Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain

 ,
,  ,
,  , ,
, ,
Abstract
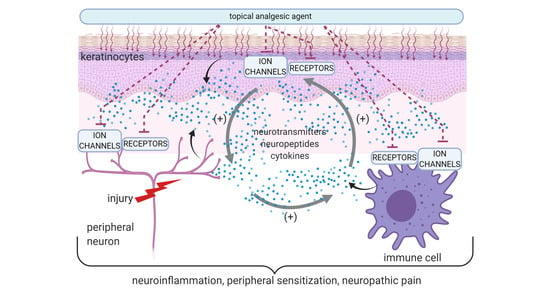
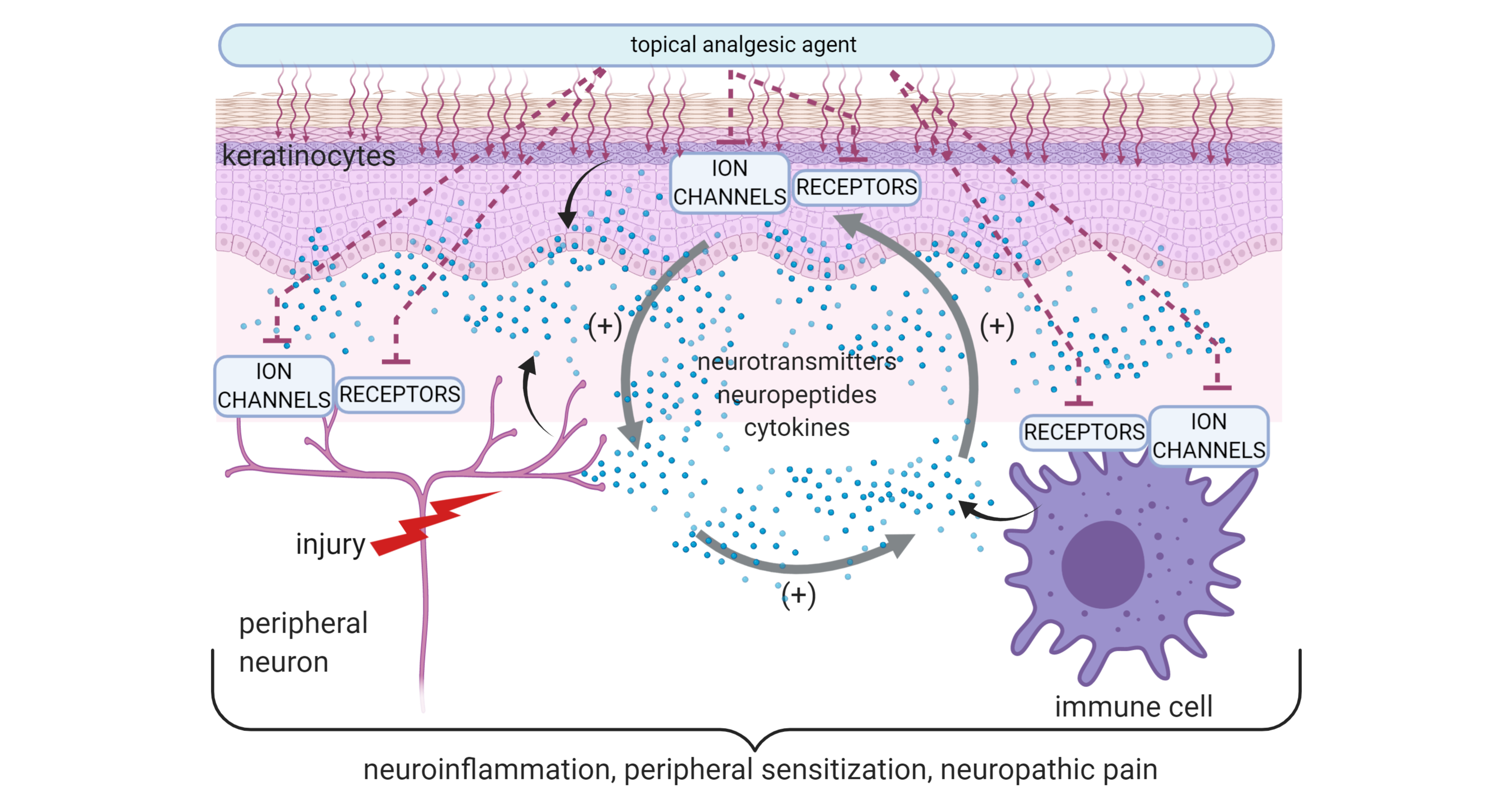
1. Introduction
2. Peripheral Mechanisms of NP
2.1. The Role of Neuronal Cells in Peripheral Mechanisms of NP
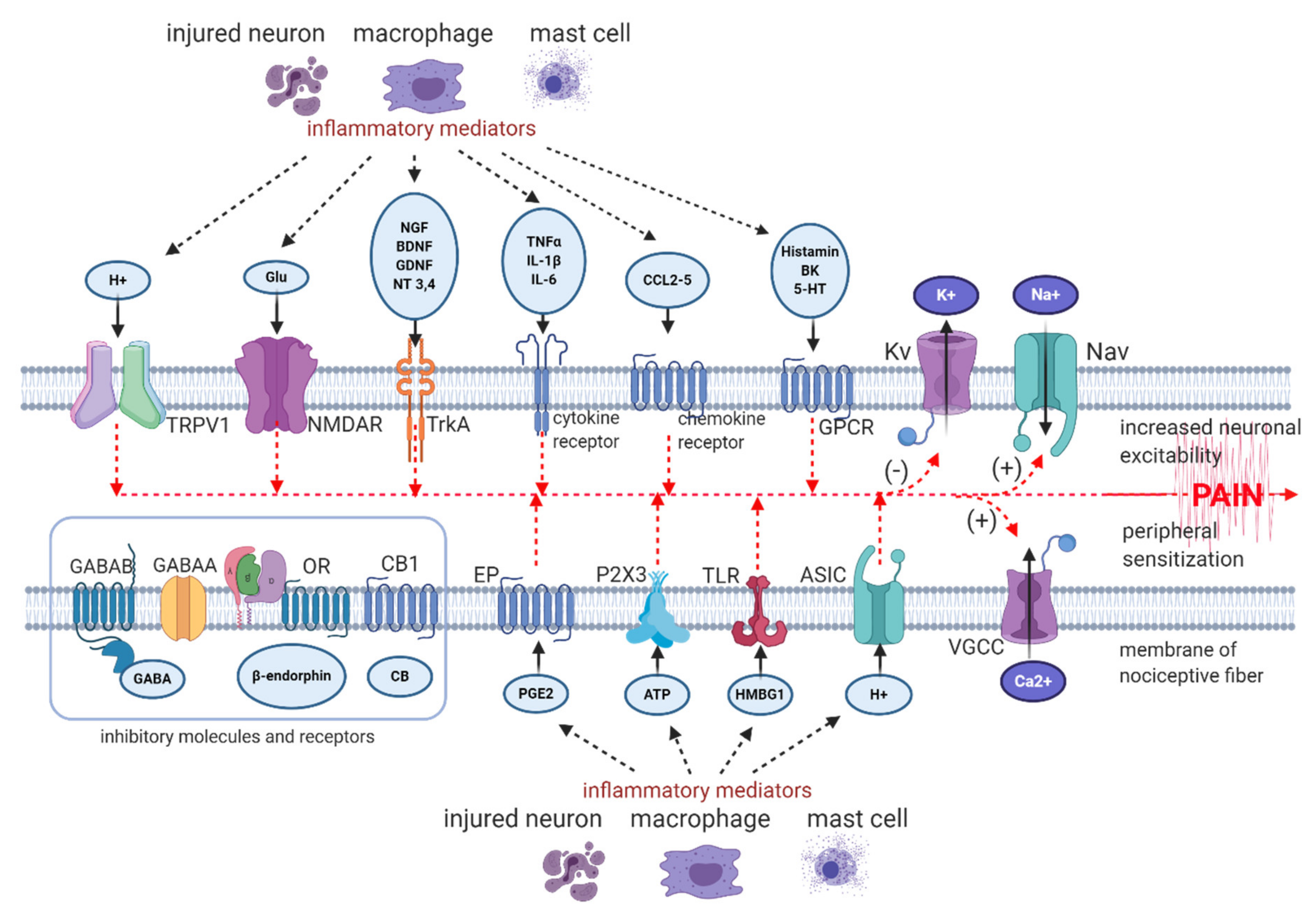
- peripheral sensitization—hyperexcitability of sensory neurons due to lowered threshold and augmented response to suprathreshold stimuli, caused by peripheral nerve or tissue injury, inflammation and subsequent release of pro-nociceptive mediators from mast cells, macrophages and from neighbouring nerve terminals, such as prostaglandins, bradykinin, histamine, serotonin, SP (substance P), extracellular ATP (adenosine triphosphate), protons, cytokines, chemokines, growth factors, peptides, acting on corresponding receptors, ion channels or altering their sensitivity to stimuli [8,29,30];
