Abstract
Immunotherapy has been established as an important area in the therapy of malignant diseases. Immunogenicity sufficient for immune recognition and subsequent elimination can be bypassed by tumors through altered and/or reduced expression levels of major histocompatibility complex class I (MHC I) molecules. Natural killer (NK) cells can eliminate tumor cells in a MHC I antigen presentation-independent manner by an array of activating and inhibitory receptors, which are promising candidates for immunotherapy. Here we summarize the latest findings in recognizing and regulating MHC I molecules that affect NK cell surveillance of glioblastoma cells.
1. Introduction
The choice of cancer treatment depends on the type of tumor and its progression, as well as the age, comorbidity, and compliance of the patient. Often a combination of treatments is considered, such as surgery combined with chemotherapy and/or radiotherapy. For many patients, surgery is the chosen form of treatment, which is applied for solid tumors. In order to be successful, surgery must ensure that the cancerous tissue is completely removed. Aside from early diagnosis and treatment, some tumor cells can remain in the body, leading to recurrence of the tumor. These cells are known as cancer stem cells (CSCs). CSCs are tumor-initiation cells, since they represent a subpopulation of tumor cells with stem cell-like characteristics, involving proliferation, differentiation, and self-renewal. Therefore, it is crucial to eliminate these CSCs with a therapeutically useful approach to avoid a relapse [1].
Astrocytoma grade IV (glioblastoma) is one of the most aggressive tumors of the central nervous system and is characterized by high vascularity, fast proliferation, and diffuse invasion of glioblastoma cells into the surrounding healthy tissue. The present therapeutic treatment comprises surgical removal of the tumor followed by radio- and chemotherapy, with the latter using the standard chemotherapeutic drug temozolomide, which prolongs survival and quality of life for only a few months [2,3]. Glioblastoma cells and glioblastoma stem cells are highly resistant to treatment. Even though several novel treatment strategies have been established, more effective and more tolerable therapies are needed, particularly for patients at an advanced age. An encouraging anti-tumor strategy is immunotherapy, which includes a variety of therapeutic options by taking advantage of the immune system, such as application of monoclonal antibodies, immunomodulatory components, chimeric antigen receptor (CAR) T cells, the CAR natural killer (NK) cell, or combined treatment with NK cells and drugs [4,5,6,7].
In particular, NK cells are considered to be promising candidates in the field of immunotherapy. Furthermore, this review will focus on mechanisms that are associated with the interaction of inhibitory receptors on NK cells with major histocompatibility complex class I (MHC I) molecules and will summarize the regulation of MHC I, the distribution of inhibitory molecules on NK cell subsets, as well as the interaction of those molecules with MHC I on tumor cells.
2. Defective Antigen Presentation of Tumor Cells Is a Result of Immune Evasion
At an early stage, immune effector cells can eliminate nascent transformed cells. Intensive and invasive growth of the tumor can lead to minor disruptions of the surrounding tissue by induction of inflammatory processes, which can be monitored by the recruitment of immune cells, such as NK cells, dendritic cells (DCs), or macrophages followed by the infiltration of tumor-specific CD4+ and CD8+ T cells to the tumor site in order to attack the remaining antigen-bearing tumor cells [8]. As a result, interactions between the tumor and the immune system take place, creating a selective pressure in the tumor microenvironment (TME), finally leading to a malignant progression or a successful elimination of tumor cells by the immune system [9].
The antigen (antigenic peptide) binding protein MHC I (major histocompatibility complex class I) is assembled by a heavy chain (α1, α2, and α3) attached to β2-microglobulin and transported to the cell surface bearing the antigen. CD8+ T cells monitor these peptides bound to MHC I molecules, compared to CD4+ T cells which distinguish antigenic peptides bound to MHC II molecules; these are normally presented by professional antigen presenting cells (APCs). MHC I molecules are encoded by the three classical loci including human leukocyte antigen A (HLA-A), HLA-B, and HLA-C, which are related to the non-classical HLA molecules (HLA-E, HLA-F, and HLA-G) [10]. Like normal cells, tumor cells present antigens on the cell surface via MHC I, which can be tumor-specific- or tumor-associated antigens. The antigen-MHC I-complex is encountered by the T cell receptor (TCR) of CD8+ T cells; however, tumor cells may lose the ability to express cell surface MHC I molecules by intracellular arrest of MHC I or by acquiring defects in the antigen presentation pathways. Consequently, CD8+ T cells cannot recognize tumor cells and evade immune recognition [11]. This would be the case if there are no alternative paths for the immune system to eliminate tumor cells. Thus, NK cells attack cells lacking sufficient MHC I cell surface expression, which is also called missing self-recognition. In order to circumvent NK cell recognition and elimination, tumor cells encompass an additional immune evasion repertoire by instantly maintaining the presence of a limited set of cell surface MHC I molecules and escaping surveillance by NK cells [12].
Additionally, tumor cells have developed further mechanisms to prevent recognition and destruction by NK cells. An important factor is the tumor environment. The conditioning of suppressive cells, such as T regulatory cells (Tregs), M2 macrophages, and myeloid-derived suppressor cells (MDSCs) favor a suppressive environment leading to tumor immune evasion [13]. Additionally, hypoxia, which frequently occurs in tumors, leads to the inhibition of NK cell function due to the down-regulation of activating receptors on NK cells. The decrease of activating ligands on tumor cells, including natural killer group 2D ligands (NKG2DL), which can bind to the natural killer group 2D activating receptor (NKG2D) of NK cells, results in tumor escape from NK cell detection. NKG2DL, like the MHC class I chain-related protein A and B (MICA and MICB) or the UL 16 binding protein 1–6 (ULBP1–6), are highly expressed on the surface of stressed cells but also by tumor cells [14]. Interestingly, tumor cells are able to shed MICA and MICB from their surface [15], which can bind to the cell surface NKG2D and provokes an internalization of NKG2D into NK cells [16]. The reduction of cell surface NKG2D, as a result of internalization without activation of NK cells, is referred to as a desensitization of NK cells and represents an additional immune evasion strategy of tumor cells [17,18]. NK cells are activated when MHC I molecules are missing on the cell surface. Therefore, the expression pattern of MHC I molecules on gliomas, including glioblastoma cells, might be highly relevant for the use of NK cells for treatment. For instance, glioma stem cells, isolated from surgical samples, have been shown to downregulate MHC I molecules associated with an activation of the Wnt/β-catenin signaling pathway leading to evasion from CD8+ T cells [19,20]. NK cells can be recruited from the blood and cross the blood-brain barrier via the CXXXCL1 gradient secreted by neurons or CCL2 and CXCL10 released by microglia and astrocytes under inflammatory conditions [21]. Thereby, NK cells might be able to target glioma stem cells. However, the downregulation of MHC I is not generally true for all gliomas and is controversially described in the literature. Generally, reduced levels of MHC I depend on the patient sample or cell lines investigated in the assays [22]. In the next section, we will discuss the different NK cell subsets and their function.
