
1. Introduction
2. Results
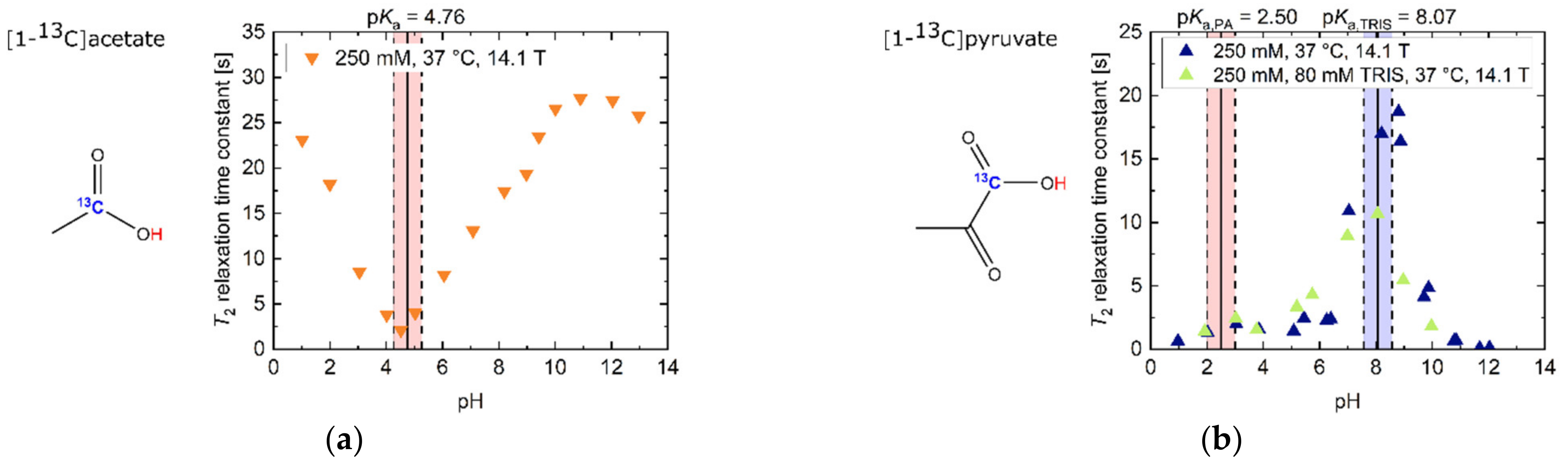
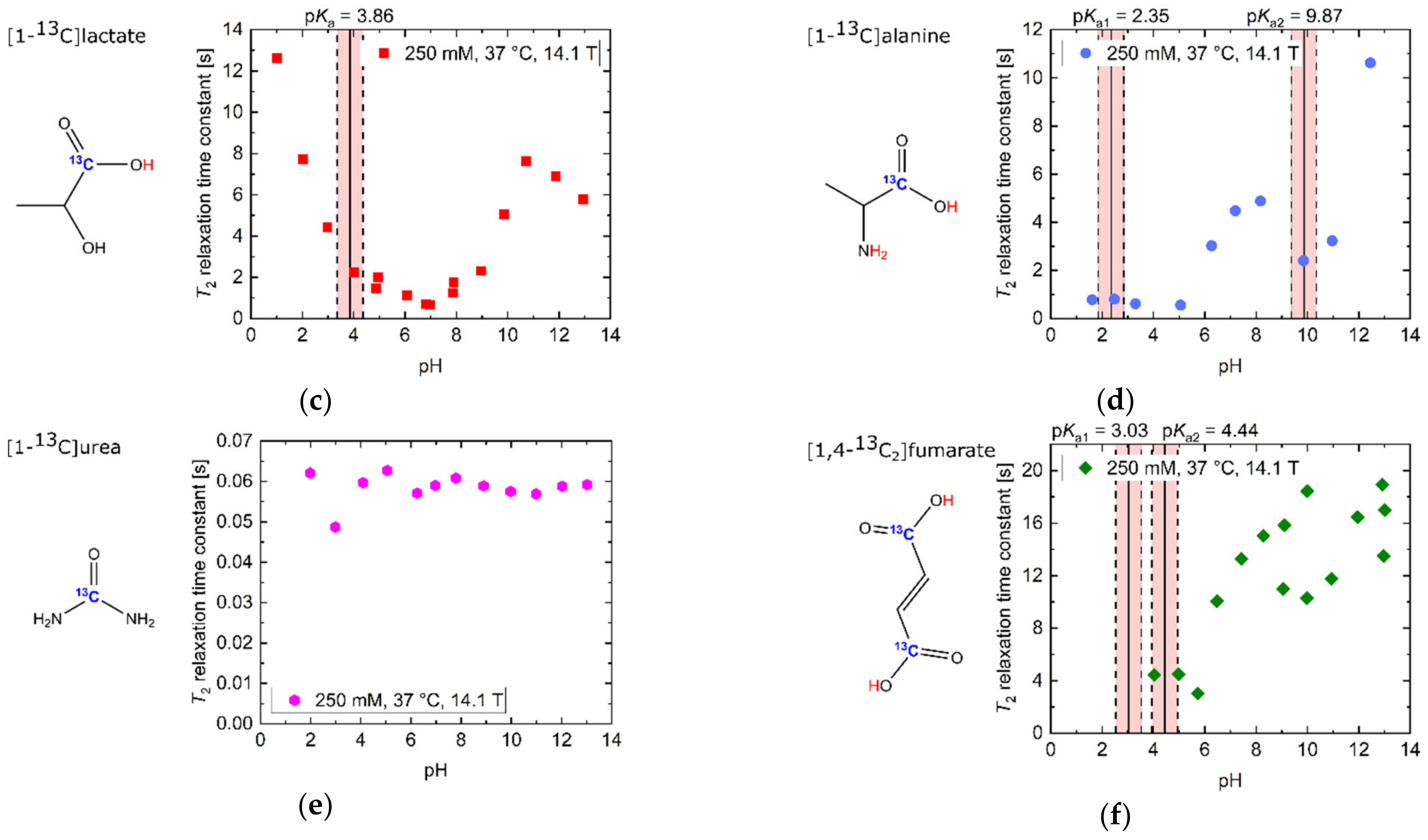
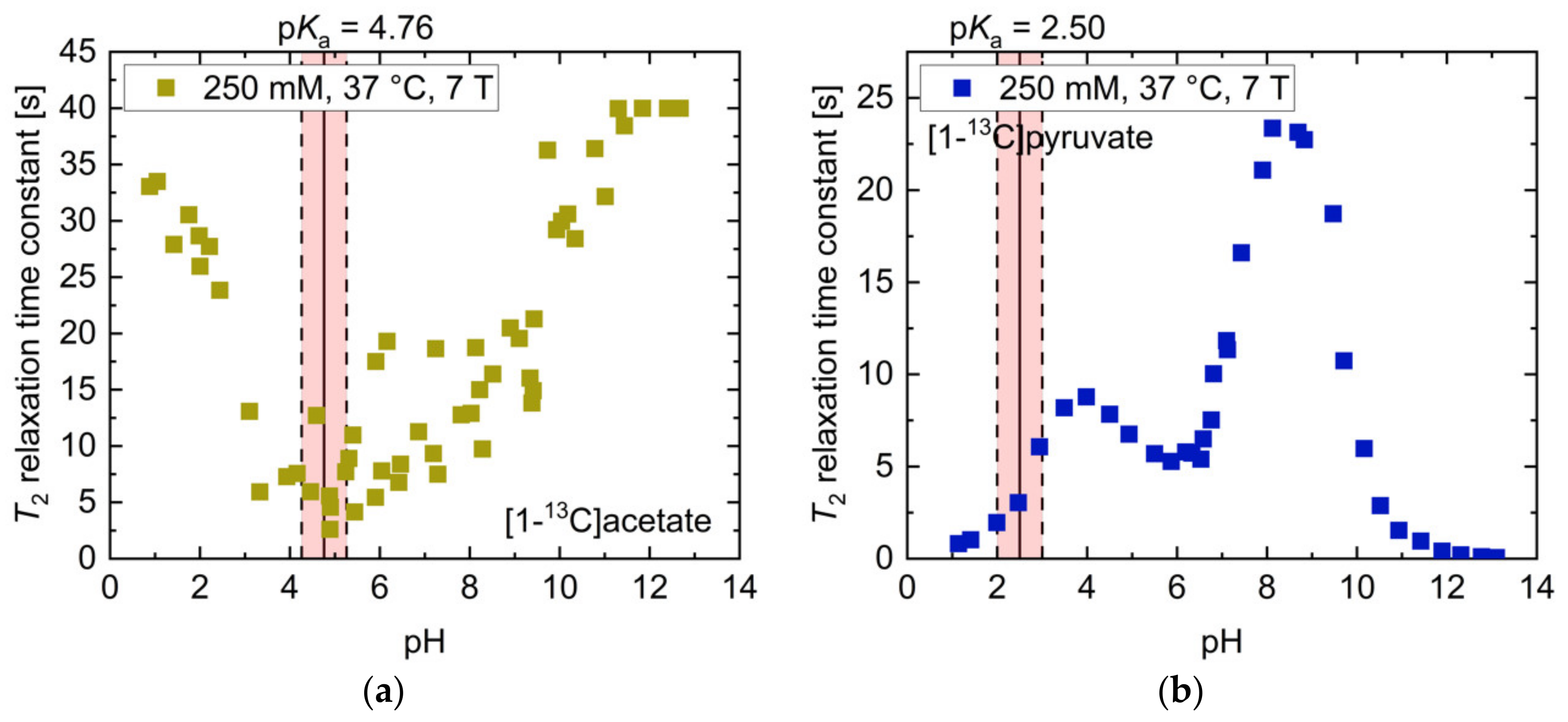
2.1. pH Dependency of T2 for Commonly Used Hyperpolarized 13C-Labelled Small Molecules
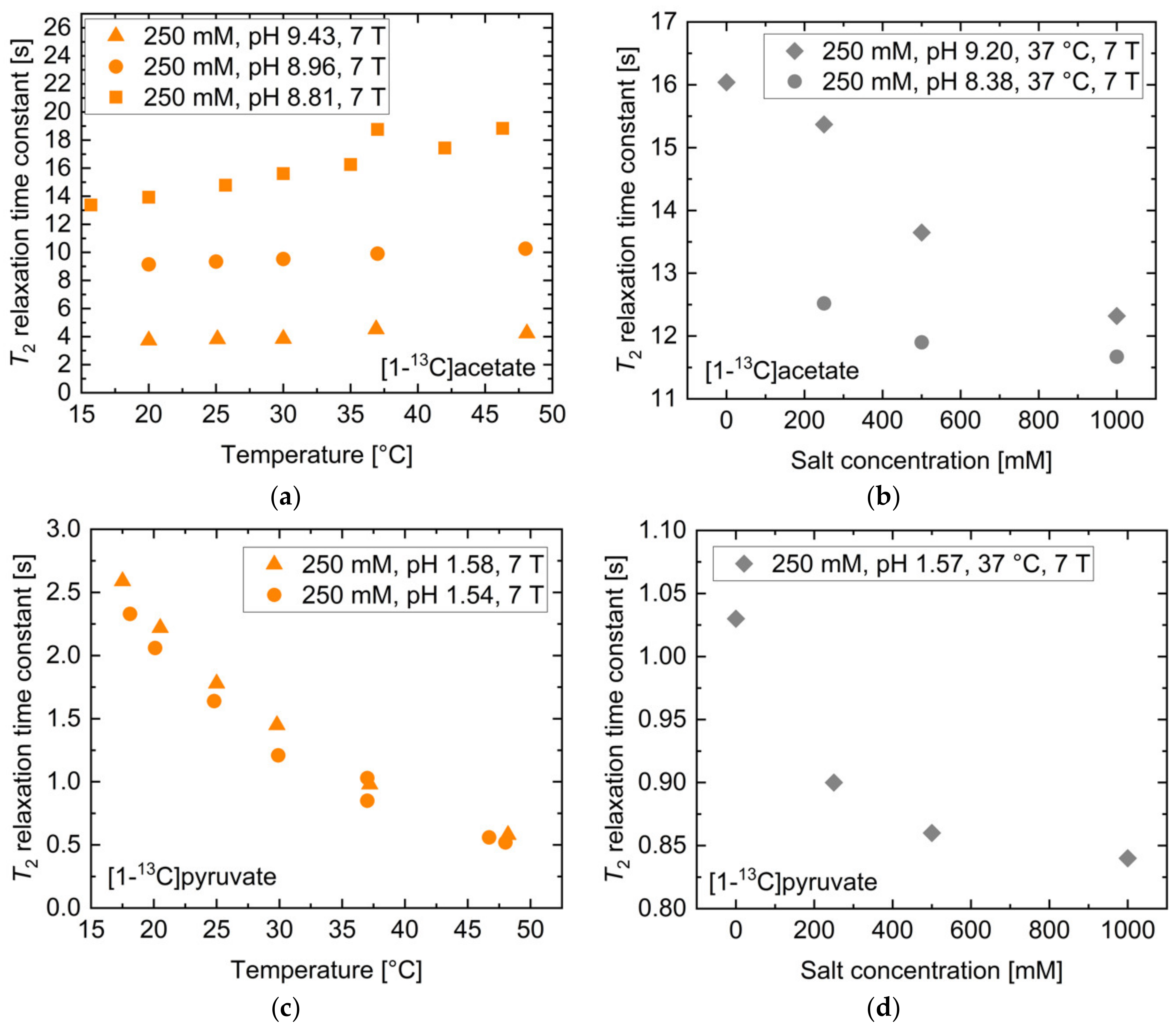
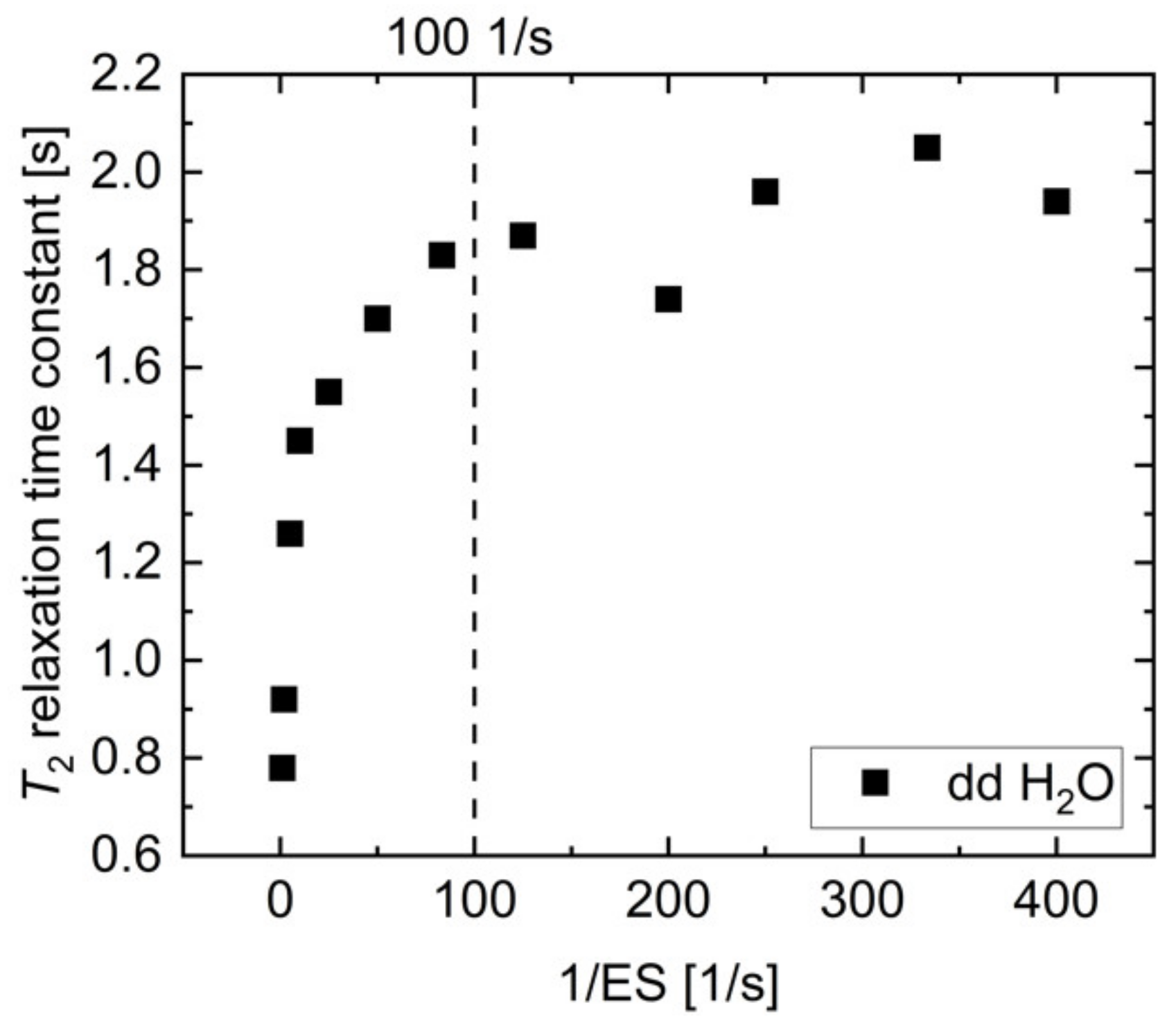
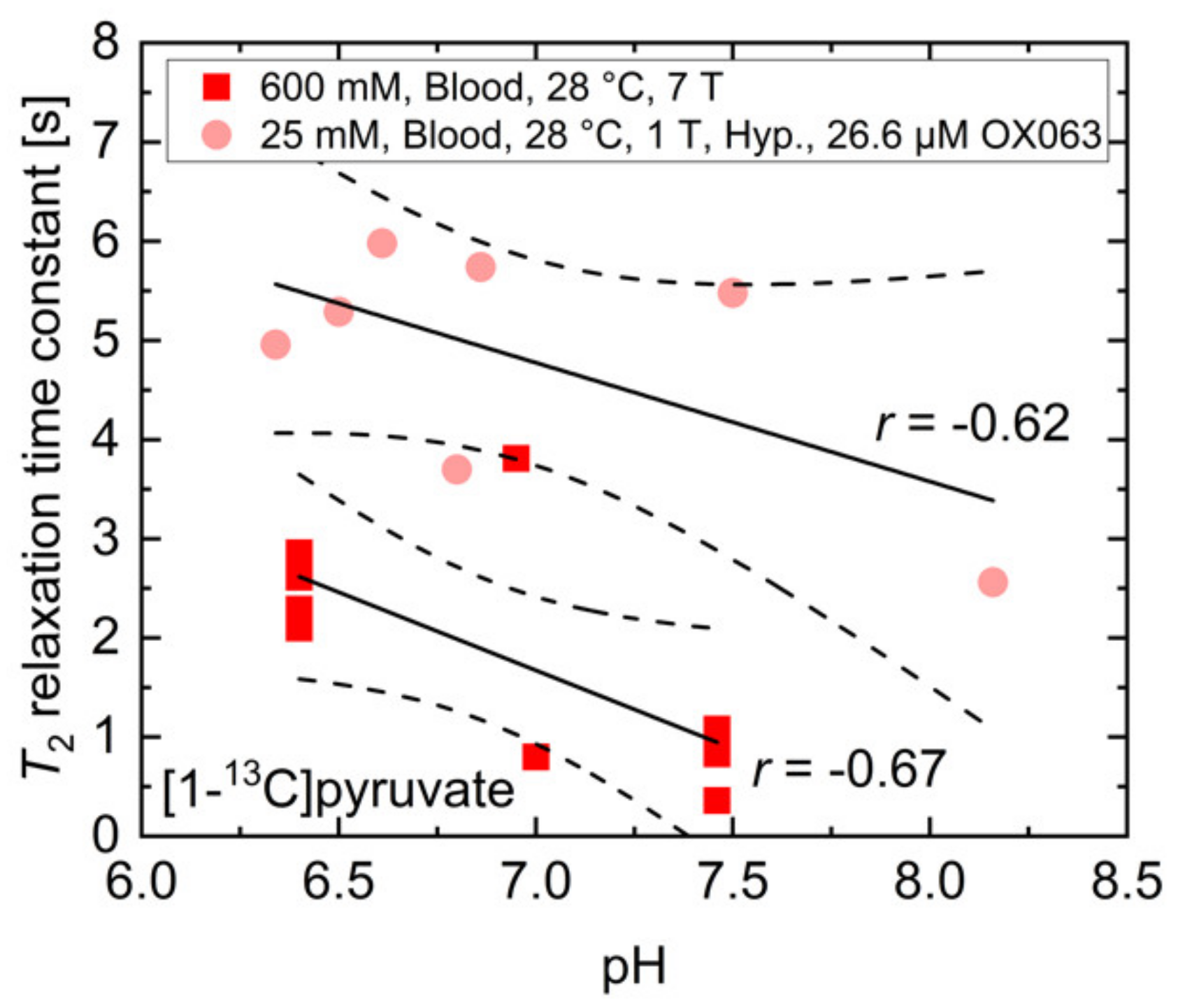
2.2. pH, Temperature and Salt Concentration Dependence of T2 for [1-13C]acetate and [1-13C]pyruvate
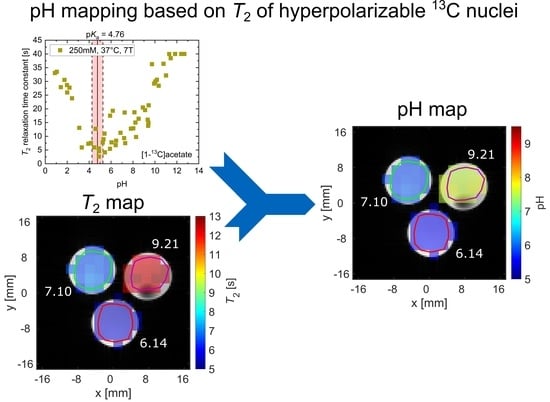
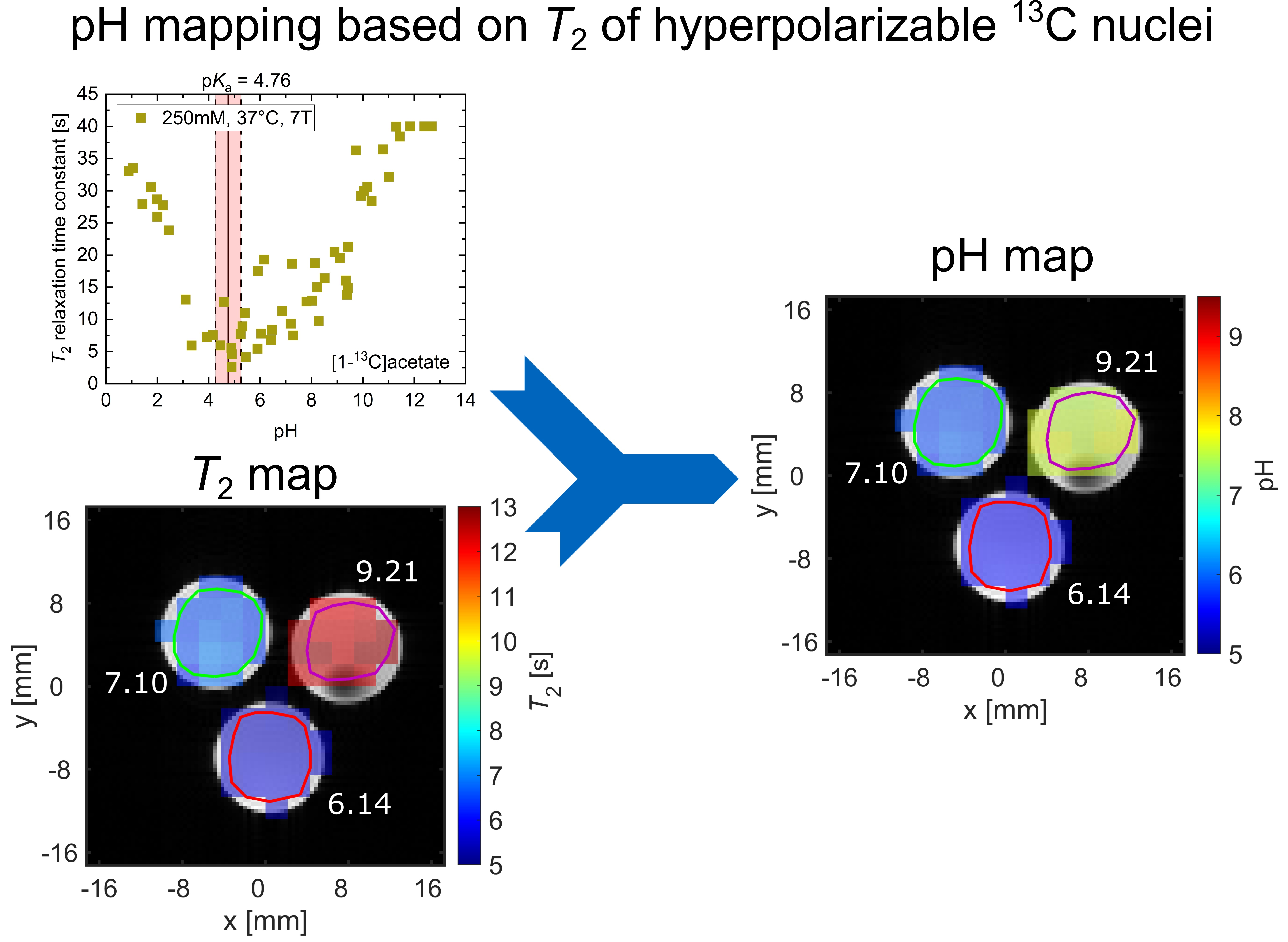
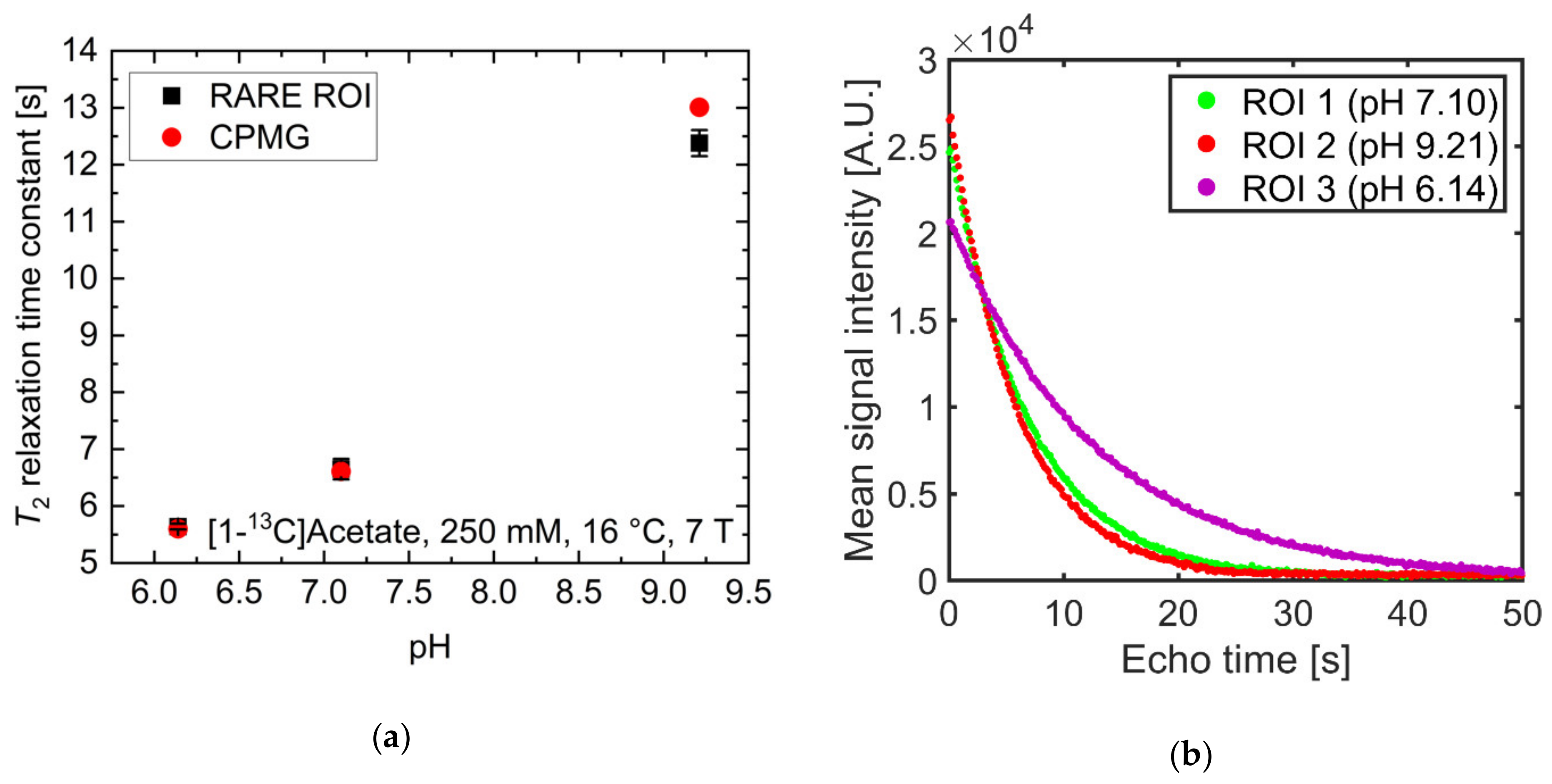
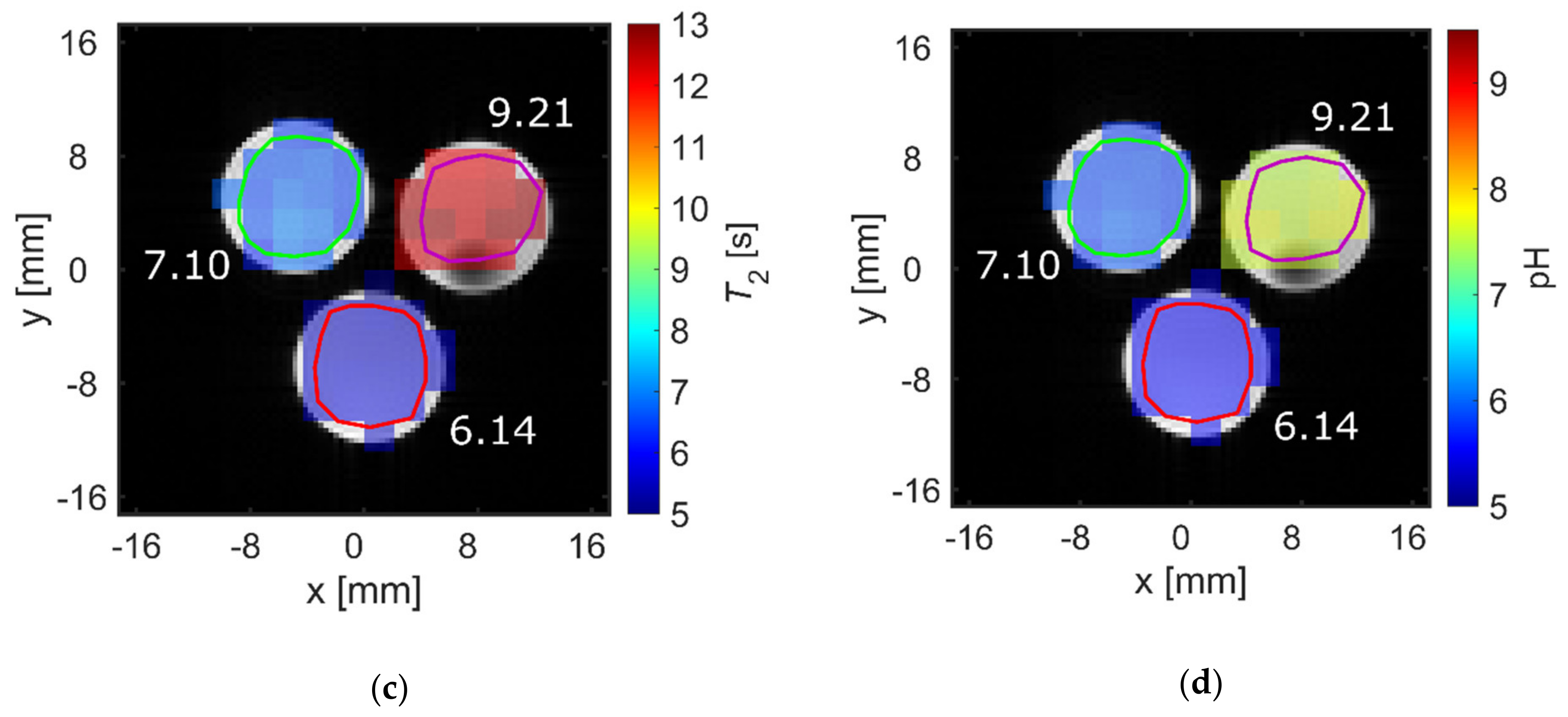
2.3. pH Imaging in Aqueous Solutions Using [1-13C]acetate
3. Discussion
4. Methods
4.1. Chemical Compounds
4.2. Sample Preparation
4.3. pH and Temperature Measurement
4.4. Hyperpolarization
4.5. C NMR Spectroscopy
4.5.1. Measurements at 1 T
4.5.2. Measurements at 7 T
4.5.3. Measurements at 14.1 T
4.6. Data Processing and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

References
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef]
- Golman, K.; Zandt, R.I.T.; Lerche, M.; Pehrson, R.; Ardenkjaer-Larsen, J.H. Metabolic Imaging by Hyperpolarized 13C Magnetic Resonance Imaging for In vivo Tumor Diagnosis. Cancer Res. 2006, 66, 10855–10860. [Google Scholar] [CrossRef]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; Van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1-13C] Pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef] [PubMed]
- Day, E.S.; Kettunen, I.M.; Gallagher, F.; Hu, D.-E.; Lerche, M.; Wolber, J.; Golman, K.; Ardenkjaer-Larsen, J.H.; Brindle, K.M. Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med. 2007, 13, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, J.D.; Yen, Y.-F.; Mayer, D.; Tropp, J.S.; Hurd, R.E.; Spielman, D.M. Detection of Inflammatory Arthritis by Using Hyperpolarized13C-Pyruvate with MR Imaging and Spectroscopy. Radiology 2011, 259, 414–420. [Google Scholar] [CrossRef]
