
Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences

 , and
, and 
Abstract
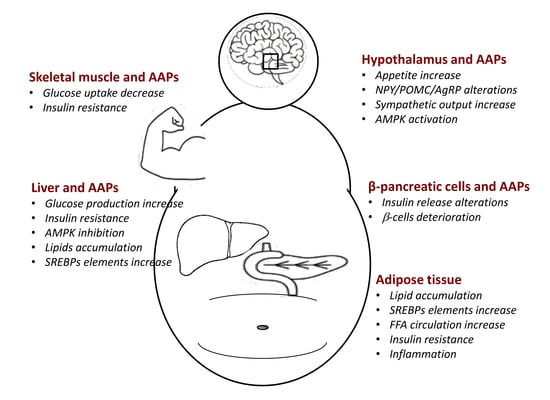
1. Introduction
2. A New Classification for AAPs: The Spectrum of Atypia
3. Role of GPCRs in the Metabolism of Peripheral Tissues
3.1. GPCRs and Pancreatic β-Cells
3.2. GPCRs and Liver
3.3. GPCRs and Adipose Tissues
3.4. GPCRs and Skeletal Muscle
4. Effects of AAPs on Hypothalamus and Peripheral Tissues
4.1. AAPs and Hypothalamus
4.2. AAPs and Liver
4.3. AAPs and Pancreatic β-Cells
4.4. AAPs and Adipose Tissue
4.5. AAPs and Skeletal Muscle
5. AAPs and MetS in Clinical Practice
6. Treatments for AAPs-Induced MetS: Pharmacological and Non-Pharmacological Interventions
6.1. Non-Pharmacological Interventions
6.2. Pharmacological Interventions
7. Towards a Personalized Therapy: AAPs Plasma Concentration Monitoring and Pharmacogenetics
8. Conclusions
Author Contributions
Conflicts of Interest
References
- De Hert, M.A.; Van Winkel, R.; Van Eyck, D.; Hanssens, L.; Wampers, M.; Scheen, A.; Peuskens, J. Prevalence of the metabolic syndrome in patients with schizophrenia treated with antipsychotic medication. Schizophr. Res. 2006, 83, 87–93. [Google Scholar] [CrossRef]
- Yevtushenko, O.O.; Cooper, S.J.; O’Neill, R.; Doherty, J.K.; Woodside, J.V.; Reynolds, G.P. Influence of 5-HT2C receptor and leptin gene polymorphisms, smoking and drug treatment on metabolic disturbances in patients with schizophrenia. Br. J. Psychiatry 2008, 192, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.M. Life expectancy among persons with schizophrenia or bipolar affective disorder. Schizophr. Res. 2011, 131, 101–104. [Google Scholar] [CrossRef]
- Taipale, H.; Tanskanen, A.; Mehtälä, J.; Vattulainen, P.; Correll, C.U.; Tiihonen, J. 20-year follow-up study of physical morbidity and mortality in relationship to antipsychotic treatment in a nationwide cohort of 62,250 patients with schizophrenia (FIN20). World Psychiatry 2020, 19, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Tiihonen, J.; Wahlbeck, K.; Lönnqvist, J.; Klaukka, T.; Ioannidis, J.P.A.; Volavka, J.; Haukka, J. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in community care after first hospitalisation due to schizophrenia and schizoaffective disorder: Observational follow-up study. Br. Med. J. 2006, 333, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.B.; Brown, C.H.; Daumit, G.L.; Lijuan, F.; Goldberg, R.W.; Wohlheiter, K.; Dixon, L.B. Health status of individuals with serious mental illness. Schizophr. Bull. 2006, 32, 584–589. [Google Scholar] [CrossRef]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 2018, 192, 20–41. [Google Scholar] [CrossRef]
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Bäckers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951. [Google Scholar] [CrossRef]
- Kowalchuk, C.; Castellani, L.N.; Chintoh, A.; Remington, G.; Giacca, A.; Hahn, M.K. Antipsychotics and glucose metabolism: How brain and body collide. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1–E15. [Google Scholar] [CrossRef] [PubMed]
- Ballon, J.S.; Pajvani, U.; Freyberg, Z.; Leibel, R.L.; Lieberman, J.A. Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol. Metab. 2014, 25, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Gründer, G.; Hippius, H.; Carlsson, A. The “atypicality” of antipsychotics: A concept re-examined and re-defined. Nat. Rev. Drug Discov. 2009, 8, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, A.L.; Samanaite, R.; Mill, J.; Egerton, A.; MacCabe, J.H. Is treatment-resistant schizophrenia categorically distinct from treatment-responsive schizophrenia? A systematic review. BMC Psychiatry 2017, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, M.; Armogida, M.; Chiacchio, S.; DeMontis, M.G.; Colzi, A.; Corsini, G.U.; Maggio, R. Reconstitution of functional dopamine D(2s) receptor by co-expression of amino- and carboxyl-terminal receptor fragments. Eur. J. Pharmacol. 2000, 397, 291–296. [Google Scholar] [CrossRef]
- Rossi, M.; Fasciani, I.; Marampon, F.; Maggio, R.; Scarselli, M. The first negative allosteric modulator for dopamine D2 and D3 receptors, SB269652 may lead to a new generation of antipsychotic drugs. Mol. Pharmacol. 2017, 91, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Maggio, R.; Scarselli, M.; Capannolo, M.; Millan, M.J. Novel dimensions of D3 receptor function: Focus on heterodimerisation, transactivation and allosteric modulation. Eur. Neuropsychopharmacol. 2015, 25, 1470–1479. [Google Scholar] [CrossRef]
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Carli, M.; Scarselli, M.; Maggio, R.; Rossi, M. Allosteric modulators of g protein-coupled dopamine and serotonin receptors: A new class of atypical antipsychotics. Pharmaceuticals 2020, 13, 388. [Google Scholar] [CrossRef]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013, 64, 393–406. [Google Scholar] [CrossRef]
- Seeman, P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin. Ther. Targets 2006, 10, 515–531. [Google Scholar] [CrossRef]
- Schmid, C.L.; Streicher, J.M.; Meltzer, H.Y.; Bohn, L.M. Clozapine acts as an agonist at serotonin 2A receptors to counter MK-801-induced behaviors through a βarrestin2-independent activation of akt. Neuropsychopharmacology 2014, 39, 1902–1913. [Google Scholar] [CrossRef]
- Fasciani, I.; Pietrantoni, I.; Rossi, M.; Mannoury la Cour, C.; Aloisi, G.; Marampon, F.; Scarselli, M.; Millan, M.J.; Maggio, R. Distinctive binding properties of the negative allosteric modulator, [3H]SB269,652, at recombinant dopamine D3 receptors. Eur. J. Pharmacol. 2018, 819, 181–189. [Google Scholar] [CrossRef]
- Carli, M.; Kolachalam, S.; Aringhieri, S.; Rossi, M.; Giovannini, L.; Maggio, R.; Scarselli, M. Dopamine D2 Receptors Dimers: How can we Pharmacologically Target Them? Curr. Neuropharmacol. 2017, 16, 222–230. [Google Scholar] [CrossRef]