- ectopic firing in peripheral nerve endings and in DRG neurons—ectopic discharge begins in Aδ fibres within hours after injury and within several days or weeks in C fibres [31,32,33,34]; the main generator of ectopic activity are hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels, belonging to the voltage-gated potassium (Kv) channels [35]; in human altered firing and ectopic activity in peripheral neurons was observed in patients holding a mutation in gene coding Nav1.7 [36];
- alterations in channel expression and composition in peripheral nerve endings, along the axon and in DRG—peripheral input via intracellular second messengers alters gene expressions, resulting in increase in protein expression of Nav (voltage-gated sodium channels), VGCC (voltage-gated calcium channels), TLR4 (toll-like receptors 4), TRP (transient receptor potential channels), α1-AR (α1 adrenergic receptors), ASIC (acid-sensing ion channels), decrease in protein expression of Kv (voltage-gated potassium channels) [37,38,39,40];
2.2. Role of Glial Activation in Peripheral Mechanisms of NP
2.3. Role of Immunocompetent Cells in Peripheral Mechanisms of NP
2.4. The Role of Skin Cells in Peripheral Mechanisms of NP
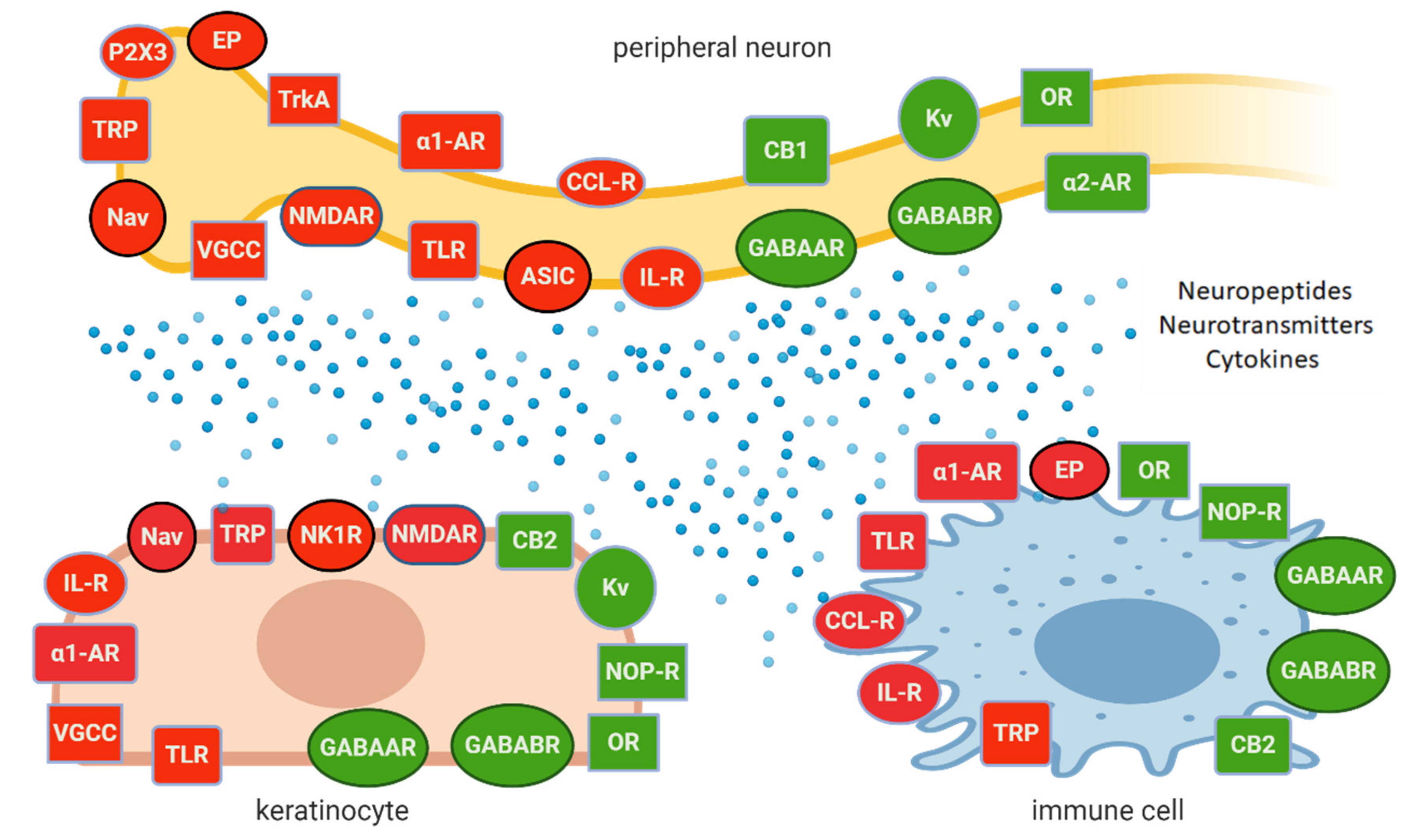
2.5. Peripheral NP as the Result of Neuronal and Non-Neuronal Mechanisms
- peripheral sensitization—hypersensitivity of primary afferent nociceptors;
- central sensitization—increased responsiveness of nociceptive neurons in the CNS to their normal or subthreshold afferent input;
- a switch in the messaging of Aβ fibers from tactile to nociceptive input.
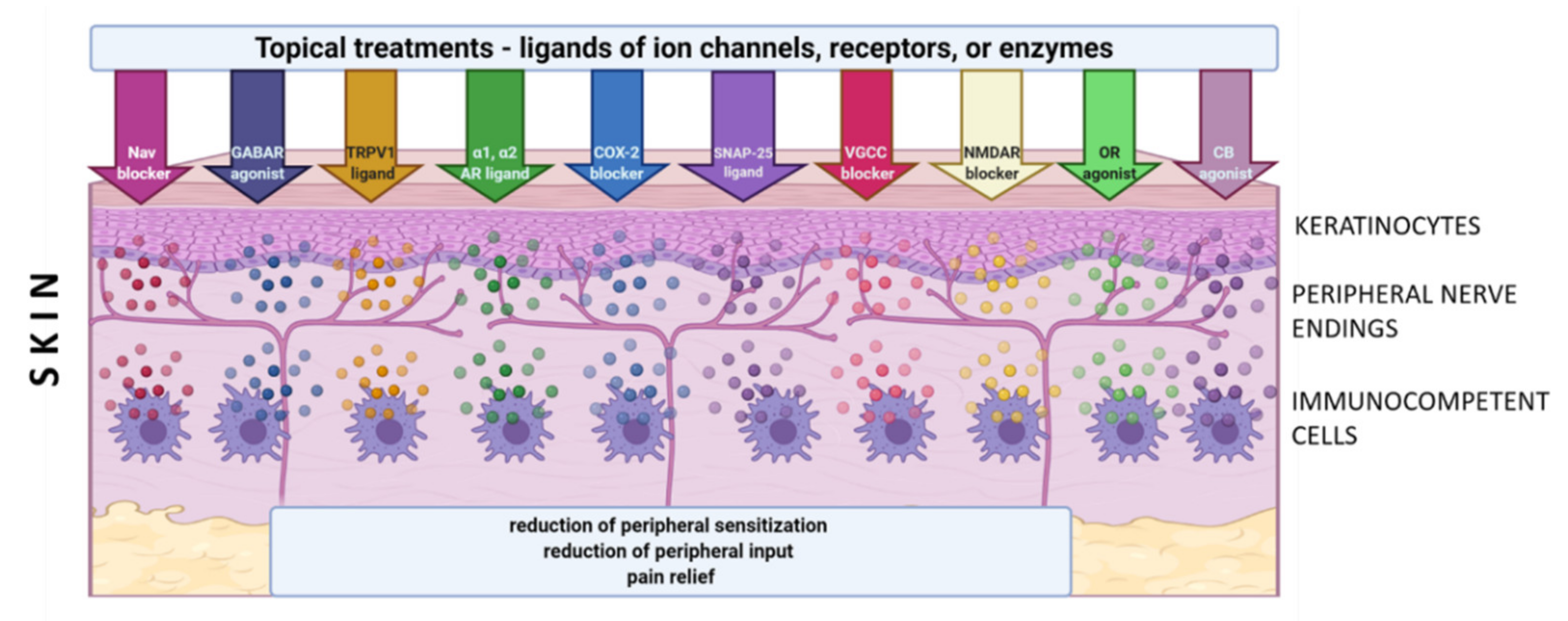
3. Topical Administration of Analgesics in LNP
- role of Nav:
- ◦
- in humans with primary erythromelalgia altered firing and ectopic activity in peripheral neurons was observed due to the mutation in gene coding Nav1.7 [36];