- Gallagher, F.A.; Kettunen, M.I.; Day, S.E.; Hu, D.-E.; Ardenkjaer-Larsen, J.H.; Zandt, R.I.T.; Jensen, P.R.; Karlsson, M.; Golman, K.; Lerche, M.H.; et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nat. Cell Biol. 2008, 453, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.Z.; Chen, A.P.; Ghugre, N.R.; Ramanan, V.; Lam, W.W.; Connelly, K.A.; Wright, G.A.; Cunningham, C.H. Rapid multislice imaging of hyperpolarized 13C pyruvate and bicarbonate in the heart. Magn. Reson. Med. 2010, 64, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.H.; Chen, A.P.; Albers, M.J.; Kurhanewicz, J.; Hurd, R.E.; Yen, Y.-F.; Pauly, J.M.; Nelson, S.J.; Vigneron, D.B. Double spin-echo sequence for rapid spectroscopic imaging of hyperpolarized 13C. J. Magn. Reson. 2007, 187, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Leupold, J.; Månsson, S.; Petersson, J.S.; Hennig, J.; Wieben, O. Fast multiecho balanced SSFP metabolite mapping of 1H and hyperpolarized 13C compounds. MAGMA 2009, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Von Morze, C.; Larson, P.E.; Hu, S.; Yoshihara, H.A.; Bok, R.A.; Goga, A.; Ardenkjaer-Larsen, J.H.; Vigneron, D.B. Investigating tumor perfusion and metabolism using multiple hyperpolarized 13C compounds: HP001, pyruvate and urea. Magn. Reson. Imaging 2012, 30, 305–311. [Google Scholar] [CrossRef]
- Mishra, A.; Pariani, G.; Oerther, T.; Schwaiger, M.; Westmeyer, G.G. Hyperpolarized Multi-Metal13C-Sensors for Magnetic Resonance Imaging. Anal. Chem. 2016, 88, 10790–10794. [Google Scholar] [CrossRef]
- Düwel, S.; Hundshammer, C.; Gersch, M.; Feuerecker, B.; Steiger, K.; Buck, A.; Walch, A.; Haase, A.; Glaser, S.J.; Schwaiger, M.; et al. Imaging of pH in vivo using hyperpolarized 13C-labelled zymonic acid. Nat. Commun. 2017, 8, 15126. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Simbrunner, J.; Stollberger, R. Analysis of Carr–Purcell Sequences with Nonideal Pulses. J. Magn. Reson. Ser. B 1995, 109, 301–309. [Google Scholar] [CrossRef]