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the most efficacious antipsychotic for activating ERK 1/2 kinases: Role of 5-HT2A receptor agonism. Eur. Neuropsychopharmacol. 2017, 27, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. (GLP-1): A gut hormone of potential interest in the treatment of diabetes. BioEssays 1998, 1, 642–651. [Google Scholar] [CrossRef]
- Ahrén, B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2009, 8, 369–385. [Google Scholar] [CrossRef]
- Sørensen, H.; Winzell, M.S.; Brand, C.L.; Fosgerau, K.; Gelling, R.W.; Nishimura, E.; Ahren, B. Glucagon receptor knockout mice display increased insulin sensitivity and impaired β-cell function. Diabetes 2006, 55, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Gavrilova, O.; Jeon, J.; Pack, S.; Jou, W.; Cui, Y.; Li, J.H.; Wess, J. Beneficial metabolic effects of M3 muscarinic acetylcholine receptor deficiency. Cell Metab. 2006, 4, 363–375. [Google Scholar] [CrossRef]
- de Azua, I.R.; Gautam, D.; Guettier, J.M.; Wess, J. Novel insights into the function of β-cell M3 muscarinic acetylcholine receptors: Therapeutic implications. Trends Endocrinol. Metab. 2011, 22, 74–80. [Google Scholar] [CrossRef]
- Gromada, J. The free fatty acid receptor GPR40 generates excitement in pancreatic β-cells. Endocrinology 2006, 147, 672–673. [Google Scholar] [CrossRef]
- Di Cairano, E.S.; Moretti, S.; Marciani, P.; Sacchi, V.F.; Castagna, M.; Davalli, A.; Folli, F.; Perego, C. Neurotransmitters and Neuropeptides: New Players in the Control of Islet of Langerhans’ Cell Mass and Function. J. Cell. Physiol. 2016, 231, 756–767. [Google Scholar] [CrossRef]
- Rubi, B.; Ljubicic, S.; Pournourmohammadi, S.; Carobbio, S.; Armanet, M.; Bartley, C.; Maechler, P. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 2005, 280, 36824–36832. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.; Maffei, A.; Freeby, M.; Burroughs, S.; Freyberg, Z.; Javitch, J.; Leibel, R.L.; Harris, P.E. Dopamine-mediated autocrine inhibitory circuit regulating human insulin secretion in vitro. Mol. Endocrinol. 2012, 26, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shang, J.; Feng, Y.; Thompson, C.M.; Horwitz, S.; Thompson, J.R.; MacIntyre, E.D.; Thornberry, N.A.; Chapman, K.; Zhou, Y.P.; et al. Identification of glucose-dependant insulin secretion targets in pancreatic β cells by combining defined-mechanism compound library screening and siRNA gene silencing. J. Biomol. Screen. 2008, 13, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Ustione, A.; Piston, D.W.; Harris, P.E. Minireview: Dopaminergic regulation of insulin secretion from the pancreatic islet. Mol. Endocrinol. 2013, 27, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Farino, Z.J.; Morgenstern, T.J.; Maffei, A.; Quick, M.; De Solis, A.J.; Wiriyasermkul, P.; Freyberg, R.J.; Aslanoglou, D.; Sorisio, D.; Inbar, B.P.; et al. New roles for dopamine D2 and D3 receptors in pancreatic beta cell insulin secretion. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Bennet, H.; Balhuizen, A.; Medina, A.; Dekker Nitert, M.; Ottosson Laakso, E.; Essén, S.; Spégel, P.; Storm, P.; Krus, U.; Wierup, N.; et al. Altered serotonin (5-HT) 1D and 2A receptor expression may contribute to defective insulin and glucagon secretion in human type 2 diabetes. Peptides 2015, 71, 113–120. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, Y.; Zhou, W.; Gao, L.; Yuan, L.; Han, X. Serotonin Receptor 2C and Insulin Secretion. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Cataldo Bascuñan, L.R.; Lyons, C.; Bennet, H.; Artner, I.; Fex, M. Serotonergic regulation of insulin secretion. Acta Physiol. 2019, 225, 1–10. [Google Scholar] [CrossRef]
- Ohta, Y.; Kosaka, Y.; Kishimoto, N.; Wang, J.; Smith, S.B.; Honig, G.; Kim, H.; Gasa, R.M.; Neubauer, N.; Liou, A.; et al. Convergence of the insulin and serotonin programs in the pancreatic β-cell. Diabetes 2011, 60, 3208–3216. [Google Scholar] [CrossRef]
- Cataldo, L.R.; Mizgier, M.L.; Sagua, R.B.; Jaña, F.; Cárdenas, C.; Llanos, P.; Busso, D.; Olmos, P.; Galgani, J.E.; Santos, J.L.; et al. Prolonged Activation of the Htr2b Serotonin Receptor Impairs Glucose Stimulated Insulin Secretion and Mitochondrial Function in MIN6 Cells. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Albrechtsen, N.J.W. Glucagon receptor signaling and glucagon resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Lv, Y.; Liang, T.; Wang, G.; Li, Z. Ghrelin, A gastrointestinal hormone, regulates energy balance and lipid metabolism. Biosci. Rep. 2018, 38, 1–13. [Google Scholar] [CrossRef]
- Sharara-Chami, R.I.; Zhou, Y.; Ebert, S.; Pacak, K.; Ozcan, U.; Majzoub, J.A. Epinephrine deficiency results in intact glucose counter-regulation, severe hepatic steatosis and possible defective autophagy in fasting mice. Int. J. Biochem. Cell Biol. 2012, 44, 905–913. [Google Scholar] [CrossRef]
- Tao, X.; Hu, Y.; Li, L.; Xu, R.; Fu, J.; Tong, Q.; Fu, Q. Genetic deletion of β 2 adrenergic receptors exacerbates hepatocellular lipid accumulation in high-fat diet mice. Biochem. Biophys. Res. Commun. 2019, 511, 73–78. [Google Scholar] [CrossRef]
- Hampson, L.J.; Agius, L. Acetylcholine exerts additive and permissive but not synergistic effects with insulin on glycogen synthesis in hepatocytes. FEBS Lett. 2007, 581, 3955–3960. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Chu, X.; Wood, C.; Bartoli, M.; Khurana, S. M3 muscarinic receptor activation reduces hepatocyte lipid accumulation via CaMKKβ/AMPK pathway. Biochem. Pharmacol. 2019, 169, 113613. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Gautam, D.; Han, S.J.; Guettier, J.M.; Cui, Y.; Lu, H.; Deng, C.; O’Hare, J.; Jou, W.; Gavrilova, O.; et al. Hepatic muscarinic acetylcholine receptors are not critically involved in maintaining glucose homeostasis in mice. Diabetes 2009, 58, 2776–2787. [Google Scholar] [CrossRef]
- Yabut, J.M.; Crane, J.D.; Green, A.E.; Keating, D.J.; Khan, W.I.; Steinberg, G.R. Emerging Roles for Serotonin in Regulating Metabolism: New Implications for an Ancient Molecule. Endocr. Rev. 2019, 40, 1092–1107. [Google Scholar] [CrossRef]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38δ in Insulin Secretion and Glucose Homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef]