3. Biology of NK Cell Subsets
Antibodies are divided into two domains, the Fab and Fc. While the Fab region of antibodies binds to a distinct antigen epitope, the Fc region is recognized by the Fc receptor. The Fc domain of IgG binds to FcγIII (CD16) which is found on NK cells and on immune cells of the innate and adaptive immune system. Simultaneous binding of cell-surface tumor antigens and CD16 of NK cells triggers antibody-dependent cell-mediated cytotoxicity (ADCC). The antibody forms a bridge between the tumor cell and the NK cell and generally enables NK cells to eradicate tumor cells [23]. A special feature of CD16 represents its ability to induce cytolytic activity of NK cells without the support of other costimulatory molecules. As a result of NK cell activation, NK cells secrete several cytokines, interferon (IFN)-γ, tumor necrosis factor-α (TNF-α), granulocyte macrophage colony-stimulating factor (GM-CSF), and chemokines (chemokine C-C motif ligand 1, CCL1, CCL2, CCL3, CCL4, CCL5, and CXCL8), thereby modulating the function of the innate and adaptive immune system. The release of cytoplasmic granules, containing perforin and granzymes, and the expression of death receptor ligands, such as FasL and TNF-related apoptosis-inducing ligand (TRAIL), can trigger apoptosis in target cells [12].
The identification of classical NK cells and their subsets has been well established over the last decades. Conventional markers used to determine NK cell subsets are CD3, CD56, and CD16 [24]. In the first approximation, NK cells can be defined by cell size and the missing expression of CD3. The cell-surface density of CD16 and CD56 can mainly be used for further characterization of NK cells into regulatory and cytotoxic subsets. The CD56+CD16™ NK cell subset has a regulatory capacity, CD56+CD16+ NK cells can be cytotoxic to target cells, and CD56™CD16+ NK cells are regarded as a kind of “defective” subset among NK cells, which have increased numbers of activating and inhibitory receptors but are deficient in their ability to be cytolytic. These cells are also impaired in cytokine production and proliferation [25,26].
Recent developments in the field of flow cytometry and phenotyping approaches by using mass cytometry by time-of-flight (CyTOF) have created a high possibility for deep profiling of NK cells. The simultaneous detection of the lysosomal marker CD107a, perforin to differentiate between newly synthesized and lytic granule-associated perforin, granzyme B, cytokines, proteolytically active proteases, and cell surface markers leads to a high-resolution phenotyping of NK cell biology [27,28,29,30]. Evidence can be gained from activation signals, like NKG2D and NKp46, which play crucial roles in anti-tumor immunity [31,32]. Moreover, several markers are used to describe the development of NK cells, among them CD27, CD57, CD62L, and CD127 [33,34], whereas CD57 is specifically used for the identification of terminally differentiated NK cells. For instance, CD57 is not expressed on NK cells from newborns, but the density of cell surface CD57 increases with age and correlates with the maturation process of NK cells, which thus have an impaired proliferation capability and increased expression of killer-cell immunoglobulin-like receptors (KIRs) [35]. Although surface markers can be used to categorize and characterize NK cells, activating and inhibitory receptors are crucial in determining the function and specific role of NK cells in the immune system and will be addressed in the following paragraph.
3.1. Activating and Inhibitory Receptors of NK Cells
In order to be able to recognize tumor cells, NK cells possess different surface receptors. The net sum of signals through these receptors determines the behavior of NK cells. These receptors are roughly divided into activating and inhibitory receptors and can recognize altered expression of self-proteins on target cells. Tumor cells have developed strategies to bypass NK cell activation by attenuating activating receptors or by stimulating inhibitory receptors [36]. The activating receptor binds to ligands whose expression is restricted to infected and transformed cells. These include NKG2D, DNAX accessory molecule 1 (DNAM-1), and the natural cytotoxicity receptors (NCR), such as p30, p44, and p46-related proteins (NKp30, NKp44, and NKp46). However, NKp44 is only expressed on previously activated NK cells and downregulation of NCRs correlates with lower cytolytic activity. Specifically, NKG2D plays a crucial role in the activation of NK cells by binding to MICA, MICB, and ULBP 1–6. In contrast to normal cells, NKG2D ligands are rarely expressed to avoid autoimmunity [37].
Transformed cells, but also virus-infected cells, down-regulate or alter the expression of cell surface MHC I molecules on tumor cells, which restricts CD8+ T cells in the recognition of tumor antigens. NK cells, on the other hand, express cell surface inhibitory receptors to specifically recognize MHC I molecules and inspect levels of MHC I which differ between normal and transformed cells. In other words, when MHC I molecules are absent, the inhibitory receptors discontinue the inhibition of the intracellular cascade of the activating receptor and provokes activation of NK cells [38,39]. Immature NK cells express the inhibitory receptor CD94:NKG2A heterodimer that binds specifically to HLA-E [40]. In comparison, late stage differentiated NK cells dominantly harbor the inhibitory receptors killer-cell immunoglobulin-like receptors (KIRs) on the cell surface [39,41]. However, the KIR receptor family represents members that have either inhibitory or activating functions. Hereby, the inhibitory KIRs show a higher avidity for MHC I compared to activating KIRs [42]. Activating and inhibitory function is in general based on the length of the cytoplasmic domain. KIRs with long cytoplasmic domains, except for KIR2DL4, show inhibitory responses including the recruitment of inhibitory protein tyrosine phosphatases in contrast to short cytoplasmic domains. Signal transduction of KIRs is mediated through an association with immunoreceptor tyrosine-based activation motif (ITAM), present in the cytoplasmic region of the DAP12 molecule [43]. Furthermore, KIRs are defined by their numbers of extracellular C2-type Ig-like domains. KIR2D contains two extracellular domains (D1 and D2) and KIR3D contains three, referred to as D0, D1, and D2 [44,45]. While ligands and functions of inhibitory KIRs are well defined and will be discussed later, a detailed classification is missing for activating KIRs. Chewning et al. showed that activating KIRs respond well when encountering allogeneic MHC I molecules [46]. Furthermore, NK cells expressing the activating molecule KIR2DS2 kill glioblastoma cells very efficiently in mice [47]. The expression of various inhibitory and activating receptors on NK cells can be linked with the function of the NK cell subset.