- Reed, G.D.; Von Morze, C.; Bok, R.; Koelsch, B.L.; Van Criekinge, M.; Smith, K.J.; Larson, P.E.Z.; Kurhanewicz, J.; Vigneron, D.B.; Shang, H. High resolution (13)C MRI with hyperpolarized urea: In vivo T(2) mapping and (15)N labeling effects. IEEE Trans. Med. Imaging 2013, 33, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, C.; Nørlinger, T.S.; Hansen, D.C.; Qi, H.; Nielsen, P.M.; Bertelsen, L.B.; Ardenkjaer-Larsen, J.H.; Jørgensen, H.S. Hyperpolarized 13 C urea relaxation mechanism reveals renal changes in diabetic nephropathy. Magn. Reson. Med. 2015, 75, 515–518. [Google Scholar] [CrossRef]
- Reed, G.D.; von Morze, C.; Verkman, A.S.; Koelsch, B.L.; Chaumeil, M.M.; Lustig, M.; Ronen, S.M.; Bok, R.A.; Sands, J.M.; Larson, P.E.Z.; et al. Imaging Renal Urea Handling in Rats at Millimeter Resolution Using Hyperpolarized Magnetic Resonance Relaxometry. Tomography 2016, 2, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.-F.; Le Roux, P.; Mayer, D.; King, R.; Spielman, D.M.; Tropp, J.; Pauly, K.B.; Pfefferbaum, A.; Vasanawala, S.S.; E Hurd, R. T2relaxation times of13C metabolites in a rat hepatocellular carcinoma model measuredin vivousing13C-MRS of hyperpolarized [1-13C]pyruvate. NMR Biomed. 2010, 23, 414–423. [Google Scholar] [CrossRef]
- Yen, Y.-F.; Le Roux, P.; Bok, R.; Tropp, J.; Chen, A.; Zhang, V.; Zierhut, M.; Albers, M.; Nelson, S. Apparent T2 of 13C-labeled Metabolites In Vivo. In Proceedings of the 16th Scientific Meeting & Exhibition ISMRM, Toronto, ON, Canada, 3–9 May 2008. [Google Scholar]
- Kettunen, M.I.; Hu, D.-E.; Witney, T.H.; McLaughlin, R.; Gallagher, F.A.; Bohndiek, S.E.; Day, S.E.; Brindle, K.M. Magnetization transfer measurements of exchange between hyperpolarized [1-13C]pyruvate and [1-13C]lactate in a murine lymphoma. Magn. Reson. Med. 2010, 63, 872–880. [Google Scholar] [CrossRef]
- Chen, A.P.; Cunningham, C.H. Single voxel localization for dynamic hyperpolarized 13C MR spectroscopy. J. Magn. Reson. 2015, 258, 81–85. [Google Scholar] [CrossRef]
- Golman, K.; Axelsson, O.; Johannesson, H.; Mansson, S.; Olofsson, C.; Petersson, J. Parahydrogen-induced polarization in imaging: Subsecond13C angiography. Magn. Reason. Med. 2001, 46, 1–5. [Google Scholar] [CrossRef]