- Osawa, Y.; Kanamori, H.; Seki, E.; Hoshi, M.; Ohtaki, H.; Yasuda, Y.; Ito, H.; Suetsugu, A.; Nagaki, M.; Moriwaki, H.; et al. L-tryptophan-mediated enhancement of susceptibility to nonalcoholic fatty liver disease is dependent on the mammalian target of rapamycin. J. Biol. Chem. 2011, 286, 34800–34808. [Google Scholar] [CrossRef]
- Baldassano, S.; Gasbjerg, L.S.; Kizilkaya, H.S.; Rosenkilde, M.M.; Holst, J.J.; Hartmann, B. Increased body weight and fat mass after subchronic GIP receptor antagonist, but not GLP-2 receptor antagonist, administration in rats. Front. Endocrinol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Thondam, S.K.; Cuthbertson, D.J.; Wilding, J.P.H. The influence of Glucose-dependent Insulinotropic Polypeptide (GIP) on human adipose tissue and fat metabolism: Implications for obesity, type 2 diabetes and Non-Alcoholic Fatty Liver Disease (NAFLD). Peptides 2020, 125, 170208. [Google Scholar] [CrossRef]
- Song, T.; Yang, Y.; Zhou, Y.; Wei, H.; Peng, J. GPR120: A critical role in adipogenesis, inflammation, and energy metabolism in adipose tissue. Cell. Mol. Life Sci. 2017, 74, 2723–2733. [Google Scholar] [CrossRef]
- Jönsson, C.; Batista, A.P.C.; Kjølhede, P.; Strålfors, P. Insulin and β-adrenergic receptors mediate lipolytic and anti-lipolytic signalling that is not altered by type 2 diabetes in human adipocytes. Biochem. J. 2019, 476, 2883–2908. [Google Scholar] [CrossRef]
- Luque, M.A.; González, N.; Márquez, L.; Acitores, A.; Redondo, A.; Morales, M.; Valverde, I.; Villanueva-Peñacarrillo, M.L. Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human myocytes. J. Endocrinol. 2002, 173, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liao, J.; Li, N.; Zhou, C.; Liu, Q.; Wang, G.; Zhang, R.; Zhang, S.; Lin, L.; Chen, K.; et al. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc. Natl. Acad. Sci. USA 2007, 104, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Jocken, J.W.E.; Blaak, E.E. Catecholamine-induced lipolysis in adipose tissue and skeletal muscle in obesity. Physiol. Behav. 2008, 94, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Maillet, C.M.; Garber, A.J. Adrenergic and serotonergic regulation of skeletal muscle metabolism in the rat: Specificity of the serotonin- and isoproterenol-stimulable adenylyl cyclase in sarcolemma. Mol. Cell. Endocrinol. 1986, 48, 179–190. [Google Scholar] [CrossRef]
- Wang, K.Y.; Tanimoto, A.; Yamada, S.; Guo, X.; Ding, Y.; Watanabe, T.; Watanabe, T.; Kohno, K.; Hirano, K.I.; Tsukada, H.; et al. Histamine regulation in glucose and lipid metabolism via histamine receptors: Model for nonalcoholic steatohepatitis in mice. Am. J. Pathol. 2010, 177, 713–723. [Google Scholar] [CrossRef]
- Grajales, D.; Ferreira, V.; Valverde, Á.M. Second-Generation Antipsychotics and Dysregulation of Glucose Metabolism: Beyond Weight Gain. Cells 2019, 8, 1336. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 2012, 9, 25–53. [Google Scholar] [CrossRef]
- Tabarean, I.V. Histamine receptor signaling in energy homeostasis. Neuropharmacology 2016, 106, 13–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vehof, J.; Risselada, A.J.; Al Hadithy, A.F.Y.; Burger, H.; Snieder, H.; Wilffert, B.; Arends, J.; Wunderink, L.; Knegtering, H.; Wiersma, D.; et al. Association of genetic variants of the histamine H1 and muscarinic M3 receptors with BMI and HbA1c values in patients on antipsychotic medication. Psychopharmacology 2011, 216, 257–265. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Kirk, S.L. Metabolic side effects of antipsychotic drug treatment—Pharmacological mechanisms. Pharmacol. Ther. 2010, 125, 169–179. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Xu, Y.; Jones, J.E.; Lauzon, D.A.; Anderson, J.G.; Balthasar, N.; Heisler, L.K.; Zinn, A.R.; Lowell, B.B.; Elmquist, J.K. A serotonin and melanocortin circuit mediates D-fenfluramine anorexia. J. Neurosci. 2010, 30, 14630–14634. [Google Scholar] [CrossRef]
- Lam, D.D.; Garfield, A.S.; Marston, O.J.; Shaw, J.; Heisler, L.K. Brain serotonin system in the coordination of food intake and body weight. Pharmacol. Biochem. Behav. 2010, 97, 84–91. [Google Scholar] [CrossRef]
- Zhou, L.; Sutton, G.M.; Rochford, J.J.; Semple, R.K.; Lam, D.D.; Oksanen, L.J.J.; Thornton-Jones, Z.D.; Clifton, P.G.; Yueh, C.Y.; Evans, M.L.; et al. Serotonin 2C Receptor Agonists Improve Type 2 Diabetes via Melanocortin-4 Receptor Signaling Pathways. Cell Metab. 2007, 6, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Bickerdike, M. 5-HT2C Receptor Agonists as Potential Drugs for the Treatment of Obesity. Curr. Top. Med. Chem. 2005, 3, 885–897. [Google Scholar] [CrossRef]
- Fernø, J.; Varela, L.; Skrede, S.; Vázquez, M.J.; Nogueiras, R.; Diéguez, C.; Vidal-Puig, A.; Steen, V.M.; López, M. Olanzapine-induced hyperphagia and weight gain associate with orexigenic hypothalamic neuropeptide signaling without concomitant AMPK phosphorylation. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Vatamaniuk, M.Z.; Horyn, O.V.; Vatamaniuk, O.K.; Doliba, N.M. Acetylcholine affects rat liver metabolism via type 3 muscarinic receptors in hepatocytes. Life Sci. 2003, 72, 1871–1882. [Google Scholar] [CrossRef]
- Breier, A.; Wolkowitz, O.M.; Roy, A.; Potter, W.Z.; Pickar, D. Plasma norepinephrine in chronic schizophrenia. Am. J. Psychiatry 1990, 147, 1467–1470. [Google Scholar] [CrossRef]
- Savoy, Y.E.; Ashton, M.A.; Miller, M.W.; Nedza, F.M.; Spracklin, D.K.; Hawthorn, M.H.; Rollema, H.; Matos, F.F.; Hajos-Korcsok, E. Differential effects of various typical and atypical antipsychotics on plasma glucose and insulin levels in the mouse: Evidence for the involvement of sympathetic regulation. Schizophr. Bull. 2010, 36, 410–418. [Google Scholar] [CrossRef]