- ◦
- gain-of-function mutation in Nav1.7, Nav1.8 or Nav1.9 coding genes was associated with small fibre neuropathy and other neuropathic and non-neuropathic pain syndromes [106];
- ◦
- loss-of-function mutation in gene coding Nav1.7 or Nav1.9 results in congenital insensitivity to pain [106];
- ◦
- increased Nav1.1, Nav1.2, Nav1.5, Nav1.6, Nav1.7 and Nav1.8 expression in the skin of patients with complex regional pain syndrome (CRPS) or postherpetic neuralgia (PHN) [91];
- role of TRPV1—in patients with small fibre neuropathy a statistically significant increase of TRPV1 expression on epidermal keratinocytes was reported [97];
- SNAP-25 (synaptosome-associated protein 25)—plasma membrane protein forming the SNARE (SNAP-receptor), involved in synaptic vesicle fusions, exocytosis, and neurotransmission. SNAP-25 modulates VGCC protein expressed on plasma membrane. Abnormal expression or function of SNAP-25 are observed in chronic pain conditions, including neuropathic pain and fibromyalgia [108].
3.1. Treatments Acting on Voltage-Gated Sodium Channels
3.2. Treatments Acting on Transient Receptor Potential Vanilloid 1 Channels
3.3. Treatments Acting on Voltage-Gated Calcium Channels
3.4. Treatments Acting on N-Methyl-D-aspartate Receptors
3.5. Treatments Acting on α1 Adrenergic Receptors
3.6. Treatments Acting on Cyclooxygenase 2
3.7. Treatments Acting on Synaptosome-Associated Protein 25
3.8. Treatments Acting on Gamma-Aminobutyric Acid Receptors
- antidepressants (amitriptylline, fluoxetine), but their antinociceptive effect has been observed after intraperitoneal administration in rats [157];
- ketamine—the agonist to GABAAR, which has been confirmed in an anesthetic model in mice [158];
- phenytoin, which potentiated GABA-induced currents in cultured rat cortical neurons through modulation of the GABAAR [159].