- Mariager, C.Ø.; Nielsen, P.M.; Qi, H.; Ringgaard, S.; Laustsen, C. Hyperpolarized 13 C,15 N2 -urea T2 relaxation changes in acute kidney injury. Magn. Reson. Med. 2018, 80, 696–702. [Google Scholar] [CrossRef]
- Larson, P.E.Z.; Kerr, A.B.; Chen, A.P.; Lustig, M.S.; Zierhut, M.L.; Hu, S.; Cunningham, C.H.; Pauly, J.M.; Kurhanewicz, J.; Vigneron, D.B. Multiband excitation pulses for hyperpolarized 13C dynamic chemical-shift imaging. J. Magn. Reson. 2008, 194, 121–127. [Google Scholar] [CrossRef]
- Milshteyn, E.; Von Morze, C.; Reed, G.D.; Shang, H.; Shin, P.J.; Zhu, Z.; Chen, H.-Y.; Bok, R.; Goga, A.; Kurhanewicz, J.; et al. Development of high resolution 3D hyperpolarized carbon-13 MR molecular imaging techniques. Magn. Reson. Imaging 2017, 38, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Varma, G.; Wang, X.; Vinogradov, E.; Bhatt, R.S.; Sukhatme, V.P.; Seth, P.; Lenkinski, R.E.; Alsop, D.C.; Grant, A.K. Selective spectroscopic imaging of hyperpolarized pyruvate and its metabolites using a single-echo variable phase advance method in balanced SSFP. Magn. Reson. Med. 2016, 76, 1102–1115. [Google Scholar] [CrossRef]
- Kettunen, M.I.; Kennedy, B.W.C.; Hu, D.; Brindle, K.M. Spin echo measurements of the extravasation and tumor cell uptake of hyperpolarized [1-13C]lactate and [1-13C]pyruvate. Magn. Reson. Med. 2012, 70, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Lee, H.; Lee, J.; Yang, S.; Choi, Y.-S.; Wang, E.; Song, H.-T.; Kim, D.-H. An indirect method forin vivo T2mapping of [1-13C] pyruvate using hyperpolarized13C CSI. NMR Biomed. 2017, 30, e3690. [Google Scholar] [CrossRef]
- Souhrada, J.; Bullard, R.W. Tissue buffering capacity: Its role in the prevention of isoproterenol induced necrosis. Life Sci. 1971, 10, 559–564. [Google Scholar] [CrossRef]
- Salenius, P. A Study of the Ph and buffer capacity of blood, Plasma and red Blood Cells. Scand. J. Clin. Lab. Investig. 1957, 9, 160–167. [Google Scholar] [CrossRef]