- Lian, J.; Huang, X.F.; Pai, N.; Deng, C. Betahistine ameliorates olanzapine-induced weight gain through modulation of histaminergic, NPY and AMPK pathways. Psychoneuroendocrinology 2014, 48, 77–86. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Kim, S.F.; Huang, A.S.; Snowman, A.M.; Teuscher, C.; Snyder, S.H. Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 3456–3459. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Chaussade, C.; Vickers, M.; Jensen, J.; Shepherd, P.R. Atypical antipsychotic drugs induce derangements in glucose homeostasis by acutely increasing glucagon secretion and hepatic glucose output in the rat. Diabetologia 2008, 51, 2309–2317. [Google Scholar] [CrossRef]
- Budick-Harmelin, N.; Anavi, S.; Madar, Z.; Tirosh, O. Fatty acids-stress attenuates gluconeogenesis induction and glucose production in primary hepatocytes. Lipids Health Dis. 2012, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- BONACCORSI, A.; GARATTINI, S.; JORI, A. Studies on the Hyperglycaemia Induced By Chlorpromazine in Rats. Br. J. Pharmacol. Chemother. 1964, 23, 93–100. [Google Scholar] [CrossRef]
- Manu, P.; Correll, C.U.; Wampers, M.; van Winkel, R.; Yu, W.; Shiffeldrim, D.; Kane, J.M.; De Hert, M. Insulin secretion in patients receiving clozapine, olanzapine, quetiapine and risperidone. Schizophr. Res. 2013, 143, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Houseknecht, K.L.; Robertson, A.S.; Zavadoski, W.; Gibbs, E.M.; Johnson, D.E.; Rollema, H. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: Implications for adverse metabolic effects. Neuropsychopharmacology 2007, 32, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Park, J.; Lee, S.Y.; Hwang, I.; Kim, J.B.; Park, T.S.; Lee, H.J.; Koo, S.H. Atypical antipsychotic drugs perturb AMPK-dependent regulation of hepatic lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhuang, X. Atypical antipsychotics-induced metabolic syndrome and nonalcoholic fatty liver disease: A critical review. Neuropsychiatr. Dis. Treat. 2019, 15, 2087–2099. [Google Scholar] [CrossRef]
- Cai, H.L.; Tan, Q.Y.; Jiang, P.; Dang, R.L.; Xue, Y.; Tang, M.M.; Xu, P.; Deng, Y.; Li, H.D.; Yao, J.K. A potential mechanism underlying atypical antipsychotics-induced lipid disturbances. Transl. Psychiatry 2015, 5. [Google Scholar] [CrossRef]
- Siafis, S.; Tzachanis, D.; Samara, M.; Papazisis, G. Antipsychotic Drugs: From Receptor-binding Profiles to Metabolic Side Effects. Curr. Neuropharmacol. 2017, 16, 1210–1223. [Google Scholar] [CrossRef]
- Melkersson, K.; Khan, A.; Hilding, A.; Hulting, A.L. Different effects of antipsychotic drugs on insulin release in vitro. Eur. Neuropsychopharmacol. 2001, 11, 327–332. [Google Scholar] [CrossRef]
- Melkersson, K. Clozapine and olanzapine, but not conventional antipsychotics, increase insulin release in vitro. Eur. Neuropsychopharmacol. 2004, 14, 115–119. [Google Scholar] [CrossRef]
- Sussman, K.E.; Pollard, H.B.; Leitner, J.W.; Nesher, R.; Adler, J.; Cerasi, E. Differential control of insulin secretion and somatostatin-receptor recruitment in isolated pancreatic islets. Biochem. J. 1983, 214, 225–230. [Google Scholar] [CrossRef]
- Teff, K.L.; Rickels, M.R.; Grudziak, J.; Fuller, C.; Nguyen, H.L.; Rickels, K. Antipsychotic-induced insulin resistance and postprandial hormonal dysregulation independent of weight gain or psychiatric disease. Diabetes 2013, 62, 3232–3240. [Google Scholar] [CrossRef]
- Rickels, M.R.; Perez, E.M.; Peleckis, A.J.; Alshehabi, E.; Nguyen, H.L.; Stefanovski, D.; Rickels, K.; Teff, K.L. Contribution of parasympathetic muscarinic augmentation of insulin secretion to olanzapine-induced hyperinsulinemia. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E250–E257. [Google Scholar] [CrossRef]
- Weston-Green, K.; Huang, X.F.; Deng, C. Second generation antipsychotic-induced type 2 diabetes: A role for the muscarinic M3 receptor. CNS Drugs 2013, 27, 1069–1080. [Google Scholar] [CrossRef]
- Nagata, M.; Yokooji, T.; Nakai, T.; Miura, Y.; Tomita, T.; Taogoshi, T.; Sugimoto, Y.; Matsuo, H. Blockade of multiple monoamines receptors reduce insulin secretion from pancreatic β-cells. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Gonçalves, P.; Araújo, J.R.; Martel, F. Antipsychotics-induced metabolic alterations: Focus on adipose tissue and molecular mechanisms. Eur. Neuropsychopharmacol. 2015, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Vestri, H.S.; Maianu, L.; Moellering, D.R.; Garvey, W.T. Atypical antipsychotic drugs directly impair insulin action in adipocytes: Effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology 2007, 32, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Gaspar, P.A.; Silva, H.; Risco, L.; Arena, P.; Cubillos-Robles, K.; Jara, B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol. Res. 2015, 101, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, V.L.; Singareddy, R.; Mauger, D.; Lynch, C.J. A double blind, placebo-controlled, randomized crossover study of the acute metabolic effects of olanzapine in healthy volunteers. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Nicol, G.E.; Yingling, M.D.; Flavin, K.S.; Schweiger, J.A.; Patterson, B.W.; Schechtman, K.B.; Newcomer, J.W. Metabolic effects of antipsychotics on adiposity and insulin sensitivity in youths a randomized clinical trial. JAMA Psychiatry 2018, 75, 788–796. [Google Scholar] [CrossRef]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. The Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- Jassim, G.; Skrede, S.; Vázquez, M.J.; Wergedal, H.; Vik-Mo, A.O.; Lunder, N.; Diéguez, C.; Vidal-Puig, A.; Berge, R.K.; López, M.; et al. Acute effects of orexigenic antipsychotic drugs on lipid and carbohydrate metabolism in rat. Psychopharmacology 2012, 219, 783–794. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, X.; Zuberi, A.; Hwang, D.; Quon, M.J.; Lefevre, M.; Ye, J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol. Endocrinol. 2004, 18, 2024–2034. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef]