3.2. NK Cell Subsets and Their Function
The expression of KIR and its interaction with MHC I molecules differs between various subsets of NK cells. CD56bright NK cells show low levels of KIR or even lack KIR completely. These subsets show a decreased cytotoxic activity in ex vivo experiments and can be found in the lymph nodes and the secondary lymphoid tissue. This is in contrast to CD56dimCD16bright NK cells expressing a high amount of KIR and are predominantly found in the peripheral blood with high levels of baseline perforin [48,49].
Immature NK cells harbor NKG2A which binds with high affinity to HLA-E and is expressed early during NK cell maturation [40]. Further maturation leads to the acquisition and co-expression of KIRs, thereby discontinuing NKG2A expression, and mainly KIR inhibitory receptors are found on mature NK cells [50,51,52]. In order to avoid autoreactivity, all mature NK subsets express at least one kind of inhibitory receptor that can detect self-MHC I molecules [53]. One exception of cell surface expression of KIR is due to unlicensed NK cells, since unlicensed NK cells lack the expression of inhibitory self-receptors per definition [54]. In the next section, we will focus on how inhibitory molecules and MHC molecules interact on the molecular level.
3.3. Molecular Interaction of the Inhibitory Receptor with MHC I
NKG2A, a heterodimer complex of CD94-NKG2A, associates with HLA-E and binds to the C-terminal end of the antigen-binding cleft at an angle of around 70°. Petrie and colleagues showed, by analyzing the crystal structure of CD94-NKG2A and HLA-E, the specific interactions between these molecules. Thereby, CD94 attaches to the α1 domain, and NKG2A to the α2 of HLA-E; α1 and α2 generate the antigen-binding groove of HLA-E. This interaction is mainly directed by electrostatic interaction based on charge complementarity. However, CD94 association is also influenced by hydrogen bonds (Gln113 to Asp69, Asn170/Glu164 to Gln72, Thr146 to Glu89, and Ser143 to Arg79 of HLA-E), hydrophobic interactions (Phe114 to Ile73 and Leu162 to Val76 of HLA-E), and salt bridges (Arg171 to Asp69 and Asp163 to Arg75 of HLA-E). The crucial interaction between NKG2A and HLA-E are based on hydrogen bonds (Arg137 to Ser151 and Ser172 to His155 of HLA-E), salt bridges (Arg137 to Glu154 and Lys217 to Asp162 of HLA-E), and Van der Waals forces (Pro171 to His155 and Gln212/Lys217 to Ala158 of HLA-E) [55]. This interaction does not provoke a conformation change and was observed previously in thermodynamic studies [56].
KIRs with long cytoplasmic domains can interact with HLA-A, -B, or -C resulting in inhibition (except for KIR2DL4). HLA-A and HLA-B are recognized by KIR3D, while HLA-C binds to KIR2D [45]. The interaction between the highly polymorphic KIR family and HLA isotypes was previously described for KIR2D, which associates to HLA-C, as well as KIR3D and its association to HLA-B. The binding of KIR2D to HLA-C was determined by analyzing the crystal structure of KIR2DL2-HLA-Cw3 [57] and KIR2DL1-HLA-Cw4 [58]. KIR2D binds HLA-C in an orthogonal orientation with six loops from D1 and D2 at the α1 and α2 helix, directly contacting the peptide at positions 7 and 8 without leading to any significant conformational changes. The interface of the KIR/HLA interaction is dominated by a complementary charge, while disrupted salt bridges can diminish the binding. Differences between KIR2DL2 and KIR2DL1 can explain the allotype specificity. An intermolecular hydrogen bond participates between Lys44 (KIR2DL2) and Asn80 (HLA-Cw3), while KIR2DL1 hosts the Lys80 residue in a pocket specific to KIR2DL1 and enables interaction with HLA-C [59,60]. Furthermore, Vivian and colleagues revealed the interaction of KIR3DL1 with HLA-B*5701 by analyzing the crystal structure and reported that the contact between both molecules is a two-step process. The D0 domain acts as an innate sensor by binding a highly preserved region of HLA-B (β2-microglobulin) with limited polymorphism. The second contact is made by D1 and D2. The interaction of D2-HLA-B*5701 is mainly driven by “complementarity”, while the D1 contacts with HLA-B*5701 are suboptimal due to sequence variation of the polymorphic region of HLA-B [61].
Differences of KIRs and their subsequent inhibition or activation explain the function of various NK cell subsets. Such observations can be obtained from unlicensed NK. Unlicensed NK cells are dependent on an activating ligand to stimulate the NK response and are triggered as soon as an activating ligand is expressed on the target cell which makes unlicensed NK cells potent protectors during viral infections or for targeting tumor cells [62,63].
4. NK Cells as a Possible Immunotherapy Approach to Treat Glioblastoma
Although it has been shown that NK cells can successfully inhibit systemic metastasis of glioblastoma in a mouse model [64], the focus of NK cells to be assessed against glioblastoma is generally based on therapies affecting the tumor within the brain. Before discussing the abilities of NK cells for immunotherapy approaches, the question remains as to how NK cells migrate across the blood brain barrier and infiltrate the glioblastoma. In addition, only 2% of immune cells infiltrating glioblastoma are NK cells [65], NK cells follow a CX3CL1 gradient to cross the blood brain barrier by expression of CX3CR1 [66]. CX3CL1 is one of the chemokines with important anti-tumoral functions [67]. Additionally, tumor-targeted polymer-conjugated checkpoint inhibitors can be applied to increase the migration of NK cells, demonstrating an effective treatment of glioblastoma by initiating a systemic and local anti-tumor immune response [68]. Successful migration of NK cells to the site of the tumor is one important part for immunotherapy, further challenges have to be addressed for an efficacious treatment option and will be described in detail in the next paragraph.
