
Antimicrobial Peptides as Potential Anti-Tubercular Leads: A Concise Review

 ,
,  ,
,  , and
, and
Abstract
1. Tuberculosis: A Brief Overview
1.1. Global Burden
1.2. Pathogenesis
1.3. Challenges to the Standard Treatment
2. Antimicrobial Peptides as a New Tool to Tackle Antibiotic-Resistant Infections
3. Animal AMP with Anti-Tubercular Activity
3.1. Cathelicidins
3.2. Human Defensins
3.3. Protegrins
3.4. Hepcidin
3.5. Lactoferrin
3.6. Ub2—A Ubiquitin-Derived Peptide
3.7. Hcl2
3.8. Cathepsin G-Derived Peptides
3.9. Venom-Derived Peptides
3.10. B1CTcu5

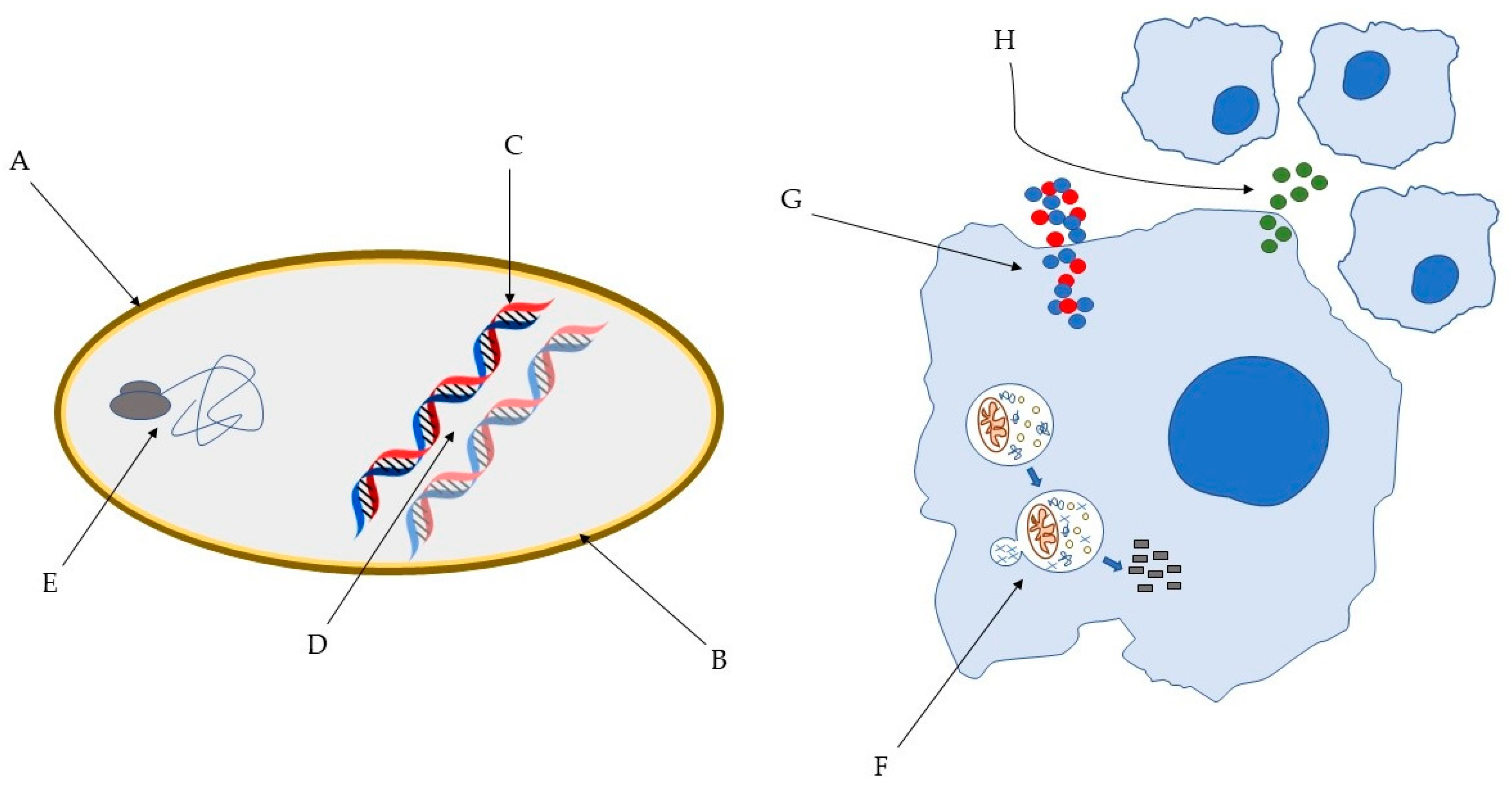{kind=link}
| Peptide | Sequence | Activity | Source | Ref. |
|---|---|---|---|---|
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | MIC (H37Rv): ~5 μg/mL | Various human immune cells | [55] |
| mCRAMP | GLLRKGGEKIGEKLKKIGQKIKNFFQKLVPQPEQ | MIC (H37Rv)—<5 μg/mL | Various mice immune cells | [55] |
| PR-39 | RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP | IC50 (H37Rv): 17 μg/mL IC50 (MDR E1380/94): 93 μg/mL | Porcine small intestine, bone marrow, neutrophils | [61] |
| Indolicidin | ILPWKWPWWPWRR | MIC (Mtb clinical isolate): 32 μg/mL | Bovine neutrophils’ granules | [62] |
| Pep-H | RRYGTCIYQGRLWAF | MIC (H37Rv): 10 μg/mL | HNP-1 in human granulocytes | [75] |
| PG-1 | RGGRLCYCRRRFCVCVGR | 68.4% CFU reduction at 64 μg/mL | Porcine leukocytes | [70] |
| Ub2 | STLHLVLRLRGG | MIC (CDC1551): 5 μM | Ubiquitin in lysosomal extracts | [99] |
| VpAmp1.0 | LPFFLLSLIPSAISAIKKI, amidated | MIC (H37Rv): 17.4 μM MIC (MDR clinical isolate): 4.2 μM | Vaejovis punctatus venom | [108] |
| VpAmp2.0 | FWGFLGKLAMKAVPSLIGGNKSSSK | MIC (H37Rv): 5.4 μM MIC (MDR clinical isolate): 8.6 μM | Vaejovis punctatus venom | [108] |
| VpAmp1.1 | FFLLSLIPSAISAIKKI, amidated | MIC (H37Rv): 21.4 μM MIC (MDR clinical isolate): 30.5 μM | Vaejovis punctatus venom | [108] |
| VpAmp2.1 | FWGFLGKLAMKAVPSLIGGNKK | MIC (H37Rv): 13.6 μM MIC (MDR clinical isolate): 8.5 μM | Vaejovis punctatus venom | [108] |
| Pin2 | FWGALAKGALKLIPSLFSSFSKKD | MIC (H37Rv): 22.1 μM MIC (MDR clinical isolate): 33.1 μM | Pandinus imperator venom | [109] |
| Pin2[14] | FWGLKGLKKFSKKL | MIC (H37Rv): 11.92 μM MIC (MDR clinical isolate): 6 μM | Pandinus imperator venom | [109] |
| Vgf-1 | ECYRKSDIVTCEPWQKFCYREVTFFPNHPVYLSGCASECTETNSKWCCTTDKCNRARGG | MIC (MDR *): 8.5 μg/mL | Naja atra venom | [110] |
| BICTcu5 | LIAGLAANFLPQILCKIARKC | MIC (H37Rv): 12.5 μg/mL | Clinotarsus curtipes skin secretion | [112] |
4. Non-Animal AMPs with Anti-Tubercular Activity
4.1. Bacterial Peptides
4.1.1. Nisin A and Lacticin 3147
4.1.2. Mutacin 1140
4.1.3. Lasso Peptides
4.1.4. Streptomyces-Derived Peptides
4.2. Fungal Peptides
4.2.1. NZX
4.2.2. Trichoderins
4.3. Plant-Derived Peptides
Capsicum Defensins
| Peptide | Sequence | Activity | Source | Ref. |
|---|---|---|---|---|
| Nisin A | I-Dhb-AI-Dha-LA-Abu-PGAK-Abu-GALMGANMK-Abu-A-Abu-ANASIHV-Dha-K | IC90 (H37Ra): >60 μg/mL | Lactococcus lactis | [116] |
| Nisin S | I-Dhb-AI-Dha-LA-Abu-PGAK-Abu-GALMGANMS-Abu-A-Abu-ANASIHV-Dha-K | Mycobacterial growth reduced in 26% compared to Nisin A | Derived from Nisin A | [117] |
| Nisin T | I-Dhb-AI-Dha-LA-Abu-PGAK-Abu-GALMGANMT-Abu-A-Abu-ANASIHV-Dha-K | Mycobacterial growth reduced in 24% compared to Nisin A | Derived from Nisin A | [117] |
| Nisin V | I-Dhb-AI-Dha-LA-Abu-PGAK-Abu-GALMGANVK-Abu-A-Abu-ANASIHV-Dha-K | Mycobacterial growth reduced in 10% compared to Nisin A | Derived from Nisin A | [117] |
| Lacticin 3147 | Equimolar mixture of Lanα [AA-Dhb-N-Dhb-F-(D-Ala)LADYWGNNGAWA-Abu-L-Abu-HEAMAWAK] and Lanβ [[CH3-CH2-CO-CO-Dhb-PA-Dhb-PAI-(D-Ala)IL-(D-Ala)AYIATNTAP-Abu-TKA-Abu-RAA] | IC90 (H37Ra): 7.5 μg/mL | Lactococcus lactis | [116] |
| Lassomycin | GLRRLFADQLVGRRNI-CO2CH3 | MICs (MDR and XDR strains *): 0.8–3 mg/mL | Lentzea kentuckyensis sp. | [122] |
| Lariatin A | cyclo-(GSQLVYRE)-WVGHSNVIKP | MIC (H37Rv): 0.39 μg/mL | Rhodococcus jostii K01-B0171 | [123] |
| Hytramycin V | cyclo-[(NMe-Ala)-(D-Pip)-L-(D-Val)-Pip-(D-Pip)] | MIC Normal (H37Rv): 11.37 μg/mL MIC Hypoxic (H37Rv): 2.4 μg/mL | Streptomyces hygroscopicus | [126] |
| Hytramycin I | cyclo-[(NMe-Ala)-(D-Pip)-L-(D-Allo-Ile)-Pip-(D-Pip)] | MIC Normal (H37Rv): 6 μg/mL MIC Hypoxic (H37Rv): 1.5 μg/mL | Streptomyces hygroscopicus | [126] |
| Cyclomarin A | cyclo-[(tert-prenylated β-hydroxy-Trp)-(NMe-5-hydroxy-Leu)-A-(β-methoxy-Phe)–V-(NMe-Leu)–(2-amino-3,5-dimethyl-4-hexeneoic acid)] | >90% of the inoculum killed in five days at 2.5 μM | Streptomyces sp. (CNB-982) | [127] |