- Hemmrich, K.; Gummersbach, C.; Pallua, N.; Luckhaus, C.; Fehsel, K. Clozapine enhances differentiation of adipocyte progenitor cells. Mol. Psychiatry 2006, 11, 980–981. [Google Scholar] [CrossRef]
- Barateiro, A.; Mahú, I.; Domingos, A.I. Leptin resistance and the neuro-adipose connection. Front. Endocrinol. 2017, 8, 8–11. [Google Scholar] [CrossRef]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Portincasa, P.; Colina, I.; Gómez-Ambrosi, J. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhou, X.; Huang, X.; Wang, C.; Li, Y. The IRS/PI3K/Akt signaling pathway mediates olanzapine-induced hepatic insulin resistance in male rats. Life Sci. 2019, 217, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, K.J.; Ward, K.M.; Sanders, E.J.; Howlett, B.H.; Seyoum, B.; Yi, Z. Atypical antipsychotics and the human skeletal muscle lipidome. Metabolites 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Cipriani, A.; Spineli, L.; Mavridis, D.; Örey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962. [Google Scholar] [CrossRef]
- Bai, Y.M.; Chen, J.Y.; Chen, T.T.; Lin, C.Y.; Chou, P.; Su, T.P.; Lin, C.C. Weight gain with clozapine: 8-year cohort naturalistic study among hospitalized Chinese schizophrenia patients. Schizophr. Res. 2009, 108, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Rummel-Kluge, C.; Komossa, K.; Schwarz, S.; Hunger, H.; Schmid, F.; Lobos, C.A.; Kissling, W.; Davis, J.M.; Leucht, S. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: A systematic review and meta-analysis. Schizophr. Res. 2010, 123, 225–233. [Google Scholar] [CrossRef]
- Bai, Y.M.; Lin, C.-C.; Chen, J.-Y.; Chen, T.T.; Su, T.-P.; Chou, P. Association of weight gain and metabolic syndrome in patients taking clozapine: An 8-year cohort study. J. Clin. Psychiatry 2011, 72, 751–756. [Google Scholar] [CrossRef]
- Maayan, L.; Correll, C.U. Management of antipsychotic-related weight gain. Expert Rev. Neurother. 2010, 10, 1175–1200. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Stroup, T.S.; McEvoy, J.P.; Swartz, M.S.; Rosenheck, R.A.; Perkins, D.O.; Keefe, R.S.E.; Davis, S.M.; Davis, C.E.; Lebowitz, B.D.; et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005, 353, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Cohen, M.; Panagides, J. Efficacy and tolerability of asenapine in acute schizophrenia: A placebo- and risperidone-controlled trial. J. Clin. Psychiatry 2007, 68, 1492–1500. [Google Scholar] [CrossRef]
- Nasrallah, H.A. Atypical antipsychotic-induced metabolic side effects: Insights from receptor-binding profiles. Mol. Psychiatry 2008, 13, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Fountaine, R.J.; Taylor, A.E.; Mancuso, J.P.; Greenway, F.L.; Byerley, L.O.; Smith, S.R.; Most, M.M.; Fryburg, D.A. Increased food intake and energy expenditure following administration of olanzapine to healthy men. Obesity (Silver Spring) 2010, 18, 1646–1651. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, J. Il Metabolic disturbances independent of body mass in patients with schizophrenia taking atypical antipsychotics. Psychiatry Investig. 2015, 12, 242–248. [Google Scholar] [CrossRef]
- Newcomer, J.W. Metabolic considerations in the use of antipsychotic medications: A review of recent evidence. J. Clin. Psychiatry 2007, 68 (Suppl. 1), 20–27. [Google Scholar]
- Kessing, L.V.; Thomsen, A.F.; Mogensen, U.B.; Andersen, P.K. Treatment with antipsychotics and the risk of diabetes in clinical practice. Br. J. Psychiatry 2010, 197, 266–271. [Google Scholar] [CrossRef]
- Hirsch, L.; Yang, J.; Bresee, L.; Jette, N.; Patten, S.; Pringsheim, T. Second-Generation Antipsychotics and Metabolic Side Effects: A Systematic Review of Population-Based Studies. Drug Saf. 2017, 40, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Ulcickas Yood, M.; DeLorenze, G.N.; Quesenberry, C.P.; Oliveria, S.A.; Tsai, A.L.; Kim, E.; Cziraky, M.J.; McQuade, R.D.; Newcomer, J.W.; L’Italien, G.J. Association between second-generation antipsychotics and newly diagnosed treated diabetes mellitus: Does the effect differ by dose? BMC Psychiatry 2011, 11, 197. [Google Scholar] [CrossRef]
- Young, S.L.; Taylor, M.; Lawrie, S.M. “first do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J. Psychopharmacol. 2015, 29, 353–362. [Google Scholar] [CrossRef]
- Saari, K.; Jokelainen, J.; Veijola, J.; Koponen, H.; Jones, P.B.; Savolainen, M.; Järvelin, M.-R.; Lauren, L.; Isohanni, M.; Lindeman, S. Serum lipids in schizophrenia and other functional psychoses: A general population northern Finland 1966 birth cohort survey. Acta Psychiatr. Scand. 2004, 110, 279–285. [Google Scholar] [CrossRef]
- Mackin, P.; Bishop, D.R.; Watkinson, H.M.O. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry 2007, 7, 1–6. [Google Scholar] [CrossRef]
- Tsan, J.Y.; Stock, E.M.; Gonzalez, J.M.; Greenawalt, D.S.; Zeber, J.E.; Rouf, E.; Copeland, L.A. Mortality and guideline-concordant care for older patients with schizophrenia: A retrospective longitudinal study. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, A.; Faulkner, G.; Cohn, T.; Remington, G. Antipsychotic switching for people with schizophrenia who have neuroleptic-induced weight or metabolic problems. Cochrane Database Syst. Rev. 2010, CD006629. [Google Scholar] [CrossRef] [PubMed]
- Corponi, F.; Fabbri, C.; Bitter, I.; Montgomery, S.; Vieta, E.; Kasper, S.; Pallanti, S.; Serretti, A. Novel antipsychotics specificity profile: A clinically oriented review of lurasidone, brexpiprazole, cariprazine and lumateperone. Eur. Neuropsychopharmacol. 2019, 29, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, S.A.; Laudani, S.; Contarini, G.; De Luca, A.; Geraci, F.; Managò, F.; Papaleo, F.; Salomone, S.; Drago, F.; Leggio, G.M. Dopamine, cognitive impairments and second-generation antipsychotics: From mechanistic advances to more personalized treatments. Pharmaceuticals 2020, 13, 365. [Google Scholar] [CrossRef]