3.9. Treatments Acting on α2 Adreno Receptors
3.10. Treatments Acting on Opioid Receptors
3.11. Treatments Acting on Cannabinoid Receptors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, T.S.; Baron, R.; Haanpää, M.; Kalso, E.; Loeser, J.D.; Rice, A.S.; Treede, R.D. A new definition of neuropathic pain. Pain 2011, 152, 2204–2205. [Google Scholar] [CrossRef] [PubMed]
- Morlion, B.; Coluzzi, F.; Aldington, D.; Kocot-Kepska, M.; Pergolizzi, J.; Mangas, A.C.; Ahlbeck, K.; Kalso, E. Pain chronification: What should a non-pain medicine specialist know? Curr. Med. Res. Opin. 2018, 34, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Cairns, B.E.; Arendt-Nielsen, L.; Sacerdote, P. Perspectives in Pain Research 2014: Neuroinflammation and glial cell activation: The cause of transition from acute to chronic pain? Scand. J. Pain 2015, 6, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M. Role of the immune system in neuropathic pain. Scand. J. Pain 2019, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Casale, R.; Mattia, C. Building a diagnostic algorithm on localized neuropathic pain (LNP) and targeted topical treatment: Focus on 5% lidocaine-medicated plaster. Ther. Clin. Risk Manag. 2014, 10, 259–268. [Google Scholar] [CrossRef][Green Version]
- Mick, G.; Baron, R.; Brix Finnerup, N.; Hans, G.; Kern, K.U.; Brett, B.; Dworkin, R.H. What is localized neuropathic pain? A first proposal to characterize and define a widely used term. Pain Manag. 2012, 2, 71–77. [Google Scholar] [CrossRef]
- Raja, S.N.; Ringkamp, M.; Guan, Y.; Campbell, J.N.; John, J. Bonica Award Lecture: Peripheral neuronal hyperexcitability: The “low-hanging” target for safe therapeutic strategies in neuropathic pain. Pain 2020, 161 (Suppl. 1), S14–S26. [Google Scholar] [CrossRef]
- Müller-Schwefe, G.; Morlion, B.; Ahlbeck, K.; Alon, E.; Coaccioli, S.; Coluzzi, F.; Huygen, F.; Jaksch, W.; Kalso, E.; Kocot-Kępska, M.; et al. Treatment for chronic low back pain: The focus should change to multimodal management that reflects the underlying pain mechanisms. Curr. Med. Res. Opin. 2017, 33, 1199–1210. [Google Scholar] [CrossRef]
- Casale, R.; Symeonidou, Z.; Bartolo, M. Topical Treatments for Localized Neuropathic Pain. Curr. Pain Headache Rep. 2017, 21, 15. [Google Scholar] [CrossRef]
- Hesselink, J.M.; Kopsky, D.J.; Bhaskar, A.K. Skin matters! The role of keratinocytes in nociception: A rational argument for the development of topical analgesics. J. Pain Res. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shipton, E.A. Skin matters: Identifying pain mechanisms and predicting treatment outcomes. Neurol. Res. Int. 2013, 2013, 329364. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J. Topical and peripherally acting analgesics. Pharmacol. Rev. 2003, 55, 1–20. [Google Scholar] [CrossRef] [PubMed]
- De Leon-Casasola, O.A. Multimodal approaches to the management of neuropathic pain: The role of topical analgesia. J. Pain Symptom Manag. 2007, 33, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652. [Google Scholar] [CrossRef]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef]
- Bouhassira, D.; Attal, N.; Alchaar, H.; Boureau, F.; Brochet, B.; Bruxelle, J.; Cunin, G.; Fermanian, J.; Ginies, P.; Grun-Overdyking, A.; et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 2005, 114, 29–36. [Google Scholar] [CrossRef]
- Haroutounian, S.; Nikolajsen, L.; Bendtsen, T.F.; Finnerup, N.B.; Kristensen, A.D.; Hasselstrøm, J.B.; Jensen, T.S. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014, 155, 1272–1279. [Google Scholar] [CrossRef]
- Colleoni, M.; Sacerdote, P. Murine models of human neuropathic pain. Biochim. Biophys. Acta. 2010, 1802, 924–933. [Google Scholar] [CrossRef]