The breakdown of immune surveillance, which might be the result of sustained immunological pressure on tumor cells developing mechanisms to become invisible to immune cells, is a critical point of tumor cells to escape immune detection [38]. Indeed, the microenvironment of glioblastoma is immunosuppressive and shows poor responses to immunotherapy [69]. This is a major challenge in developing approaches to support the immune system in combating glioblastoma cells. The engagement of NK cells for immunotherapy is very encouraging (Figure 1). However, the immunosuppressive environment and resident cells as well as the relative expression of MHC I molecules, particularly HLA-E, on gliomas have a strongly inhibitory function for NK cells [65,70,71]. Although various studies have shown that NK cells efficiently combat tumor cells [72,73,74], untreated or freshly isolated NK cells might not be sufficient to attack glioblastoma cells [71,75,76]. Instead, upregulation of activating molecules like NKG2D on NK cells [77] or the use of allogeneic and autologous cytokine-activated (IL-2 or IL-15) NK cells are important to prime NK cells to eliminate glioblastoma cells and overcome the failure of immune surveillance. In particular, utilizing NK cells activated with HSP70 and IL-2 is promising since these cells can cross the blood brain barrier and infiltrate to the site of glioblastoma [78].
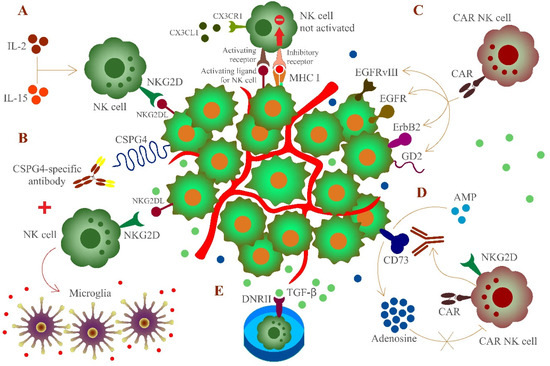
Figure 1.
Targeting of glioblastoma cells expressing major histocompatibility complex class I (MHC I) molecules by activated or engineered natural killer (NK) cells. The chemokine receptor CX3CR1 is expressed on NK cells. NK cells can follow a CX3CL1 gradient to cross the blood brain barrier to the site of glioblastoma. However, glioblastoma cells partly express MHC I molecules to impair recognition by NK cells as an immune evasion strategy. To circumvent this obstacle in NK cell-mediated immunotherapy, several procedures were developed. (A) Induced upregulation of NKG2D or cytokine (IL-2 and IL-15) -activated NK cells can overcome immune surveillance, (B) combined use of activated NK cells and monoclonal antibodies recognizing CSPG-4 leads to the recruitment of macrophages and differentiate to microglia, provoking a proinflammatory environment, (C) chimeric antigen receptor (CAR)-engineered NK cells can target epidermal growth factor receptor variant III (EGFRvIII), epidermal growth factor receptor (EGFR), ErbB2, or disialoganglioside GD2 on glioblastoma cells, (D) engineered NK cells expressing the GD2- and NKG2D-based CARs can locally release an antibody fragment which blocks the activity of CD73 and decreases adenosine concentration, which in turn is not able to inhibit NK cell function, and (E) umbilical cord blood (UCB)-derived NK cells expressing the TGF-β-dominant-negative receptor II (DNRII) that is able to consume TGF-β molecules, thereby counteracting the immune-suppressive tumor environment of glioblastoma. MHC I molecules and inhibitory receptors are not shown for all cells to improve the overview.
The heterogeneity of tumor-associated antigens expressed on glioblastoma cells is a major obstacle for an effective immunotherapy which indicates the significance of searching for adequate markers. Previous studies indicated that chondroitin sulfate proteoglycan 4 (CSPG4, also known as NG2) a surface type I transmembrane core proteoglycan [79] being involved in several pathways regulating cell survival, angiogenesis, and migration [80], is highly expressed on glioblastoma cells (67%) with restricted intratumoral heterogeneity [81]. Long-term activation of NK cells, combined with monoclonal antibodies recognizing specific cell surface markers on glioblastoma cells, can improve a NK cell response and making them less susceptible to the tumor microenvironment. Recent evidence suggests that a combination of activated NK cells and a monoclonal antibody (mAb9.2.27), which targets CSPG4, is an encouraging strategy [82]. Moreover, the combined treatment of activated NK cells and mAb9.2.27 led to a recruitment of ED1+CCR2low macrophages, followed by differentiation to ED1+ED2lowMHC II+ microglia secreting pro-inflammatory cytokines [82]. However, this strategy does not bypass the issue of expression of MHC I molecules on glioblastoma cells and the subsequent inhibition of NK cells. Yet, such a combined treatment induces NK cells to create a pro-inflammatory environment and improves the survival rate of glioblastoma patients [83] by recruiting immune cells [84].
Aside from joint treatment with NK cells and antibodies, more studies of CAR-engineered NK cells to attack glioblastoma cells were designed to interfere with epidermal growth factor receptor variant III (EGFRvIII) [85], EGFRvIII and epidermal growth factor receptor (EGFR), which increased cytotoxicity and IFN-γ secretion [86], EGFR and ErbB2 [87], or disialoganglioside GD2 [88]. On the other hand, CAR-NK cell strategies have been constrained to the application of NK cell lines while showing a minimal and unreliable overall response [86,89,90], which is based on the fact that heterogeneity and diversity of glioblastoma are accompanied by various genetic and epigenetic manifestations [91]. Thus, novel treatment strategies are indicated for clinical needs, amongst them CD73, an ecto-5′-nucleotidase that has been reported to be associated with a negative survival prognosis of patients, which emerged as an effective approach to combating cancer [92]. The activity of CD73 can provoke an immunometabolic dysregulation of NK cell function by the accumulation of extracellular adenosine [93]. Glioblastoma cells expressing disialoganglioside GD2 and ligands binding to NKG2D are attacked by engineered NK cells expressing the GD2- and NKG2D-based CARs, which then locally release an antibody fragment blocking the activity of CD73 to decrease the concentration of adenosine. This combination addresses several aspects of tumor immune evasion outcomes, namely tumor antigen heterogeneity, the immunosuppressive environment, and insufficient intratumoral NK cell presence (https://www.biorxiv.org/content/10.1101/2020.10.07.330043v1, accessed on 1 March 2021). This procedure is applicable to other markers that are pro-tumorigenic, such as CD155 [94], and might offer possible treatment strategies for further investigations.