| Sansanmycin A | Not available | MIC (H37Rv): 16 μg/mL MIC (2199): 8 μg/mL | Streptomyces sp. SS | [128] |
| NZX | GFGCNGPWSEDDIQCHNHCKSIKGYKGGYCARGGFVCKCY | MIC (H37Rv and clinical MDR isolate): 6.3 μM MIC (clinical isolates): 3.2–6.3 μM | Derived from plecstasin of Pseudoplectania nigrella | [129] |
| Trichoderin A | (MDA)-P-(AHMOD)-Aib-Aib-IL-Aib-Aib-(AMAE) | MIC (H37Rv): 0.12 μg/mL | Trichoderma sp. 05FI48 | [130] |
| Trichoderin A1 | (MDA)-P-(AHMOD2)-Aib-Aib-IL-Aib-Aib-(AMAE) | MIC (H37Rv): 2 μg/mL | Trichoderma sp. 05FI48 | [130] |
| Trichoderin B | (MDA)-P-(AHMOD)-Aib-Aib-LL-Aib-Aib-(AMAE) | MIC (H37Rv): 0.13 μg/mL | Trichoderma sp. 05FI48 | [130] |
| CaDef2.1 | ICEALSGNFKGLCLSSRREGFTDGSCIGFRLQCFCTKPCA | MIC (H37Rv): 39.2 μg/mL | Capsicum annuum | [134] |
| CaDef2.2 | SKYFTGLCWTDSSCRKVCIEKDFQDGHCSKIQR |
5. Synthetic AMPs with Anti-Tubercular Activity
5.1. De Novo Designed Peptides
5.2. Nature-Inspired Synthetic Peptides
| Peptide | Sequence | Activity | Source | Ref. |
|---|---|---|---|---|
| 1-C134mer | H-Ntridec-NLys-Nspe-Nspe-NLys, amidated | MIC (H37Rv): 6.6 μM | De novo | [138] |
| 1 | Ac-GF-(A6c)-G-(A6c)-KK-(A6c)-G-(A6c)-F-(A6c)-G-(A6c)-GKK-(A6c)-KKKK, amidated | MIC (H37Ra, MDR, XDR): 4.92 μM | De novo | [139] |
| M(LLKK)2M | MLLKKLLKKM | MIC (H37Rv): 125 μg/mL MIC (MDR CSU87): 62.5 μg/mL | De novo | [140] |
| WKWLKKWIK | WKWLKKWIK | IC90 (lux): 1 μM | De novo | [143] |
| Cin+CAMP1 | (Cin1)-KWLKKWIK, amidated | IC50 (H37Rv): 0.69 μM IC50 (MDR *): 2.77 μM | De novo | [144] |
| Cin+CAMP3 | (Cin2)-ARLWWWWRRK, amidated | IC50 (H37Rv): 2.41 μM IC50 (MDR *): 0.60 μM | De novo | [144] |
| IP-1 | KFLNRFWHWLQLKPGQPMY | IC50 (H37Rv): 99.27 μM IC50 (MDR CIBIN99): 92.66 μM | De novo | [146] |
| E2 | RIWVIWRR, amidated | MIC (H37Rv, MDR clinical isolate): 2.6 μg/mL | Bactenicin | [55] |
| E6 | RRWRIVVIRVRR, amidated | MIC (H37Rv, MDR clinical isolate): 3.2 μg/mL | Bactenicin | [55] |
| C26 | KWKSFIKKLTSAAKKVVTTAKPLISS | MIC (H37Rv, MDR clinical isolate): 2.1 μg/mL | Bactenicin | [55] |
| IDR-HH2 | VQLRIRVAVIRA, amidated | MIC (H37Rv): 29.3 μg/mL ** | MCP-1 | [147] |
| IDR-1002 | VQRWLIVWRIRK, amidated | MIC (H37Rv): 29.3 μg/mL ** | MCP-1 | [147] |
| IDR-1018 | VRLIVAVRIWRR, amidated | MIC (H37Rv): 16 μg/mL ** | MCP-1 | [147] |
| Sansanmycin A derivative 1d | Not available | MIC (H37Rv, MDR 2199): 8 μg/mL | Sansanmycin A | [128] |
| AK15-6 | AVKKLLRWWSRWWKK | MIC (H37Rv): 18.75 μg/mL | AK15 | [148] |
6. Taking AMPs from Bench to the Clinic
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef]
- Bruchfeld, J.; Correia-Neves, M.; Kallenius, G. Tuberculosis and HIV Coinfection. Cold Spring Harb. Perspect. Med. 2015, 5, a017871. [Google Scholar] [CrossRef]
- Bourzac, K. Infectious disease: Beating the big three. Nature 2014, 507, S4–S7. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2020. Available online: https://www.who.int/publications/i/item/9789240013131 (accessed on 28 January 2021).
- Kurz, S.G.; Furin, J.J.; Bark, C.M. Drug-Resistant Tuberculosis: Challenges and Progress. Infect. Dis. Clin. N. Am. 2016, 30, 509–522. [Google Scholar] [CrossRef]
- Chan, E.D.; Iseman, M.D. Multidrug-resistant and extensively drug-resistant tuberculosis: A review. Curr. Opin. Infect. Dis. 2008, 21, 587–595. [Google Scholar] [CrossRef]
- Usmani, S.S.; Kumar, R.; Kumar, V.; Singh, S.; Raghava, G.P.S. AntiTbPdb: A knowledgebase of anti-tubercular peptides. Database 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Appelberg, R.; Gama, F.M. Antimicrobial peptides as novel anti-tuberculosis therapeutics. Biotechnol. Adv. 2016, 34, 924–940. [Google Scholar] [CrossRef]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 2011, 240, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Desvignes, L.; Linas, B.; Banaiee, N.; Tamura, T.; Takatsu, K.; Ernst, J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2008, 205, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Samstein, M.; Schreiber, H.A.; Leiner, I.M.; Susac, B.; Glickman, M.S.; Pamer, E.G. Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife 2013, 2, e01086. [Google Scholar] [CrossRef] [PubMed]
- Silva Miranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 2012, 139127. [Google Scholar] [CrossRef] [PubMed]
- Qualls, J.E.; Murray, P.J. Immunometabolism within the tuberculosis granuloma: Amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 2016, 38, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Jarlier, V.; Nikaido, H. Mycobacterial cell wall: Structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar] [CrossRef]