- Caraci, F.; Santagati, M.; Caruso, G.; Cannavò, D.; Leggio, G.M.; Salomone, S.; Drago, F. New antipsychotic drugs for the treatment of agitation and psychosis in Alzheimer’s disease: Focus on brexpiprazole and pimavanserin. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Citrome, L.; Vreeland, B. Schizophrenia, obesity, and antipsychotic medications: What can we do? Postgrad. Med. 2008, 120, 18–33. [Google Scholar] [CrossRef]
- Bonfioli, E.; Berti, L.; Goss, C.; Muraro, F.; Burti, L. Health promotion lifestyle interventions for weight management in psychosis: A systematic review and meta-analysis of randomised controlled trials. BMC Psychiatry 2012, 12. [Google Scholar] [CrossRef]
- Ball, M.P.; Coons, V.B.; Buchanan, R.W. A program for treating olanzapine-related weight gain. Psychiatr. Serv. 2001, 52, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.A.; Cottingham, E.M.; Barton, B.A. Metformin for weight loss in pediatric patients taking psychotropic drugs. Am. J. Psychiatry 2002, 159, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Littrell, K.H.; Hilligoss, N.M.; Kirshner, C.D.; Petty, R.G.; Johnson, C.G. The effects of an educational intervention on antipsychotic-induced weight gain. J. Nurs. Scholarsh. Off. Publ. Sigma Theta Tau Int. Honor Soc. Nurs. 2003, 35, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.; Castrogiovanni, S.; Simoncini, M.; Iovieno, N.; Miniati, M.; Rossi, A.; Dell’Agnello, G.; Fagiolini, A.; Donda, P.; Cassano, G.B. Effects of an educational intervention on weight gain in patients treated with antipsychotics. J. Clin. Psychopharmacol. 2006, 26, 462–466. [Google Scholar] [CrossRef]
- Curtis, J.; Watkins, A.; Rosenbaum, S.; Teasdale, S.; Kalucy, M.; Samaras, K.; Ward, P.B. Evaluating an individualized lifestyle and life skills intervention to prevent antipsychotic-induced weight gain in first-episode psychosis. Early Interv. Psychiatry 2016, 10, 267–276. [Google Scholar] [CrossRef]
- Weber, M.; Wyne, K. A cognitive/behavioral group intervention for weight loss in patients treated with atypical antipsychotics. Schizophr. Res. 2006, 83, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A. Metabolic changes associated with antipsychotic use. Prim. Care Companion J. Clin. Psychiatry 2002, 4, 239. [Google Scholar]
- Boyle, J.G.; Logan, P.J.; Jones, G.C.; Small, M.; Sattar, N.; Connell, J.M.C.; Cleland, S.J.; Salt, I.P. AMP-activated protein kinase is activated in adipose tissue of individuals with type 2 diabetes treated with metformin: A randomised glycaemia-controlled crossover study. Diabetologia 2011, 54, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Jesus, C.; Jesus, I.; Agius, M. A review of the evidence for the use of metformin in the treatment of metabolic syndrome caused by antipsychotics. Psychiatr. Danub. 2015, 27 (Suppl. 1), S489–S491. [Google Scholar]
- Rado, J.; Von Ammon Cavanaugh, S. A naturalistic randomized placebo-controlled trial of extended-release metformin to prevent weight gain associated with olanzapine in a US community-dwelling population. J. Clin. Psychopharmacol. 2016, 36, 163–168. [Google Scholar] [CrossRef]
- Wu, R.R.; Zhao, J.P.; Guo, X.F.; He, Y.Q.; Fang, M.S.; Guo, W.B.; Chen, J.D.; Li, L.H. Metformin addition attenuates olanzapine-induced weight gain in drug-naive first-episode schizophrenia patients: A double-blind, placebo-controlled study. Am. J. Psychiatry 2008, 165, 352–358. [Google Scholar] [CrossRef]
- Baptista, T.; Rangel, N.; Fernández, V.; Carrizo, E.; El Fakih, Y.; Uzcátegui, E.; Galeazzi, T.; Gutiérrez, M.A.; Servigna, M.; Dávila, A.; et al. Metformin as an adjunctive treatment to control body weight and metabolic dysfunction during olanzapine administration: A multicentric, double-blind, placebo-controlled trial. Schizophr. Res. 2007, 93, 99–108. [Google Scholar] [CrossRef]
- Baptista, T.; Martínez, J.; Lacruz, A.; Rangel, N.; Beaulieu, S.; Serrano, A.; Arapé, Y.; Martinez, M.; De Mendoza, S.; Teneud, L.; et al. Metformin for prevention of weight gain and insulin resistance with olanzapine: A double-blind placebo-controlled trial. Can. J. Psychiatry 2006, 51, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Huang, M.-C.; Kao, C.-F.; Lin, S.-K.; Kuo, P.-H.; Chiu, C.-C.; Lu, M.-L. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: A 24-week, randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry 2013, 74, e424–e430. [Google Scholar] [CrossRef]
- Carrizo, E.; Fernández, V.; Connell, L.; Sandia, I.; Prieto, D.; Mogollón, J.; Valbuena, D.; Fernández, I.; Araujo de Baptista, E.; Baptista, T. Extended release metformin for metabolic control assistance during prolonged clozapine administration: A 14 week, double-blind, parallel group, placebo-controlled study. Schizophr. Res. 2009, 113, 19–26. [Google Scholar] [CrossRef]
- Baptista, T.; Rangel, N.; El Fakih, Y.; Uzcátegui, E.; Galeazzi, T.; Beaulieu, S.; Araujo De Baptista, E. Rosiglitazone in the assistance of metabolic control during olanzapine administration in schizophrenia: A pilot double-blind, placebo-controlled, 12-week trial. Pharmacopsychiatry 2009, 42, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.C.; Fan, X.; Sharma, B.; Copeland, P.M.; Borba, C.P.; Boxill, R.; Freudenreich, O.; Cather, C.; Eden Evins, A.; Goff, D.C. A double-blind, placebo-controlled trial of rosiglitazone for clozapine-induced glucose metabolism impairment in patients with schizophrenia. Acta Psychiatr. Scand. 2009, 119, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.R.; Vedtofte, L.; Jakobsen, M.S.L.; Jespersen, H.R.; Jakobsen, M.I.; Svensson, C.K.; Koyuncu, K.; Schjerning, O.; Oturai, P.S.; Kjaer, A.; et al. Effect of liraglutide treatment on prediabetes and overweight or obesity in clozapine- or olanzapine-treated patients with schizophrenia spectrum disorder: A randomized clinical trial. JAMA Psychiatry 2017, 74, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Mayfield, K.; Siskind, D.; Winckel, K.; Russell, A.W.; Kisely, S.; Smith, G.; Hollingworth, S. Glucagon-like peptide-1 agonists combating clozapine-associated obesity and diabetes. J. Psychopharmacol. 2016, 30, 227–236. [Google Scholar] [CrossRef]