In general, the advantage of CAR-NK cells is that NK cells can be administered from a different donor to the respective recipient even with an HLA-mismatch [95]. It is possible to adopt NK cells from any blood donor to apply these cells in CAR-NK cell therapy, which makes CAR-NK cells a cost-effective treatment option without the barriers of an activation state or HLA matching [96]. An essential element for the procedure of CAR-engineered NK cells for therapy is the availability of NK cells, which is one reason for the increasing interest in assessing umbilical cord blood (UCB)-derived NK cells [97]. Additionally, the use of UCB is an improvement since the proportion of NK cells is much higher; while peripheral blood contains around 15 % NK cells of lymphocytes in contrast to UCB having about 25 % NK cells of lymphocytes [98]. Approaches with proliferative cytokines like IL-2 and IL-15 combined with genetically engineered feeder cells enable an effective expansion of UCB-derived NK cells in vitro and maintaining their cytotoxic activity [99,100]. Another strategy encompasses the application of UCB-derived NK cells expressing the TGF-β-dominant-negative receptor II (DNRII) which can consume TGF-β molecules to reduce the immune-suppressive capacity of TGF-β in the tumor environment of glioblastoma [101]. Thus, combining accessible NK cells with glioblastoma targeted approaches is a promising therapeutical tool.
5. Proteolytic Regulation of MHC I Molecules
It is common that cell surface receptors are internalized and recycled back to the cell surface. Internalization of MHC I from the cell surface is constitutively performed to reach the endocytic compartment for degradation or recycled back to the cell surface to present a new round of antigenic peptides to T cells [102]. Several recycling pathways (comprehensively summarized in [103]) are proposed for MHC I molecules, which are internalized to a so-called early sorting endosome in non-immune cells. First, MHC I molecules transit the early sorting endosome before being driven straight back to the cell surface, a process called the “fast recycling” pathway. Second, MHC I molecules from the early sorting endosome are directed to the trans-Golgi network (TGN) to consequently reach the cell surface. This process is termed the “retrograde transport” pathway. “Slow recycling” is the third pathway where MHC I molecules from the early sorting endosome are trafficked to the endocytic recycling compartment and then to the cell surface. The fourth pathway postulates that MHC I molecules move from the early sorting endosome to the endocytic recycling compartment followed by being maneuvered to the so-called tubular recycling endosome and finally appear at the cell surface. Another possibility is that MHC I molecules from early sorting endosomes are forced to late endosomes and then to the endocytic recycling compartment or are possibly degraded in late endosomes or lysosomes [103]. The proteolytic environment in lysosomes is responsible for the turnover of MHC I molecules [104]. It was postulated that cysteine proteases degrade MHC I molecules since the cysteine protease inhibitor E64 impaired the decrease of MHC I molecules in the endosomal/lysosomal compartment [105].
Cathepsin G (CatG), which harbors serine amino acid residue at the catalytic center (serine protease) [106], CatD (aspartic protease), and CatS (cysteine protease) proteolytically digest MHC I molecules in vitro [107]. In order to rescue MHC I from degradation, we treated peripheral blood mononuclear cells (PBMCs) with cell permeable protease inhibitors including the selective inhibitor for CatG, cysteine protease inhibitor E64d, or the aspartic protease inhibitor pepstatin A and levels of cell surface expression of MHC I were determined. The CatG inhibitor increased levels of cell surface MHC I molecules, but neither E64d nor pepstatin A altered the amount of cell surface MHC I molecules [107]. In general, human embryonic kidney 293 cells, glioblastoma cell line U87, or glioblastoma stem cells do not harbor CatG, which is suitable for insertion of CatG into the cell in order to analyze MHC I processing. Therefore, HIV Nef (Nef), which is known to provoke an internalization of cell surface MHC I [108], and CatG were overexpressed in these cells. Both CatG and Nef induced a significant downregulation of cell surface MHC I molecules. Intracellular breakdown of MHC I molecules occurred only with overexpression of CatG, but not when Nef was introduced to the respective cells because an accumulation of intracellular MHC I molecules was detected [107]. These findings suggest that CatG degrades MHC I intracellularly and Nef-driven internalization of MHC I molecules accumulate in the cell. Whether such a reduction of cell surface MHC I molecules in glioblastoma cells become a target of NK cells remains unsolved.
Based on these data, might it be possible to use CatG as a therapeutic reagent for NK cell immunotherapy? Serine proteases are commercially used to treat sepsis, hemophilia, stroke, or muscle spasms [109]. Neutrophils, when activated, release neutrophil serine proteases (NSPs), including CatG, and additional mediators at the site of inflammation. However, dysregulated proteolytic activity is dependable for many complications, such as chronic inflammatory disorders and cardiovascular diseases [106]. Moreover, it was shown that the extent of infiltrating neutrophils correlates with a severe clinical outcome of glioblastoma [110] and neutrophilia (increase of neutrophil count) is a poor prognosis of glioblastoma patients undergoing chemoradiation [111]. In general, a high level of CatG correlates with tumor grade and favors tumor progression [112]. Thus, CatG is not suitable to be delivered to the surrounding glioblastoma area.
Nanoparticles are promising delivery agents [113] also in glioblastoma [68,114]. One might speculate that CatG-loaded magnetic mesoporous silica nanoparticles (MMSN) could be delivered into glioblastoma cells to reduce MHC I molecules and sensitizing NK cells. Interestingly, neutrophils can be used as a “Trojan horse” by using immune cells as a “living” drug delivery vehicle. Thereby, neutrophils are loaded with chemotherapeutic agents and can transport the content to the site of glioma, since neutrophils can cross the blood brain barrier and infiltrate the tumor [115]. Neutrophils internalize doxorubicin-loaded MMSN (D-MMSN), whereby doxorubicin is a model anti-cancer drug, Fe3O2 for contrast in MRI, and the mesoporous silica shell is important for the encapsulation and sustained release of the chemotherapeutic agent doxorubicin. After injection of D-MMSN-loaded neutrophils to an animal model, neutrophils are attracted to the site of the tumor, since surgical resection causes inflamed tissue where neutrophils are recruited; thereby, neutrophils are highly activated and can spread neutrophil extracellular traps (NETs) containing D-MMSN. D-MMSNs are up-taken by the surrounding glioma cells and, as a result, this maximizes drug bioavailability to eliminate remaining glioma cells [116]. Notably, catalytically active NSPs are not a prerequisite for the formation of NETs, as NSPs are released as inactive proteases [117].