- Kolattukudy, P.E.; Fernandes, N.D.; Azad, A.K.; Fitzmaurice, A.M.; Sirakova, T.D. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol. Microbiol. 1997, 24, 263–270. [Google Scholar] [CrossRef]
- Mangtani, P.; Abubakar, I.; Ariti, C.; Beynon, R.; Pimpin, L.; Fine, P.E.; Rodrigues, L.C.; Smith, P.G.; Lipman, M.; Whiting, P.F.; et al. Protection by BCG vaccine against tuberculosis: A systematic review of randomized controlled trials. Clin. Infect. Dis. 2014, 58, 470–480. [Google Scholar] [CrossRef]
- Onyebujoh, P.; Zumla, A.; Ribeiro, I.; Rustomjee, R.; Mwaba, P.; Gomes, M.; Grange, J.M. Treatment of tuberculosis: Present status and future prospects. Bull. World Health Organ. 2005, 83, 857–865. [Google Scholar] [CrossRef]
- Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 2020, 10, 2278. [Google Scholar] [CrossRef]
- Wehrli, W. Rifampin: Mechanisms of action and resistance. Rev. Infect. Dis. 1983, 5 (Suppl. 3), S407–S411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Carcaboso, A.M.; Glisoni, R.J.; Moretton, M.A.; Chiappetta, D.A. New old challenges in tuberculosis: Potentially effective nanotechnologies in drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Loddenkemper, R.; Sagebiel, D.; Brendel, A. Strategies against multidrug-resistant tuberculosis. Eur. Respir. J. Suppl. 2002, 36, 66s–77s. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.G.; Chung, J.H. Diagnosis and treatment of multidrug-resistant tuberculosis. Yeungnam Univ. J. Med. 2020, 37, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef]
- Padhi, A.; Sengupta, M.; Sengupta, S.; Roehm, K.H.; Sonawane, A. Antimicrobial peptides and proteins in mycobacterial therapy: Current status and future prospects. Tuberculosis 2014, 94, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Gaglione, R.; Pizzo, E.; Notomista, E.; de la Fuente-Nunez, C.; Arciello, A. Host Defence Cryptides from Human Apolipoproteins: Applications in Medicinal Chemistry. Curr. Top. Med. Chem. 2020, 20, 1324–1337. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E. Antimicrobial Peptides: An Introduction. Methods Mol. Biol. 2017, 1548, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Aguiar, L.; Gomes, P. Antimicrobial peptides: A new class of antimalarial drugs? Front. Pharmacol. 2014, 5, 275. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Gomes, M.S. Immuno-Stimulatory Peptides as a Potential Adjunct Therapy against Intra-Macrophagic Pathogens. Molecules 2017, 22, 1297. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Hemshekhar, M.; Anaparti, V.; Mookherjee, N. Functions of Cationic Host Defense Peptides in Immunity. Pharmaceuticals 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Kosciuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzalkowska, N.; Jozwik, A.; Horbanczuk, J.; Krzyzewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. The role of cathelicidins in the innate host defenses of mammals. Curr. Issues Mol. Biol. 2005, 7, 179–196. [Google Scholar]
- Sonawane, A.; Santos, J.C.; Mishra, B.B.; Jena, P.; Progida, C.; Sorensen, O.E.; Gallo, R.; Appelberg, R.; Griffiths, G. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol. 2011, 13, 1601–1617. [Google Scholar] [CrossRef]
- Durr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Yang, B.; Good, D.; Mosaiab, T.; Liu, W.; Ni, G.; Kaur, J.; Liu, X.; Jessop, C.; Yang, L.; Fadhil, R.; et al. Significance of LL-37 on Immunomodulation and Disease Outcome. Biomed. Res. Int. 2020, 2020, 8349712. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Mily, A.; Rekha, R.S.; Kamal, S.M.; Arifuzzaman, A.S.; Rahim, Z.; Khan, L.; Haq, M.A.; Zaman, K.; Bergman, P.; Brighenti, S.; et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0138340. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Hernandez-Pando, R.; Carranza, C.; Juarez, E.; Contreras, J.L.; Aguilar-Leon, D.; Torres, M.; Sada, E. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect. Immun. 2008, 76, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Rivas Santiago, C.E.; Castaneda-Delgado, J.E.; Leon-Contreras, J.C.; Hancock, R.E.; Hernandez-Pando, R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2013, 41, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Torres-Juarez, F.; Cardenas-Vargas, A.; Montoya-Rosales, A.; Gonzalez-Curiel, I.; Garcia-Hernandez, M.H.; Enciso-Moreno, J.A.; Hancock, R.E.; Rivas-Santiago, B. LL-37 immunomodulatory activity during Mycobacterium tuberculosis infection in macrophages. Infect. Immun. 2015, 83, 4495–4503. [Google Scholar] [CrossRef] [PubMed]