- Viikari, S.J.; Harjola, P.; Maamies, T. Clinical Studies on the Buffer Capacity of the Blood. Scand. J. Clin. Lab. Investig. 1954, 6, 122–128. [Google Scholar] [CrossRef]
- Von Oettingen, J.; Wolfsdorf, J.; Feldman, H.A.; Rhodes, E.T. Use of Serum Bicarbonate to Substitute for Venous pH in New-Onset Diabetes. Pediatry 2015, 136, e371–e377. [Google Scholar] [CrossRef] [PubMed]
- Granja, S.; Tavares-Valente, D.; Queirós, O.; Baltazar, F. Value of pH regulators in the diagnosis, prognosis and treatment of cancer. Semin. Cancer Biol. 2017, 43, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Adrogué, H.J.; Madias, N.E. Management of Life-Threatening Acid–Base Disorders. New Engl. J. Med. 1998, 338, 107–111. [Google Scholar] [CrossRef]
- Hundshammer, C.; Grashei, M.; Greiner, A.; Glaser, S.J.; Schilling, F. pH Dependence ofT1for13C-Labelled Small Molecules Commonly Used for Hyperpolarized Magnetic Resonance Imaging. Chem. Phys. Chem. 2019, 20, 798–802. [Google Scholar] [CrossRef]
- Arcis, H.; Ferguson, J.P.; Cox, J.S.; Tremaine, P.R. The Ionization Constant of Water at Elevated Temperatures and Pressures: New Data from Direct Conductivity Measurements and Revised Formulations from T = 273 K to 674 K and p = 0.1 MPa to 31 MPa. J. Phys. Chem. Ref. Data 2020, 49, 033103. [Google Scholar] [CrossRef]
- Yaprak, M.; Kemec, Z.; Cetinkaya, E.; Dayanan, R.; Celiker, O.; Sünger, O. Is a pH of 6.73 compatible with life? Internet J. Nephrol. 2015, 10. [Google Scholar] [CrossRef]
- Tripathy, S. Extreme metabolic alkalosis in intensive care. Indian J. Crit. Care Med. 2009, 13, 217–220. [Google Scholar] [CrossRef][Green Version]
- Griffiths, J.R. Are cancer cells acidic? Br. J. Cancer 1991, 64, 425–427. [Google Scholar] [CrossRef]
- Reynolds, R.M.; Padfield, P.L.; Seckl, J.R. Disorders of sodium balance. BMJ 2006, 332, 702–705. [Google Scholar] [CrossRef]
- Wallerstein, J.; Weininger, U.; Khan, M.A.I.; Linse, S.; Akke, M. Site-Specific Protonation Kinetics of Acidic Side Chains in Proteins Determined by pH-Dependent Carboxyl 13C NMR Relaxation. J. Am. Chem. Soc. 2015, 137, 3093–3101. [Google Scholar] [CrossRef]