6. Conclusions
Immune evasion of tumor cells by down regulation of MHC I molecules activates NK cell function since NK cells are specialized to eliminate tumor cells in an MHC I antigen presentation independent pathway. Therefore, activating and inhibitory receptors expressed on the cell surface of NK cells are responsible and are promising candidates for immunotherapy. Taken together, NK cells harbor tumor cell surveillance tasks and play an important role in the anti-tumor response by interfering with tumor cell-mediated immune evasion. However, tumor cells have developed further strategies to mask their recognition by NK cells. Understanding the mechanisms of immune evasion provides the basics to assess NK cells in immunotherapy and how to potentiate their anti-tumor efficiency.
Author Contributions
T.B., F.G., C.B., A.Z., A.G., and U.K.: writing, reviewing, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
T.B. was funded by the Nazarbayev University Faculty-Development Competitive Research Grants Program, reference: 280720FD1907.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| APCs | antigen-presenting cells |
| Cat | cathepsin |
| CatG | cathepsin G |
| CCL1 | chemokine C-C motif ligand 1 |
| CSPG4 | chondroitin sulfate proteoglycan 4 |
| DCs | dendritic cells |
| D-MMSN | doxorubicin-loaded magnetic mesoporous silica nanoparticles |
| DNAM-1 | DNAX accessory molecule 1 |
| DNRII | TGF-β-dominant-negative receptor II |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor variant III |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| HLA | human leukocyte antigen |
| IL-2 | interleukin 2 |
| ITAM | immunoreceptor tyrosine-based activation motif |
| KIRs | killer immunoglobulin-like receptors |
| MDSCs | myeloid-derived suppressor cells |
| MHC I | major histocompatibility complex class I |
| NETs | neutrophil extracellular traps |
| MICA | MHC class I chain-related protein A |
| MICB | MHC class I chain-related protein B |
| NCR | natural cytotoxicity receptor |
| NK cells | natural killer cells |
| NKG2D | natural killer group 2D activating receptor |
| NKG2DL | natural killer group 2D ligands |
| NSPs | neutrophil serine proteases |
| PBMCs | peripheral blood mononuclear cells |
| TCR | T cell receptor |
| TGF-β | transforming growth factor beta |
| TGN | trans-Golgi network |
| Th cells | T helper cells |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor α |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Tregs | T regulatory cells |
| ULBP1–6 | UL 16 binding protein 1–6 |
References
- Bischof, J.; Westhoff, M.A.; Wagner, J.E.; Halatsch, M.E.; Trentmann, S.; Knippschild, U.; Wirtz, C.R.; Burster, T. Cancer stem cells: The potential role of autophagy, proteolysis, and cathepsins in glioblastoma stem cells. Tumour Biol. 2017, 39, 1010428317692227. [Google Scholar] [CrossRef] [PubMed]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; et al. Considering the Experimental use of Temozolomide in Glioblastoma Research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Kos, J.; Kaur, K.; Safaei, T.; Sutanto, C.; Chen, W.; Wong, P.; Namagerdi, A.K.; Fang, C.; Fong, Y.; et al. Natural Killer Cells: Diverse Functions in Tumor Immunity and Defects in Pre-neoplastic and Neoplastic Stages of Tumorigenesis. Mol. Ther. Oncolytics 2020, 16, 41–52. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2020. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- McCoach, C.E.; Bivona, T.G. The evolving understanding of immunoediting and the clinical impact of immune escape. J. Thorac. Dis. 2018, 10, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Jurewicz, M.M.; Stern, L.J. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics 2019, 71, 171–187. [Google Scholar] [CrossRef]
- Dersh, D.; Holly, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Jelencic, V.; Sestan, M.; Kavazovic, I.; Lenartic, M.; Marinovic, S.; Holmes, T.D.; Prchal-Murphy, M.; Lisnic, B.; Sexl, V.; Bryceson, Y.T.; et al. NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat. Immunol. 2018, 19, 1083–1092. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Baragano Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, 2040. [Google Scholar] [CrossRef]
- Narni-Mancinelli, E.; Vivier, E. Shed NKG2D ligand boosts NK cell immunity. Cell Res. 2015, 25, 651–652. [Google Scholar] [CrossRef][Green Version]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/beta-catenin signaling pathway. Oncogene 2020, 39, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Shi, F.D.; Ljunggren, H.G.; La Cava, A.; Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 2011, 11, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Wildes, T.J.; Dyson, K.A.; Francis, C.; Wummer, B.; Yang, C.; Yegorov, O.; Shin, D.; Grippin, A.; Dean, B.D.; Abraham, R.; et al. Immune Escape After Adoptive T-cell Therapy for Malignant Gliomas. Clin. Cancer Res. 2020, 26, 5689–5700. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Bjorkstrom, N.K.; Ljunggren, H.G.; Michaelsson, J. Emerging insights into natural killer cells in human peripheral tissues. Nat. Rev. Immunol. 2016, 16, 310–320. [Google Scholar] [CrossRef]
- Sondergaard, S.R.; Ullum, H.; Pedersen, B.K. Proliferative and cytotoxic capabilities of CD16+CD56™ and CD16+/™CD56+ natural killer cells. APMIS 2000, 108, 831–837. [Google Scholar] [CrossRef]
- Alter, G.; Suscovich, T.J.; Kleyman, M.; Teigen, N.; Streeck, H.; Zaman, M.T.; Meier, A.; Altfeld, M. Low perforin and elevated SHIP-1 expression is associated with functional anergy of natural killer cells in chronic HIV-1 infection. AIDS 2006, 20, 1549–1551. [Google Scholar] [CrossRef]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.T.; Orange, J.S. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, I.; Sonnerborg, I.; Strunz, B.; Friberg, D.; Cornillet, M.; Hertwig, L.; Ivarsson, M.A.; Bjorkstrom, N.K. 29-Color Flow Cytometry: Unraveling Human Liver NK Cell Repertoire Diversity. Front. Immunol. 2019, 10, 2692. [Google Scholar] [CrossRef]
- Poreba, M.; Groborz, K.M.; Rut, W.; Pore, M.; Snipas, S.J.; Vizovisek, M.; Turk, B.; Kuhn, P.; Drag, M.; Salvesen, G.S. Multiplexed Probing of Proteolytic Enzymes Using Mass Cytometry-Compatible Activity-Based Probes. J. Am. Chem. Soc. 2020, 142, 16704–16715. [Google Scholar] [CrossRef]