- Wessely-Szponder, J.; Majer-Dziedzic, B.; Smolira, A. Analysis of antimicrobial peptides from porcine neutrophils. J. Microbiol. Methods 2010, 83, 8–12. [Google Scholar] [CrossRef]
- Storici, P.; Zanetti, M. A cDNA derived from pig bone marrow cells predicts a sequence identical to the intestinal antibacterial peptide PR-39. Biochem. Biophys. Res. Commun. 1993, 196, 1058–1065. [Google Scholar] [CrossRef]
- Shi, J.; Ross, C.R.; Chengappa, M.M.; Blecha, F. Identification of a proline-arginine-rich antibacterial peptide from neutrophils that is analogous to PR-39, an antibacterial peptide from the small intestine. J. Leukoc. Biol. 1994, 56, 807–811. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [CrossRef]
- Linde, C.M.; Hoffner, S.E.; Refai, E.; Andersson, M. In vitro activity of PR-39, a proline-arginine-rich peptide, against susceptible and multi-drug-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2001, 47, 575–580. [Google Scholar] [CrossRef]
- Portell-Buj, E.; Vergara, A.; Alejo, I.; Lopez-Gavin, A.; Monte, M.R.; San Nicolas, L.; Gonzalez-Martin, J.; Tudo, G. In vitro activity of 12 antimicrobial peptides against Mycobacterium tuberculosis and Mycobacterium avium clinical isolates. J. Med. Microbiol. 2019, 68, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Lv, Y.; Zhao, D.; Barrow, P.; Zhou, X. Defensins: The Case for Their Use against Mycobacterial Infections. J. Immunol. Res. 2016, 2016, 7515687. [Google Scholar] [CrossRef] [PubMed]
- Amerikova, M.; Pencheva El-Tibi, I.; Maslarska, V.; Bozhanov, S.; Tachkov, K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol. Biotechnol. Equip. 2019, 33, 671–682. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Machado, L.R.; Ottolini, B. An evolutionary history of defensins: A role for copy number variation in maximizing host innate and adaptive immune responses. Front. Immunol. 2015, 6, 115. [Google Scholar] [CrossRef]
- Rivas-Santiago, B.; Schwander, S.K.; Sarabia, C.; Diamond, G.; Klein-Patel, M.E.; Hernandez-Pando, R.; Ellner, J.J.; Sada, E. Human {beta}-defensin 2 is expressed and associated with Mycobacterium tuberculosis during infection of human alveolar epithelial cells. Infect. Immun. 2005, 73, 4505–4511. [Google Scholar] [CrossRef]
- Rivas-Santiago, C.E.; Rivas-Santiago, B.; Leon, D.A.; Castaneda-Delgado, J.; Hernandez Pando, R. Induction of beta-defensins by l-isoleucine as novel immunotherapy in experimental murine tuberculosis. Clin. Exp. Immunol. 2011, 164, 80–89. [Google Scholar] [CrossRef]
- Fattorini, L.; Gennaro, R.; Zanetti, M.; Tan, D.; Brunori, L.; Giannoni, F.; Pardini, M.; Orefici, G. In vitro activity of protegrin-1 and beta-defensin-1, alone and in combination with isoniazid, against Mycobacterium tuberculosis. Peptides 2004, 25, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Ashitani, J.; Mukae, H.; Hiratsuka, T.; Nakazato, M.; Kumamoto, K.; Matsukura, S. Elevated levels of alpha-defensins in plasma and BAL fluid of patients with active pulmonary tuberculosis. Chest 2002, 121, 519–526. [Google Scholar] [CrossRef]
- Jacobsen, M.; Repsilber, D.; Gutschmidt, A.; Neher, A.; Feldmann, K.; Mollenkopf, H.J.; Ziegler, A.; Kaufmann, S.H. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J. Mol. Med. 2007, 85, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Verma, I.; Khuller, G.K. Therapeutic potential of human neutrophil peptide 1 against experimental tuberculosis. Antimicrob. Agents Chemother. 2001, 45, 639–640. [Google Scholar] [CrossRef]
- Kalita, A.; Verma, I.; Khuller, G.K. Role of human neutrophil peptide-1 as a possible adjunct to antituberculosis chemotherapy. J. Infect. Dis. 2004, 190, 1476–1480. [Google Scholar] [CrossRef]
- Sharma, R.; Raghav, R.; Priyanka, K.; Rishi, P.; Sharma, S.; Srivastava, S.; Verma, I. Exploiting chitosan and gold nanoparticles for antimycobacterial activity of in silico identified antimicrobial motif of human neutrophil peptide-1. Sci. Rep. 2019, 9, 7866. [Google Scholar] [CrossRef]
- Zhao, C.; Ganz, T.; Lehrer, R.I. The structure of porcine protegrin genes. FEBS Lett. 1995, 368, 197–202. [Google Scholar] [CrossRef]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef]