- Carver, J.; Richards, R. A general two-site solution for the chemical exchange produced dependence of T2 upon the carr-Purcell pulse separation. J. Magn. Reson. 1972, 6, 89–105. [Google Scholar] [CrossRef]
- Bloch, F.; Hansen, W.W.; Packard, M. The Nuclear Induction Experiment. Phys. Rev. 1946, 70, 474–485. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Rahman, H.M.A.; Hefter, G.; Buchner, R. Hydration of Formate and Acetate Ions by Dielectric Relaxation Spectroscopy. J. Phys. Chem. B 2011, 116, 314–323. [Google Scholar] [CrossRef]
- Valadbeigi, Y.; Farrokhpour, H. Theoretical study on keto-enol tautomerism and isomerization in pyruvic acid. Int. J. Quantum Chem. 2013, 113, 2372–2378. [Google Scholar] [CrossRef]
- Liepinsh, E.; Otting, G. Proton exchange rates from amino acid side chains—implications for image contrast. Magn. Reson. Med. 1996, 35, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Pocker, Y.; Meany, J.E.; Nist, B.J.; Zadorojny, C. Reversible hydration of pyruvic acid. I. Equilibrium studies. J. Phys. Chem. 1969, 73, 2879–2882. [Google Scholar] [CrossRef]
- Strehlow, H. Die Kinetik der Hydratation von a-Ketocarbonsäuren. Zeitschrift für Elektrochemie Ber. Bunsenges. Phys. Chem 1962, 66, 392–396. [Google Scholar] [CrossRef]
- Losada, M.; Tran, H.; Xu, Y. Lactic acid in solution: Investigations of lactic acid self-aggregation and hydrogen bonding interactions with water and methanol using vibrational absorption and vibrational circular dichroism spectroscopies. J. Chem. Phys. 2008, 128, 014508. [Google Scholar] [CrossRef]
- Cassanas, G.; Morssli, M.; Fabrègue, E.; Bardet, L. Vibrational spectra of lactic acid and lactates. J. Raman Spectrosc. 1991, 22, 409–413. [Google Scholar] [CrossRef]
- Achilli, E.; Vertova, A.; Visibile, A.; Locatelli, C.; Minguzzi, A.; Rondinini, S.; Ghigna, P. Structure and Stability of a Copper(II) Lactate Complex in Alkaline Solution: A Case Study by Energy-Dispersive X-ray Absorption Spectroscopy. Inorg. Chem. 2017, 56, 6982–6989. [Google Scholar] [CrossRef]