- Gärtner, F.; Knippschild, U.; Burster, T. Application of an Activity-Based Probe to Determine Proteolytic Activity of Cell Surface Cathepsin G by Mass Cytometry Data Acquisition. ACS Omega 2020, 5, 28233–28238. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Hudspeth, K.; Silva-Santos, B.; Mavilio, D. Natural cytotoxicity receptors: Broader expression patterns and functions in innate and adaptive immune cells. Front. Immunol. 2013, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Montaldo, E.; Del Zotto, G.; Della Chiesa, M.; Mingari, M.C.; Moretta, A.; De Maria, A.; Moretta, L. Human NK cell receptors/markers: A tool to analyze NK cell development, subsets and function. Cytom. Part A 2013, 83, 702–713. [Google Scholar] [CrossRef]
- Cichocki, F.; Miller, J.S.; Anderson, S.K.; Bryceson, Y.T. Epigenetic regulation of NK cell differentiation and effector functions. Front. Immunol. 2013, 4, 55. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Verges, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Pietra, G.; Vacca, P.; Pende, D.; Moretta, F.; Bertaina, A.; Mingari, M.C.; Locatelli, F.; Moretta, A. Human NK cells: From surface receptors to clinical applications. Immunol. Lett. 2016, 178, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.P.; Villa-Alvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez, S. NK Cells in the Treatment of Hematological Malignancies. J. Clin. Med. 2019, 8, 1557. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Moretta, L. Dissecting CD56dim human NK cells. Blood 2010, 116, 3689–3691. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Laugier-Anfossi, F.; Vely, F.; Saulquin, X.; Riedmuller, J.; Tisserant, A.; Gauthier, L.; Romagne, F.; Ferracci, G.; Arosa, F.A.; et al. Recognition of peptide-MHC class I complexes by activating killer immunoglobulin-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 13224–13229. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef]
- Marsh, S.G.; Parham, P.; Dupont, B.; Geraghty, D.E.; Trowsdale, J.; Middleton, D.; Vilches, C.; Carrington, M.; Witt, C.; Guethlein, L.A.; et al. Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Immunogenetics 2003, 55, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.G.; Sun, P.D. The structural basis of ligand recognition by natural killer cell receptors. J. Biomed. Biotechnol. 2011, 2011, 203628. [Google Scholar] [CrossRef]
- Chewning, J.H.; Gudme, C.N.; Hsu, K.C.; Selvakumar, A.; Dupont, B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J. Immunol. 2007, 179, 854–868. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, O.; Zimmer, J.; Enger, P.O.; Chekenya, M. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 2014, 193, 6192–6206. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Freud, A.G.; Caligiuri, M.A. Human natural killer cell development. Immunol. Rev. 2006, 214, 56–72. [Google Scholar] [CrossRef]
- Sivori, S.; Cantoni, C.; Parolini, S.; Marcenaro, E.; Conte, R.; Moretta, L.; Moretta, A. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur. J. Immunol. 2003, 33, 3439–3447. [Google Scholar] [CrossRef]
- Romagnani, C.; Juelke, K.; Falco, M.; Morandi, B.; D’Agostino, A.; Costa, R.; Ratto, G.; Forte, G.; Carrega, P.; Lui, G.; et al. CD56brightCD16− killer Ig-like receptor− NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J. Immunol. 2007, 178, 4947–4955. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Clements, C.S.; Lin, J.; Sullivan, L.C.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H.; et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef]
- Kaiser, B.K.; Barahmand-Pour, F.; Paulsene, W.; Medley, S.; Geraghty, D.E.; Strong, R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005, 174, 2878–2884. [Google Scholar] [CrossRef] [PubMed]
- Boyington, J.C.; Motyka, S.A.; Schuck, P.; Brooks, A.G.; Sun, P.D. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 2000, 405, 537–543. [Google Scholar] [CrossRef]
- Fan, Q.R.; Long, E.O.; Wiley, D.C. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol. 2001, 2, 452–460. [Google Scholar] [CrossRef]
- Hilton, H.G.; Guethlein, L.A.; Goyos, A.; Nemat-Gorgani, N.; Bushnell, D.A.; Norman, P.J.; Parham, P. Polymorphic HLA-C Receptors Balance the Functional Characteristics of KIR Haplotypes. J. Immunol. 2015, 195, 3160–3170. [Google Scholar] [CrossRef]
- Hilton, H.G.; Vago, L.; Older Aguilar, A.M.; Moesta, A.K.; Graef, T.; Abi-Rached, L.; Norman, P.J.; Guethlein, L.A.; Fleischhauer, K.; Parham, P. Mutation at positively selected positions in the binding site for HLA-C shows that KIR2DL1 is a more refined but less adaptable NK cell receptor than KIR2DL3. J. Immunol. 2012, 189, 1418–1430. [Google Scholar] [CrossRef]
- Vivian, J.P.; Duncan, R.C.; Berry, R.; O’Connor, G.M.; Reid, H.H.; Beddoe, T.; Gras, S.; Saunders, P.M.; Olshina, M.A.; Widjaja, J.M.; et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 2011, 479, 401–405. [Google Scholar] [CrossRef]
- Orr, M.T.; Murphy, W.J.; Lanier, L.L. ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat. Immunol. 2010, 11, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Mahmoud, A.B.; Makrigiannis, A.P. Licensed and Unlicensed NK Cells: Differential Roles in Cancer and Viral Control. Front. Immunol. 2016, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Hertwig, L.; Hamann, I.; Romero-Suarez, S.; Millward, J.M.; Pietrek, R.; Chanvillard, C.; Stuis, H.; Pollok, K.; Ransohoff, R.M.; Cardona, A.E.; et al. CX3CR1-dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur. J. Immunol. 2016, 46, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Siminska, D.; Kojder, K.; Grochans, S.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Fractalkine/CX3CL1 in Neoplastic Processes. Int. J. Mol. Sci. 2020, 21, 3723. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Mittelbronn, M.; Simon, P.; Loffler, C.; Capper, D.; Bunz, B.; Harter, P.; Schlaszus, H.; Schleich, A.; Tabatabai, G.; Goeppert, B.; et al. Elevated HLA-E levels in human glioblastomas but not in grade I to III astrocytomas correlate with infiltrating CD8+ cells. J. Neuroimmunol. 2007, 189, 50–58. [Google Scholar] [CrossRef]