- Sousa, A.M.; Lages, G.V.; Pereira, C.L.; Slullitel, A. Experimental models for the study of neuropathic pain. Modelos experimentais para o estudo da dor neuropática. Rev. Dor. São Paulo 2016, 17 (Suppl. 1), S27–S30. [Google Scholar]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wodarski, R.; Bagdas, D.; Paris, J.J.; Pheby, T.; Toma, W.; Xu, R.; Damaj, M.I.; Knapp, P.E.; Rice, A.S.C.; Hauser, K.F. Reduced intraepidermal nerve fibre density, glial activation, and sensory changes in HIV type-1 Tat-expressing female mice: Involvement of Tat during early stages of HIV-associated painful sensory neuropathy. Pain Rep. 2018, 14, e654. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.M.; Vornov, J.J.; Wu, Y.; Liu, Y.; Carozzi, V.A.; Rodriguez-Menendez, V.; Ballarini, E.; Alberti, P.; Pozzi, E.; Semperboni, S.; et al. Peripheral Neuropathy Induced by Microtubule-Targeted Chemotherapies: Insights into Acute Injury and Long-term Recovery. Cancer Res. 2018, 78, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.J.; Rinaldi, S.; Costigan, M.; Oh, S.B. Cytotoxic Immunity in Peripheral Nerve Injury and Pain. Front. Neurosci. 2020, 14, 142. [Google Scholar] [CrossRef]
- Ratte, S.; Prescott, S.A. Afferent hyperexcitability in neuropathic pain and the inconvenient truth about its degeneracy. Curr. Opin. Neurobiol. 2016, 36, 31–37. [Google Scholar] [CrossRef]
- Dubový, P. Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Ann. Anat. Anat. Anz. 2011, 193, 267–275. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian degeneration: Gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflamm. 2011, 8, 110. [Google Scholar] [CrossRef]
- Ochoa, J.L.; Campero, M.; Serra, J.; Bostock, H. Hyperexcitable polymodal and insensitive nociceptors in painful human neuropathy. Muscle Nerve 2005, 32, 459–472. [Google Scholar] [CrossRef]
- Si-Qi, W.; Zhuo-Ying, T.; Yang, X.; Dong-Yuan, C. Peripheral Sensitization. In Peripheral Nerve Disorders and Treatment; Turker, H., Benavides, L.G., Gallardo, G.R., del Villar, M.M., Eds.; IntechOpen: London, UK, 4 December 2019; Available online: https://www.intechopen.com/books/peripheral-nerve-disorders-and-treatment/peripheral-sensitization (accessed on 2 December 2020). [CrossRef]
- Liu, X.; Chung, K.; Chung, J.M. Ectopic discharges and adrenergic sensitivity of sensory neurons after spinal nerve injury. Brain Res. 1999, 849, 244–247. [Google Scholar] [CrossRef]
- Truini, A. A review of neuropathic pain: From diagnostic tests to mechanisms. Pain Ther. 2017, 6, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Ringkamp, M.; Hartke, T.V.; Chien, H.F.; Flavahan, N.A.; Campbell, J.N.; Meyer, R.A. Uninjured C-fiber nociceptors develop spontaneous activity and alpha-adrenergic sensitivity following L6 spinal nerve ligation in monkey. J. Neurophysiol. 1999, 81, 455–466. [Google Scholar] [CrossRef] [PubMed]
- North, R.Y.; Lazaro, T.T.; Dougherty, P.M. Ectopic spontaneous afferent activity and neuropathic pain. Neurosurgery 2018, 65, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wuni, G.Y.; Bahuva, R. Pacemaking Activity in the Peripheral Nervous System: Physiology and Roles of Hyperpolarization Activated and Cyclic Nucleotide-Gated Channels in Neuropathic Pain. Cureus 2020, 12, e11111. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef]
- Novakovic, S.D.; Tzoumaka, E.; McGivern, J.G.; Haraguchi, M.; Sangameswaran, L.; Gogas, K.R.; Eglen, R.M.; Hunter, J.C. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J. Neurosci. 1998, 15, 2174–2187. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Fjell, J.; Cummins, T.R.; Zheng, Z.; Fried, K.; LaMotte, R.; Black, J.A.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar] [CrossRef]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Zhang, H.; Kosturakis, A.K.; Jawad, A.B.; Dougherty, P.M. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J. Pain 2014, 15, 712–725. [Google Scholar] [CrossRef]
- Woolf, C.J.; Shortland, P.; Coggeshall, R.E. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 1992, 355, 75–78. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Kwiecien, J.M.; Dabrowski, W.; Ungard, R.; Zhu, K.L.; Huizinga, J.D.; Henry, J.L.; Singh, G. Cancer pain and neuropathic pain are associated with A β sensory neuronal plasticity in dorsal root ganglia and abnormal sprouting in lumbar spinal cord. Mol. Pain 2018, 14, 1744806918810099. [Google Scholar] [CrossRef] [PubMed]