- Dudás, C.; Kutus, B.; Böszörményi, É.; Peintler, G.; Attia, A.A.; Lupan, A.; Kele, Z.; Sipos, P.; Pálinkó, I. Calcium complexing behaviour of lactate in neutral to highly alkaline medium. J. Mol. Struct. 2019, 1180, 491–498. [Google Scholar] [CrossRef]
- Schnitzler, E.G.; Seifert, N.A.; Ghosh, S.; Thomas, J.; Xu, Y.; Jäger, W. Hydration of the simplest α-keto acid: A rotational spectroscopic and ab initio study of the pyruvic acid–water complex. Phys. Chem. Chem. Phys. 2017, 19, 4440–4446. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, D.; Luo, M.; Grassian, V.H.; Gerber, R.B. Absorption spectra of pyruvic acid in water: Insights from calculations for small hydrates and comparison to experiment. Phys. Chem. Chem. Phys. 2020, 22, 12658–12670. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.; Dastrù, W.; Digilio, G.; Keupp, J.; Langereis, S.; Lanzardo, S.; Prestigio, S.; Steinbach, O.; Terreno, E.; Uggeri, F.; et al. Iopamidol as a responsive MRI-chemical exchange saturation transfer contrast agent for pH mapping of kidneys: In vivo studies in mice at 7 T. Magn. Reson. Med. 2010, 65, 202–211. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D.R. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Tsukiashi, A.; Min, K.S.; Kitayama, H.; Terasawa, H.; Yoshinaga, S.; Takeda, M.; Lindoy, L.F.; Hayami, S. Application of spin-crossover water soluble nanoparticles for use as MRI contrast agents. Sci. Rep. 2018, 8, 14911. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T2,max [s] (pH) | T2,min [s] (pH) | T2 [s] (pH 7.4) | Ratio T2,max/T2,min | Mean ΔT2 [s/0.1pH] (pH Range 6.8–7.8) | |
|---|---|---|---|---|---|
| [1-13C]acetate | 27.7 (10.90) | 2.1 (4.52) | 14.7 | 13.2 | 0.44 |
| [1-13C]alanine | 10.6 (12.45) | 0.6 (5.06) | 4.6 | 19.0 | 0.09 |
| [1,4-13C2]fumarate | 18.9 (12.91) | 3.0 (5.73) | 13.9 | 6.3 | 0.19 |
| [1-13C]lactate | 12.6 (1.03) | 0.7 (6.99) | 1.1 | 18.9 | −0.05 (pH 6.8–7)/ 0.08 (pH 7–7.8) |
| [1-13C]pyruvate | 18.7 (8.81) | 0.1 (11.69) | 13.9 | 208.1 | 0.95 |