- Boyda, H.N.; Procyshyn, R.M.; Pang, C.C.Y.; Hawkes, E.; Wong, D.; Jin, C.H.; Honer, W.G.; Barr, A.M. Metabolic Side-Effects of the Novel Second-Generation Antipsychotic Drugs Asenapine and Iloperidone: A Comparison with Olanzapine. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef]
- Farilla, L.; Hui, H.; Bertolotto, C.; Kang, E.; Bulotta, A.; Di Mario, U.; Perfetti, R. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 2002, 143, 4397–4408. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Lykkegaard, K.; Larsen, P.J.; Vrang, N.; Bock, C.; Bock, T.; Knudsen, L.B. The once-daily human GLP-1 analog, liraglutide, reduces olanzapine-induced weight gain and glucose intolerance. Schizophr. Res. 2008, 103, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Ishøy, P.L.; Knop, F.K.; Vilsbøll, T.; Glenthøj, B.Y.; Ebdrup, B.H. Sustained weight loss after treatment with a glucagon-like peptide-1 receptor agonist in an obese patient with schizophrenia and type 2 diabetes. Am. J. Psychiatry 2013, 170, 681–682. [Google Scholar] [CrossRef]
- Jackson, H.C.; Bearham, M.C.; Hutchins, L.J.; Mazurkiewicz, S.E.; Needham, A.M.; Heal, D.J. Investigation of the mechanisms underlying the hypophagic effects of the 5-HT and noradrenaline reuptake inhibitor, sibutramine, in the rat. Br. J. Pharmacol. 1997, 121, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Poyurovsky, M.; Isaacs, I.; Fuchs, C.; Schneidman, M.; Faragian, S.; Weizman, R.; Weizman, A. Attenuation of olanzapine-induced weight gain with reboxetine in patients with schizophrenia: A double-blind, placebo-controlled study. Am. J. Psychiatry 2003, 160, 297–302. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, M.; Wiles, P.G.; Wales, J.K. Fluoxetine in the Treatment of Obese Type 2 Diabetic Patients. Diabet. Med. 1994, 11, 105–110. [Google Scholar] [CrossRef]
- Floris, M.; Lejeune, J.; Deberdt, W. Effect of amantadine on weight gain during olanzapine treatment. Eur. Neuropsychopharmacol. 2001, 11, 181–182. [Google Scholar] [CrossRef]
- Deberdt, W.; Winokur, A.; Cavazzoni, P.A.; Trzaskoma, Q.N.; Carlson, C.D.; Bymaster, F.P.; Wiener, K.; Floris, M.; Breier, A. Amantadine for weight gain associated with olanzapine treatment. Eur. Neuropsychopharmacol. 2005, 15, 13–21. [Google Scholar] [CrossRef]
- Ben-Menachem, E.; Axelsen, M.; Johanson, E.H.; Stagge, A.; Smith, U. Predictors of weight loss in adults with topiramate-treated epilepsy. Obes. Res. 2003, 11, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Scaparrotta, A.; Agostinelli, S.; Di Pillo, S.; Chiarelli, F.; Grosso, S. Topiramate-induced weight loss: A review. Epilepsy Res. 2011, 95, 189–199. [Google Scholar] [CrossRef]
- McElroy, S.L.; Hudson, J.I.; Capece, J.A.; Beyers, K.; Fisher, A.C.; Rosenthal, N.R. Topiramate for the treatment of binge eating disorder associated with obesity: A placebo-controlled study. Biol. Psychiatry 2007, 61, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Feucht, M.; Hauser, E.; Freilinger, M.; Seidl, R. Changes in body mass index during long-term topiramate therapy in paediatric epilepsy patients—A retrospective analysis. Seizure 2004, 13, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, M.; Kuloglu, M.; Tezcan, E.; Ustundag, B. Nizatidine treatment and its relationship with leptin levels in patients with olanzapine-induced weight gain. Hum. Psychopharmacol. 2003, 18, 457–461. [Google Scholar] [CrossRef]
- Melkersson, K.I.; Hulting, A.L.; Brismar, K.E. Elevated levels of insulin, leptin, and blood lipids in olanzapine-treated patients with schizophrenia or related psychoses. J. Clin. Psychiatry 2000, 61, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-M.; Zhao, X.-M.; Deng, C.; Zeng, Y.-P.; Hu, C.-H. Simvastatin improves olanzapine-induced dyslipidemia in rats through inhibiting hepatic mTOR signaling pathway. Acta Pharmacol. Sin. 2019, 40, 1049–1057. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Rosendo, A.B.; Lima, L.O.; Dal-Pizzol, F.; Almeida, S. Lipid and C-reactive protein levels, cardiovascular disease risk factors and simvastatin treatment in brazilian individuals. Inflammation 2010, 33, 244–250. [Google Scholar] [CrossRef]
- Werneke, U.; Taylor, D.; Sanders, T.A.B. Options for pharmacological management of obesity in patients treated with atypical antipsychotics. Int. Clin. Psychopharmacol. 2002, 17, 145–160. [Google Scholar] [CrossRef]
- Kolachalam, S.; Aringhieri, S.; Scarselli, M. Therapeutic Drug Monitoring in Neuropsychiatric Disorders. In Brain and Heart Dynamics; Govoni, S., Politi, P., Vanoli, E., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–11. [Google Scholar] [CrossRef]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Kacirova, I.; Urinovska, R. Therapeutic drug monitoring of atypical antipsychotic drugs. Acta Pharm. 2014, 64, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Lako, I.M.; Van Den Heuvel, E.R.; Knegtering, H.; Bruggeman, R.; Taxis, K. Estimating dopamine d2 receptor occupancy for doses of 8 antipsychotics: A meta-analysis. J. Clin. Psychopharmacol. 2013, 33, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Alberati, D.; Moreau, J.L.; Lengyel, J.; Hauser, N.; Mory, R.; Borroni, E.; Pinard, E.; Knoflach, F.; Schlotterbeck, G.; Hainzl, D.; et al. Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology 2012, 62, 1152–1161. [Google Scholar] [CrossRef]
- Spina, E.; de Leon, J. Metabolic drug interactions with newer antipsychotics: A comparative review. Basic Clin. Pharmacol. Toxicol. 2007, 100, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.C.; Volonteri, L.S.; Colasanti, A.; Fiorentini, A.; De Gaspari, I.F.; Bareggi, S.R. Clinical pharmacokinetics of atypical antipsychotics: A critical review of the relationship between plasma concentrations and clinical response. Clin. Pharmacokinet. 2007, 46, 359–388. [Google Scholar] [CrossRef]