- Aumelas, A.; Mangoni, M.; Roumestand, C.; Chiche, L.; Despaux, E.; Grassy, G.; Calas, B.; Chavanieu, A. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 1996, 237, 575–583. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Malyszko, J.; Tesar, V.; Macdougall, I.C. Neutrophil gelatinase-associated lipocalin and hepcidin: What do they have in common and is there a potential interaction? Kidney Blood Press Res. 2010, 33, 157–165. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol. 2009, 122, 78–86. [Google Scholar] [CrossRef]
- Gomes, A.C.; Moreira, A.C.; Mesquita, G.; Gomes, M.S. Modulation of Iron Metabolism in Response to Infection: Twists for All Tastes. Pharmaceuticals 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Harrington-Kandt, R.; Stylianou, E.; Eddowes, L.A.; Lim, P.J.; Stockdale, L.; Pinpathomrat, N.; Bull, N.; Pasricha, J.; Ulaszewska, M.; Beglov, Y.; et al. Hepcidin deficiency and iron deficiency do not alter tuberculosis susceptibility in a murine M.tb infection model. PLoS ONE 2018, 13, e0191038. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Pellegrino, N.M.; Altamura, M.; Rigon, A.; Amati, L.; Amoroso, A.; Jirillo, E. Antimicrobial and immunoregulatory functions of lactoferrin and its potential therapeutic application. J. Endotoxin Res. 2002, 8, 403–417. [Google Scholar] [CrossRef]
- Bruni, N.; Capucchio, M.T.; Biasibetti, E.; Pessione, E.; Cirrincione, S.; Giraudo, L.; Corona, A.; Dosio, F. Antimicrobial Activity of Lactoferrin-Related Peptides and Applications in Human and Veterinary Medicine. Molecules 2016, 21, 752. [Google Scholar] [CrossRef]
- Koo, H.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111. [Google Scholar] [CrossRef]
- Silva, T.; Moreira, A.C.; Nazmi, K.; Moniz, T.; Vale, N.; Rangel, M.; Gomes, P.; Bolscher, J.G.M.; Rodrigues, P.N.; Bastos, M.; et al. Lactoferricin Peptides Increase Macrophages’ Capacity To Kill Mycobacterium avium. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Silva, T.; Magalhaes, B.; Maia, S.; Gomes, P.; Nazmi, K.; Bolscher, J.G.; Rodrigues, P.N.; Bastos, M.; Gomes, M.S. Killing of Mycobacterium avium by lactoferricin peptides: Improved activity of arginine- and D-amino-acid-containing molecules. Antimicrob. Agents Chemother. 2014, 58, 3461–3467. [Google Scholar] [CrossRef]
- Fornili, S.L.; Pizzi, R.; Rebeccani, D. Conformational Analysis of a Synthetic Antimicrobial Peptide in Water and Membrane-Mimicking Solvents: A Molecular Dynamics Simulation Study. Int. J. Pept. Res. Ther. 2010, 16, 223–231. [Google Scholar] [CrossRef]
- Welsh, K.J.; Hwang, S.A.; Boyd, S.; Kruzel, M.L.; Hunter, R.L.; Actor, J.K. Influence of oral lactoferrin on Mycobacterium tuberculosis induced immunopathology. Tuberculosis 2011, 91 (Suppl. 1), S105–S113. [Google Scholar] [CrossRef]
- Hwang, S.A.; Wilk, K.M.; Bangale, Y.A.; Kruzel, M.L.; Actor, J.K. Lactoferrin modulation of IL-12 and IL-10 response from activated murine leukocytes. Med. Microbiol. Immunol. 2007, 196, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.A.; Kruzel, M.L.; Actor, J.K. CHO expressed recombinant human lactoferrin as an adjuvant for BCG. Int. J. Immunopathol. Pharmacol. 2015, 28, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Thom, R.E.; Elmore, M.J.; Williams, A.; Andrews, S.C.; Drobniewski, F.; Marsh, P.D.; Tree, J.A. The expression of ferritin, lactoferrin, transferrin receptor and solute carrier family 11A1 in the host response to BCG-vaccination and Mycobacterium tuberculosis challenge. Vaccine 2012, 30, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, H.; Patidar, A.; Boradia, V.M.; Kumar, R.; Nimbalkar, R.D.; Kumar, A.; Gani, Z.; Kaur, R.; Garg, P.; Raje, M.; et al. Mycobacterium tuberculosis Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Functions as a Receptor for Human Lactoferrin. Front. Cell. Infect. Microbiol. 2017, 7, 245. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef]
- Teng, T.; Liu, J.; Wei, H. Anti-mycobacterial peptides: From human to phage. Cell. Physiol. Biochem. 2015, 35, 452–466. [Google Scholar] [CrossRef]
- Alonso, S.; Pethe, K.; Russell, D.G.; Purdy, G.E. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. USA 2007, 104, 6031–6036. [Google Scholar] [CrossRef]
- Foss, M.H.; Powers, K.M.; Purdy, G.E. Structural and functional characterization of mycobactericidal ubiquitin-derived peptides in model and bacterial membranes. Biochemistry 2012, 51, 9922–9929. [Google Scholar] [CrossRef][Green Version]