| 13C-urea | 0.1 (7.79) | 0.1 (2.99) | 0.1 | 1.2 | <0.001 |
| [1-13C]acetate | T2,ref [s] | T2,lower bound [s] | T2,upper bound [s] | Rel. Change Lower Bound [%] | Rel. Change Upper Bound [%] |
| Concentration | 20.8 | 26.1 | 11.4 | 25.2 | −45.4 |
| Salt concentration | 14.8 | 15.1 | 14.5 | 2.0 | −1.8 |
| Temperature | 11.4 | 11.3 | 11.6 | −1.6 | 1.5 |
| pH | 11.7 | 9.7 | 13.0 | −16.9 | 11.2 |
| [1-13C]pyruvate. | T2,ref [s] | T2,lower bound [s] | T2,upper bound [s] | Rel. Change Lower Bound [%] | Rel. Change Upper Bound [%] |
|---|---|---|---|---|---|
| Concentration | 2.1 | 2.1 | 1.6 | 1.9 | −23.6 |
| Salt concentration | 0.9 | 1.0 | 0.9 | 8.5 | −2.1 |
| Temperature | 0.9 | 1.0 | 0.9 | 10.6 | −9.6 |
| pH | 15.4 | 9.1 | 19.7 | −41.2 | 27.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grashei, M.; Hundshammer, C.; van Heijster, F.H.A.; Topping, G.J.; Schilling, F. pH Dependence of T2 for Hyperpolarizable 13C-Labelled Small Molecules Enables Spatially Resolved pH Measurement by Magnetic Resonance Imaging. Pharmaceuticals 2021, 14, 327. https://doi.org/10.3390/ph14040327
Grashei M, Hundshammer C, van Heijster FHA, Topping GJ, Schilling F. pH Dependence of T2 for Hyperpolarizable 13C-Labelled Small Molecules Enables Spatially Resolved pH Measurement by Magnetic Resonance Imaging. Pharmaceuticals. 2021; 14(4):327. https://doi.org/10.3390/ph14040327
Chicago/Turabian StyleGrashei, Martin, Christian Hundshammer, Frits H. A. van Heijster, Geoffrey J. Topping, and Franz Schilling. 2021. "pH Dependence of T2 for Hyperpolarizable 13C-Labelled Small Molecules Enables Spatially Resolved pH Measurement by Magnetic Resonance Imaging" Pharmaceuticals 14, no. 4: 327. https://doi.org/10.3390/ph14040327
APA StyleGrashei, M., Hundshammer, C., van Heijster, F. H. A., Topping, G. J., & Schilling, F. (2021). pH Dependence of T2 for Hyperpolarizable 13C-Labelled Small Molecules Enables Spatially Resolved pH Measurement by Magnetic Resonance Imaging. Pharmaceuticals, 14(4), 327. https://doi.org/10.3390/ph14040327