- Mandrioli, R.; Protti, M.; Mercolini, L. Metabolic Syndrome in Schizophrenia: Focus on the Role of Antipsychotic Medications and Indications for Therapeutic Drug Monitoring (TDM) Methods. In Frontiers in Clinical Drug Research—Diabetes and Obesity; Bentham Science: Sharjah, United Arab Emirates, 2020; pp. 1–65. ISBN 9781681087535. [Google Scholar]
- Pouget, J.G.; Shams, T.A.; Tiwari, A.K.; Müller, D.J. Pharmacogenetics and outcome with antipsychotic drugs. Dialogues Clin. Neurosci. 2014, 16, 555–566. [Google Scholar] [CrossRef]
- Thorn, C.F.; Aklillu, E.; Klein, T.E.; Altman, R.B. PharmGKB summary: Very important pharmacogene information for CYP1A2. Pharmacogenet. Genomics 2012, 22, 73–77. [Google Scholar] [CrossRef]
- Leggio, G.M.; Torrisi, S.A.; Mastrogiacomo, R.; Mauro, D.; Chisari, M.; Devroye, C.; Scheggia, D.; Nigro, M.; Geraci, F.; Pintori, N.; et al. The epistatic interaction between the dopamine D3 receptor and dysbindin-1 modulates higher-order cognitive functions in mice and humans. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Gressier, F.; Porcelli, S.; Calati, R.; Serretti, A. Pharmacogenetics of clozapine response and induced weight gain: A comprehensive review and meta-analysis. Eur. Neuropsychopharmacol. 2016, 26, 163–185. [Google Scholar] [CrossRef]
- de Leon, J.; Spina, E. Possible Pharmacodynamic and Pharmacokinetic Drug-Drug Interactions That Are Likely to Be Clinically Relevant and/or Frequent in Bipolar Disorder. Curr. Psychiatry Rep. 2018, 20, 17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| AAPs | Negative/Cognitive Symptoms Improvement | Weight Gain | Diabetes (T2D) | Dyslipidemia | Parkinsonism |
|---|---|---|---|---|---|
| Clozapine | ++++ | +++ | +++ | +++ | 0 |
| Olanzapine | +++ | +++ | +++ | +++ | +/− |
| Quetiapine | ++ | ++ | ++ | ++ | +/− |
| Risperidone | ++ | ++ | ++ | ++ | ++ |
| Amisulpride | ++ | ++ | ++ | ++ | ++ |
| Asenapine | ++ | + | + | + | + |
| Lurasidone | ++ | + | + | + | + |
| Ziprasidone | ++ | + | + | + | + |
| Aripiprazole | ++ | + | + | + | + |
| Added Treatment | AAP | Study | Pharmacological Response | Reference |
|---|---|---|---|---|
| Metformin | Olanzapine | Double-blind study: 25 patients randomly assigned to olanzapine plus metformin or olanzapine plus placebo for 24 weeks. | Metformin-group gained 3% of body weight compared to 7% for placebo group. BMI change was 0.85 in metformin-group vs. 2.02 in placebo-group. | [140] |
| 40 patients randomly assigned treatment with olanzapine plus metformin or olanzapine plus placebo for 12 weeks. | Metformin-group vs. placebo group resulted in lower increase in body weight (1.90 vs. 6.87), fasting insulin level (0.81 vs. 6.78) and insulin resistance index (0.22 vs. 1.49). | [141] | ||
| 80 patients taking olanzapine were randomized metformin or placebo comedication treatment for 12 weeks. | Body weight change was −1.4 in metformin-group and non-significant in placebo. Insulin resistance increased after placebo and not after metformin. | [142] | ||
| 40 patients taking olanzapine were assigned to metformin or placebo for 14 weeks. | No significant improvements for treated vs. placebo group. | [143] | ||
| Clozapine | 55 subjects taking clozapine for at least 3 months, were assigned to metformin or placebo for 24 weeks. | Body weight, BMI, fasting plasma glucose, HDL, insulin level had significant changes in the metformin-group. | [144] | |
| 61 patients treated with clozapine were randomly assigned to metformin extended release or placebo for 14 weeks. | Mean change in body weight was −1.87 kg for metformin-group and 0.16kg for placebo-group Insulin and the triglyceride/HDL ratio significantly decreased after metformin. | [145] | ||
| Rosiglitazione | Olanzapine | 12-week double blind study on 30 patients treated with olanzapine were allocated to rosiglitazione or placebo. | Insulin and the insulin resistance significantly decreased after rosiglitazone, while no effect was seen on weight gain and lipid profile. | [146] |
| Clozapine | 8-week double blind, placebo-controlled trial of rosiglitazone 4 mg/day in 18 clozapine-treated schizophrenia subjects with insulin resistance. | Non-significant improvement on glucose utilization and insulin sensitivity index; significant reduction in LDL level in rosiglitazone group. | [147] | |
| Liraglutide | Olanzapine or clozapine | 103 patients with a BMI > 27 and prediabetes randomly assigned to liraglutide or placebo for 16 weeks. | Liraglutide-group (63.8%) developed normal glucose tolerance compared with placebo-group (16%). Liraglutide induced a placebo-subtracted body weight loss of 5.3 kg. | [148] |
| Exenatide | Clozapine | 28 patients treated with clozapine randomly assigned to exenatide extended release or standard care for 24 weeks. | 6 people on exenatide achieved >5% weight loss vs. 1 usual care. Participants on exenatide had greater weight loss (−5.29 kg vs. −1.12 kg), BMI reduction (−1.78 vs. −0.39), reduced fasting glucose (−0.34 vs. 0.39) and HbA1c (−0.21 vs. 0.03) compared to control. | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals 2021, 14, 238. https://doi.org/10.3390/ph14030238
Carli M, Kolachalam S, Longoni B, Pintaudi A, Baldini M, Aringhieri S, Fasciani I, Annibale P, Maggio R, Scarselli M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals. 2021; 14(3):238. https://doi.org/10.3390/ph14030238
Chicago/Turabian StyleCarli, Marco, Shivakumar Kolachalam, Biancamaria Longoni, Anna Pintaudi, Marco Baldini, Stefano Aringhieri, Irene Fasciani, Paolo Annibale, Roberto Maggio, and Marco Scarselli. 2021. "Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences" Pharmaceuticals 14, no. 3: 238. https://doi.org/10.3390/ph14030238
APA StyleCarli, M., Kolachalam, S., Longoni, B., Pintaudi, A., Baldini, M., Aringhieri, S., Fasciani, I., Annibale, P., Maggio, R., & Scarselli, M. (2021). Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals, 14(3), 238. https://doi.org/10.3390/ph14030238