- Purdy, G.E.; Niederweis, M.; Russell, D.G. Decreased outer membrane permeability protects mycobacteria from killing by ubiquitin-derived peptides. Mol. Microbiol. 2009, 73, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Samuchiwal, S.K.; Tousif, S.; Singh, D.K.; Kumar, A.; Ghosh, A.; Bhalla, K.; Prakash, P.; Kumar, S.; Bhattacharyya, M.; Moodley, P.; et al. A peptide fragment from the human COX3 protein disrupts association of Mycobacterium tuberculosis virulence proteins ESAT-6 and CFP10, inhibits mycobacterial growth and mounts protective immune response. BMC Infect. Dis. 2014, 14, 355. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Ahmed, A.; Parveen, N.; Jha, V.; Valluri, V.L.; Ghosh, S.; Mukhopadhyay, S. The ESAT-6 protein of Mycobacterium tuberculosis interacts with beta-2-microglobulin (beta2M) affecting antigen presentation function of macrophage. PLoS Pathog. 2014, 10, e1004446. [Google Scholar] [CrossRef] [PubMed]
- Stapels, D.A.; Geisbrecht, B.V.; Rooijakkers, S.H. Neutrophil serine proteases in antibacterial defense. Curr. Opin. Microbiol. 2015, 23, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Marrero, C.A.; Stewart, J.; Shafer, W.M.; Roman, J. The down-regulation of cathepsin G in THP-1 monocytes after infection with Mycobacterium tuberculosis is associated with increased intracellular survival of bacilli. Infect. Immun. 2004, 72, 5712–5721. [Google Scholar] [CrossRef]
- Hmed, B.; Serria, H.T.; Mounir, Z.K. Scorpion peptides: Potential use for new drug development. J. Toxicol. 2013, 2013, 958797. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Carreto, S.; Jimenez-Vargas, J.M.; Rivas-Santiago, B.; Corzo, G.; Possani, L.D.; Becerril, B.; Ortiz, E. Peptides from the scorpion Vaejovis punctatus with broad antimicrobial activity. Peptides 2015, 73, 51–59. [Google Scholar] [CrossRef]
- Rodriguez, A.; Villegas, E.; Montoya-Rosales, A.; Rivas-Santiago, B.; Corzo, G. Characterization of antibacterial and hemolytic activity of synthetic pandinin 2 variants and their inhibition against Mycobacterium tuberculosis. PLoS ONE 2014, 9, e101742. [Google Scholar] [CrossRef]
- Xie, J.P.; Yue, J.; Xiong, Y.L.; Wang, W.Y.; Yu, S.Q.; Wang, H.H. In vitro activities of small peptides from snake venom against clinical isolates of drug-resistant Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2003, 22, 172–174. [Google Scholar] [CrossRef]
- Morikawa, N.; Hagiwara, K.; Nakajima, T. Brevinin-1 and -2, unique antimicrobial peptides from the skin of the frog, Rana brevipoda porsa. Biochem. Biophys. Res. Commun. 1992, 189, 184–190. [Google Scholar] [CrossRef]
- Abraham, P.; Jose, L.; Maliekal, T.T.; Kumar, R.A.; Kumar, K.S. B1CTcu5: A frog-derived brevinin-1 peptide with anti-tuberculosis activity. Peptides 2020, 132, 170373. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, J. Lantibiotics as prospective antimycobacterial agents. Bioeng. Bugs 2010, 1, 437–439. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Arauz, L.J.; Jozala, A.F.; Mazzola, P.G.; Vessoni Penna, T.C. Nisin biotechnological production and application: A review. Trends Food Sci. Technol. 2009, 20, 146–154. [Google Scholar] [CrossRef]
- Suda, S.; Cotter, P.D.; Hill, C.; Ross, R.P. Lacticin 3147--biosynthesis, molecular analysis, immunity, bioengineering and applications. Curr. Protein Pept. Sci. 2012, 13, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Draper, L.A.; O’Connor, P.M.; Coffey, A.; Hill, C.; Ross, R.P.; Cotter, P.D.; O’Mahony, J. Comparison of the activities of the lantibiotics nisin and lacticin 3147 against clinically significant mycobacteria. Int. J. Antimicrob. Agents 2010, 36, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Field, D.; O’Connor, P.M.; Cotter, P.D.; Coffey, A.; Hill, C.; Ross, R.P.; O’Mahony, J. Gene encoded antimicrobial peptides, a template for the design of novel anti-mycobacterial drugs. Bioeng. Bugs 2010, 1, 408–412. [Google Scholar] [CrossRef]
- Ghobrial, O.; Derendorf, H.; Hillman, J.D. Pharmacokinetic and pharmacodynamic evaluation of the lantibiotic MU1140. J. Pharm. Sci. 2010, 99, 2521–2528. [Google Scholar] [CrossRef]
- Hillman, J.D.; Novak, J.; Sagura, E.; Gutierrez, J.A.; Brooks, T.A.; Crowley, P.J.; Hess, M.; Azizi, A.; Leung, K.; Cvitkovitch, D.; et al. Genetic and biochemical analysis of mutacin 1140, a lantibiotic from Streptococcus mutans. Infect. Immun. 1998, 66, 2743–2749. [Google Scholar] [CrossRef]
- Smith, L.; Hasper, H.; Breukink, E.; Novak, J.; Cerkasov, J.; Hillman, J.D.; Wilson-Stanford, S.; Orugunty, R.S. Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry 2008, 47, 3308–3314. [Google Scholar] [CrossRef]