- Wischhusen, J.; Friese, M.A.; Mittelbronn, M.; Meyermann, R.; Weller, M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: Implications for immune escape in vivo. J. Neuropathol. Exp. Neurol. 2005, 64, 523–528. [Google Scholar] [CrossRef]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Domingues, P.; Gonzalez-Tablas, M.; Otero, A.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Goncalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.H.; Teodosio, C.; Ortiz, J.; Sousa, P.; Otero, A.; Maillo, A.; Barcena, P.; Garcia-Macias, M.C.; Lopes, M.C.; de Oliveira, C.; et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am. J. Pathol. 2012, 181, 1749–1761. [Google Scholar] [CrossRef]
- Friese, M.A.; Platten, M.; Lutz, S.Z.; Naumann, U.; Aulwurm, S.; Bischof, F.; Buhring, H.J.; Dichgans, J.; Rammensee, H.G.; Steinle, A.; et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003, 63, 8996–9006. [Google Scholar] [PubMed]
- Wu, A.; Wiesner, S.; Xiao, J.; Ericson, K.; Chen, W.; Hall, W.A.; Low, W.C.; Ohlfest, J.R. Expression of MHC I and NK ligands on human CD133+ glioma cells: Possible targets of immunotherapy. J. Neurooncol. 2007, 83, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [PubMed]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Los, M.J.; Azizi, Z.; et al. HSP70/IL-2 Treated NK Cells Effectively Cross the Blood Brain Barrier and Target Tumor Cells in a Rat Model of Induced Glioblastoma Multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef] [PubMed]
- Pluschke, G.; Vanek, M.; Evans, A.; Dittmar, T.; Schmid, P.; Itin, P.; Filardo, E.J.; Reisfeld, R.A. Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc. Natl. Acad. Sci. USA 1996, 93, 9710–9715. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol. Biol. Lett. 2017, 22, 4. [Google Scholar] [CrossRef]
- Pellegatta, S.; Savoldo, B.; Di Ianni, N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Wang, J.; Domingues, O.; Planaguma, J.; Yan, T.; Rygh, C.B.; Skaftnesmo, K.O.; Thorsen, F.; McCormack, E.; Hentges, F.; et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget 2013, 4, 1527–1546. [Google Scholar] [CrossRef]
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology 2013, 2, e23401. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel Human NK Cell Line Carrying CAR Targeting EGFRvIII Induces Antitumor Effects in Glioblastoma Cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Longee, D.C.; Wikstrand, C.J.; Mansson, J.E.; He, X.; Fuller, G.N.; Bigner, S.H.; Fredman, P.; Svennerholm, L.; Bigner, D.D. Disialoganglioside GD2 in human neuroectodermal tumor cell lines and gliomas. Acta Neuropathol. 1991, 82, 45–54. [Google Scholar] [CrossRef]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Wang, J.; Matosevic, S. NT5E/CD73 as Correlative Factor of Patient Survival and Natural Killer Cell Infiltration in Glioblastoma. J. Clin. Med. 2019, 8, 1526. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J. Hematol. Oncol. 2020, 13, 76. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Waller-Wise, R. Umbilical cord blood: Information for childbirth educators. J. Perinat. Educ. 2011, 20, 54–60. [Google Scholar] [CrossRef]
- Dalle, J.H.; Menezes, J.; Wagner, E.; Blagdon, M.; Champagne, J.; Champagne, M.A.; Duval, M. Characterization of cord blood natural killer cells: Implications for transplantation and neonatal infections. Pediatr. Res. 2005, 57, 649–655. [Google Scholar] [CrossRef]
- Phan, M.T.; Lee, S.H.; Kim, S.K.; Cho, D. Expansion of NK Cells Using Genetically Engineered K562 Feeder Cells. Methods Mol. Biol. 2016, 1441, 167–174. [Google Scholar] [CrossRef]
- Vasu, S.; Berg, M.; Davidson-Moncada, J.; Tian, X.; Cullis, H.; Childs, R.W. A novel method to expand large numbers of CD56(+) natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy 2015, 17, 1582–1593. [Google Scholar] [CrossRef]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Montealegre, S.; van Endert, P.M. Endocytic Recycling of MHC Class I Molecules in Non-professional Antigen Presenting and Dendritic Cells. Front. Immunol. 2018, 9, 3098. [Google Scholar] [CrossRef]
- Machold, R.P.; Ploegh, H.L. Intermediates in the assembly and degradation of class I major histocompatibility complex (MHC) molecules probed with free heavy chain-specific monoclonal antibodies. J. Exp. Med. 1996, 184, 2251–2259. [Google Scholar] [CrossRef]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.G.; Hengel, H.; Koszinowski, U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed]
- Palesch, D.; Wagner, J.; Meid, A.; Molenda, N.; Sienczyk, M.; Burkhardt, J.; Munch, J.; Prokop, L.; Stevanovic, S.; Westhoff, M.A.; et al. Cathepsin G-mediated proteolytic degradation of MHC class I molecules to facilitate immune detection of human glioblastoma cells. Cancer Immunol. Immunother. 2016, 65, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Craik, C.S.; Page, M.J.; Madison, E.L. Proteases as therapeutics. Biochem. J. 2011, 435, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Schernberg, A.; Nivet, A.; Dhermain, F.; Ammari, S.; Escande, A.; Pallud, J.; Louvel, G.; Deutsch, E. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin. Transl. Radiat. Oncol. 2018, 10, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Nannuru, K.C.; Futakuchi, M.; Sadanandam, A.; Singh, R.K. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res. 2008, 68, 5803–5811. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.J.; Lo, J.H.; Bhatia, S.N. Smart nanosystems: Bio-inspired technologies that interact with the host environment. Proc. Natl. Acad. Sci. USA 2015, 112, 14460–14466. [Google Scholar] [CrossRef] [PubMed]
- Alphandery, E. Nano-Therapies for Glioblastoma Treatment. Cancers 2020, 12, 242. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, H.; Tie, C.; Yan, C.; Deng, Z.; Wan, Q.; Liu, X.; Yan, F.; Zheng, H. MR imaging tracking of inflammation-activatable engineered neutrophils for targeted therapy of surgically treated glioma. Nat. Commun. 2018, 9, 4777. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Hempel, A.; Janiszewski, T.; Kolt, S.; Snipas, S.J.; Drag, M.; Salvesen, G.S. NETosis Occurs Independently of Neutrophil Serine Proteases. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).