- Zhu, S.; Su, Y.; Shams, S.; Feng, Y.; Tong, Y.; Zheng, G. Lassomycin and lariatin lasso peptides as suitable antibiotics for combating mycobacterial infections: Current state of biosynthesis and perspectives for production. Appl. Microbiol. Biotechnol. 2019, 103, 3931–3940. [Google Scholar] [CrossRef]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Uchida, R.; Takakusagi, Y.; Matsumoto, A.; Jiang, C.L.; Takahashi, Y.; Arai, M.; Kobayashi, S.; Matsumoto, M.; Inokoshi, J.; et al. Lariatins, novel anti-mycobacterial peptides with a lasso structure, produced by Rhodococcus jostii K01-B0171. J. Antibiot. 2007, 60, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Shoji, J.; Sakazaki, R. A new peptide antibiotic complex S-520. II. Further characterization and degradative studies. J. Antibiot. 1970, 23, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Stott, K.E.; Munsamy, V.; Manson, A.L.; Earl, A.M.; Pym, A.S. Evidence for Expanding the Role of Streptomycin in the Management of Drug-Resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Cai, G.; Napolitano, J.G.; McAlpine, J.B.; Wang, Y.; Jaki, B.U.; Suh, J.W.; Yang, S.H.; Lee, I.A.; Franzblau, S.G.; Pauli, G.F.; et al. Hytramycins V and I, anti-Mycobacterium tuberculosis hexapeptides from a Streptomyces hygroscopicus strain. J. Nat. Prod. 2013, 76, 2009–2018. [Google Scholar] [CrossRef]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. Engl. 2011, 50, 5889–5891. [Google Scholar] [CrossRef]
- Li, Y.B.; Xie, Y.Y.; Du, N.N.; Lu, Y.; Xu, H.Z.; Wang, B.; Yu, Y.; Liu, Y.X.; Song, D.Q.; Chen, R.X. Synthesis and in vitro antitubercular evaluation of novel sansanmycin derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 6804–6807. [Google Scholar] [CrossRef]
- Tenland, E.; Krishnan, N.; Ronnholm, A.; Kalsum, S.; Puthia, M.; Morgelin, M.; Davoudi, M.; Otrocka, M.; Alaridah, N.; Glegola-Madejska, I.; et al. A novel derivative of the fungal antimicrobial peptide plectasin is active against Mycobacterium tuberculosis. Tuberculosis 2018, 113, 231–238. [Google Scholar] [CrossRef]
- Pruksakorn, P.; Arai, M.; Kotoku, N.; Vilcheze, C.; Baughn, A.D.; Moodley, P.; Jacobs, W.R., Jr.; Kobayashi, M. Trichoderins, novel aminolipopeptides from a marine sponge-derived Trichoderma sp., are active against dormant mycobacteria. Bioorg. Med. Chem. Lett. 2010, 20, 3658–3663. [Google Scholar] [CrossRef]
- Pruksakorn, P.; Arai, M.; Liu, L.; Moodley, P.; Jacobs, W.R., Jr.; Kobayashi, M. Action-mechanism of trichoderin A, an anti-dormant mycobacterial aminolipopeptide from marine sponge-derived Trichoderma sp. Biol. Pharm. Bull. 2011, 34, 1287–1290. [Google Scholar] [CrossRef]
- Koffi-Nevry, R.; Kouassi, K.C.; Nanga, Z.Y.; Koussémon, M.; Loukou, G.Y. Antibacterial Activity of Two Bell Pepper Extracts: Capsicum annuum L. and Capsicum frutescens. Int. J. Food Prop. 2012, 15, 961–971. [Google Scholar] [CrossRef]
- Santos, M.M.; Vieira-da-Motta, O.; Vieira, I.J.; Braz-Filho, R.; Goncalves, P.S.; Maria, E.J.; Terra, W.S.; Rodrigues, R.; Souza, C.L. Antibacterial activity of Capsicum annuum extract and synthetic capsaicinoid derivatives against Streptococcus mutans. J. Nat. Med. 2012, 66, 354–356. [Google Scholar] [CrossRef] [PubMed]
- da Silva Gebara, R.; Taveira, G.B.; de Azevedo Dos Santos, L.; Calixto, S.D.; Simao, T.; Lassounskaia, E.; Muzitano, M.F.; Teixeira-Ferreira, A.; Perales, J.; Rodrigues, R.; et al. Identification and Characterization of Two Defensins from Capsicum annuum Fruits that Exhibit Antimicrobial Activity. Probiotics Antimicrob. Proteins 2020, 12, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Costa, F.; Teixeira, C.; Gomes, P.; Martins, M.C.L. Clinical Application of AMPs. Adv. Exp. Med. Biol. 2019, 1117, 281–298. [Google Scholar] [CrossRef]
- Dohm, M.T.; Kapoor, R.; Barron, A.E. Peptoids: Bio-inspired polymers as potential pharmaceuticals. Curr. Pharm. Des. 2011, 17, 2732–2747. [Google Scholar] [CrossRef]
- Kapoor, R.; Eimerman, P.R.; Hardy, J.W.; Cirillo, J.D.; Contag, C.H.; Barron, A.E. Efficacy of antimicrobial peptoids against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 3058–3062. [Google Scholar] [CrossRef]
- Hicks, R.P. Antibacterial and anticancer activity of a series of novel peptides incorporating cyclic tetra-substituted C(alpha) amino acids. Bioorg. Med. Chem. 2016, 24, 4056–4065. [Google Scholar] [CrossRef]
- Khara, J.S.; Wang, Y.; Ke, X.Y.; Liu, S.; Newton, S.M.; Langford, P.R.; Yang, Y.Y.; Ee, P.L. Anti-mycobacterial activities of synthetic cationic alpha-helical peptides and their synergism with rifampicin. Biomaterials 2014, 35, 2032–2038. [Google Scholar] [CrossRef]
- Vermeer, L.S.; Lan, Y.; Abbate, V.; Ruh, E.; Bui, T.T.; Wilkinson, L.J.; Kanno, T.; Jumagulova, E.; Kozlowska, J.; Patel, J.; et al. Conformational flexibility determines selectivity and antibacterial, antiplasmodial, and anticancer potency of cationic alpha-helical peptides. J. Biol. Chem. 2012, 287, 34120–34133. [Google Scholar] [CrossRef]
- Lan, Y.; Lam, J.T.; Siu, G.K.; Yam, W.C.; Mason, A.J.; Lam, J.K. Cationic amphipathic D-enantiomeric antimicrobial peptides with in vitro and ex vivo activity against drug-resistant Mycobacterium tuberculosis. Tuberculosis 2014, 94, 678–689. [Google Scholar] [CrossRef]
- Ramon-Garcia, S.; Mikut, R.; Ng, C.; Ruden, S.; Volkmer, R.; Reischl, M.; Hilpert, K.; Thompson, C.J. Targeting Mycobacterium tuberculosis and other microbial pathogens using improved synthetic antibacterial peptides. Antimicrob. Agents Chemother. 2013, 57, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Santos-Silva, A.; da Costa, J.M.C.; Vale, N. Potent cationic antimicrobial peptides against Mycobacterium tuberculosis in vitro. J. Glob. Antimicrob. Resist. 2019, 19, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Ellerby, H.M.; Bredesen, D.E.; Fujimura, S.; John, V. Hunter-killer peptide (HKP) for targeted therapy. J. Med. Chem. 2008, 51, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Pelaez Coyotl, E.A.; Barrios Palacios, J.; Mucino, G.; Moreno-Blas, D.; Costas, M.; Montiel Montes, T.; Diener, C.; Uribe-Carvajal, S.; Massieu, L.; Castro-Obregon, S.; et al. Antimicrobial Peptide against Mycobacterium Tuberculosis That Activates Autophagy Is an Effective Treatment for Tuberculosis. Pharmaceutics 2020, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Castaneda-Delgado, J.E.; Rivas Santiago, C.E.; Waldbrook, M.; Gonzalez-Curiel, I.; Leon-Contreras, J.C.; Enciso-Moreno, J.A.; del Villar, V.; Mendez-Ramos, J.; Hancock, R.E.; et al. Ability of innate defence regulator peptides IDR-1002, IDR-HH2 and IDR-1018 to protect against Mycobacterium tuberculosis infections in animal models. PLoS ONE 2013, 8, e59119. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; He, X.; Yang, J.; Wu, J.; Yang, H.; Li, M.; Qian, Q.; Lai, R.; Xu, W.; et al. A small mycobacteriophage-derived peptide and its improved isomer restrict mycobacterial infection via dual mycobactericidal-immunoregulatory activities. J. Biol. Chem. 2019, 294, 7615–7631. [Google Scholar] [CrossRef]
- Mendez-Samperio, P. Role of antimicrobial peptides in host defense against mycobacterial infections. Peptides 2008, 29, 1836–1841. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef]
- Martin-Serrano, A.; Gomez, R.; Ortega, P.; de la Mata, F.J. Nanosystems as Vehicles for the Delivery of Antimicrobial Peptides (AMPs). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef]
- Patel, A.; Cholkar, K.; Mitra, A.K. Recent developments in protein and peptide parenteral delivery approaches. Ther. Deliv. 2014, 5, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Shukla, P. Antimicrobial Peptides: Recent Insights on Biotechnological Interventions and Future Perspectives. Protein Pept. Lett. 2019, 26, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Yevtushenko, D.; Misra, S. Transgenic Expression of Antimicrobial Peptides in Plants: Strategies for Enhanced Disease Resistance, Improved Productivity, and Production of Therapeutics. ACS Symp. Ser. 2012, 1095, 445–458. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, G.S.; Costa, R.P.; Gomes, P.; Gomes, M.S.; Silva, T.; Teixeira, C. Antimicrobial Peptides as Potential Anti-Tubercular Leads: A Concise Review. Pharmaceuticals 2021, 14, 323. https://doi.org/10.3390/ph14040323
Oliveira GS, Costa RP, Gomes P, Gomes MS, Silva T, Teixeira C. Antimicrobial Peptides as Potential Anti-Tubercular Leads: A Concise Review. Pharmaceuticals. 2021; 14(4):323. https://doi.org/10.3390/ph14040323
Chicago/Turabian StyleOliveira, Gabriel S., Raquel P. Costa, Paula Gomes, Maria Salomé Gomes, Tânia Silva, and Cátia Teixeira. 2021. "Antimicrobial Peptides as Potential Anti-Tubercular Leads: A Concise Review" Pharmaceuticals 14, no. 4: 323. https://doi.org/10.3390/ph14040323
APA StyleOliveira, G. S., Costa, R. P., Gomes, P., Gomes, M. S., Silva, T., & Teixeira, C. (2021). Antimicrobial Peptides as Potential Anti-Tubercular Leads: A Concise Review. Pharmaceuticals, 14(4), 323. https://doi.org/10.3390/ph14040323