- Drummond, P.D. Neuronal changes resulting in up-regulation of alpha-1 adrenoceptors after peripheral nerve injury. Neural. Regen. Res. 2014, 15, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.P.; Magnussen, C.; Yousefpour, N.; Ribeiro-da-Silva, A. Sympathetic fibre sprouting in the skin contributes to pain-related behaviour in spared nerve injury and cuff models of neuropathic pain. Mol. Pain 2015, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.D.; Bennett, G.J.; Ribeiro-da-Silva, A. Sympathetic sprouting and changes in nociceptive sensory innervation in the glabrous skin of the rat hind paw following partial peripheral nerve injury. J. Comp. Neurol. 2006, 495, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Ramer, M.S.; Bisby, M.A. Adrenergic innervation of rat sensory ganglia following proximal or distal painful sciatic neuropathy: Distinct mechanisms revealed by anti-NGF treatment. Eur. J. Neurosci. 1999, 11, 837–846. [Google Scholar] [CrossRef]
- Hsieh, M.T.; Donaldson, L.F.; Lumb, B.M. Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. Pain 2015, 156, 1074–1083. [Google Scholar] [CrossRef]
- Hulse, R.P. Identification of mechano-sensitive C fibre sensitization and contribution to nerve injury-induced mechanical hyperalgesia. Eur. J. Pain 2016, 20, 615–625. [Google Scholar] [CrossRef]
- Forstenpointner, J.; Naleschinski, D.; Wasner, G.; Hüllemann, P.; Binder, A.; Baron, R. Sensitized vasoactive C-nociceptors: Key fibers in peripheral neuropathic pain. PAIN Rep. 2019, 4, e709. [Google Scholar] [CrossRef]
- Devor, M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp. Brain Res. 2009, 196, 115–128. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Wu, Q.; Henry, J.L. Changes in functional properties of A-type but not C-type sensory neurons in vivo in a rat model of peripheral neuropathy. J. Pain Res. 2012, 5, 175–192. [Google Scholar] [CrossRef][Green Version]
- Djouhri, L.; Zeidan, A.; Abd El-Aleem, S.A.; Smith, T. Cutaneous Aβ-Non-nociceptive, but Not C-Nociceptive, Dorsal Root Ganglion Neurons Exhibit Spontaneous Activity in the Streptozotocin Rat Model of Painful Diabetic Neuropathy in vivo. Front. Neurosci. 2020, 25, 530. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Henry, J.L. Excitability of Aβ sensory neurons is altered in an animal model of peripheral neuropathy. BMC Neurosci. 2012, 30, 15. [Google Scholar] [CrossRef] [PubMed]
- Djouhri, L. L5 spinal nerve axotomy induces sensitization of cutaneous L4 Aβ-nociceptive dorsal root ganglion neurons in the rat in vivo. Neurosci. Lett. 2016, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. 1), S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 15, 106–119. [Google Scholar] [CrossRef]
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Biology of Schwann cells. Handb. Clin. Neurol. 2013, 115, 55–79. [Google Scholar] [CrossRef]
- Yajima, Y.; Narita, M.; Usui, A.; Kaneko, C.; Miyatake, M.; Narita, M.; Yamaguchi, T.; Tamaki, H.; Wachi, H.; Seyama, Y.; et al. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J. Neurochem. 2005, 93, 584–594. [Google Scholar] [CrossRef]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Front. Cell Neurosci. 2019, 27, 116. [Google Scholar] [CrossRef]
- Lee, H.; Baek, J.; Min, H.; Cho, I.H.; Yu, S.W.; Lee, S.J. Toll-Like Receptor 3 Contributes to Wallerian Degeneration after Peripheral Nerve Injury. Neuroimmunomodulation 2016, 23, 209–216. [Google Scholar] [CrossRef]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Ristoiu, V. Contribution of macrophages to peripheral neuropathic pain pathogenesis. Life Sci. 2013, 93, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Peng, J.; Han, G.H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of macrophages in peripheral nerve injury and repair. Neural. Regen. Res. 2019, 14, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.A.; Dumlao, D.S.; Stokes, J.A.; Dennis, E.A.; Svensson, C.I.; Corr, M.; Yaksh, T.L. Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain 2011, 152, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Woster, A.P.; Dahlman, J.; Sauter, E.R.; Combs, C.K.; Porter, J.E. α1-adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. J. Pharmacol. Exp. Ther. 2011, 338, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Interactions between the immune and nervous systems in pain. Nat. Med. 2010, 16, 1267–1276. [Google Scholar] [CrossRef]
- Zhang, N.; Inan, S.; Cowan, A.; Sun, R.; Wang, J.M.; Rogers, T.J.; Caterina, M.; Oppenheim, J.J. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. USA 2005, 102, 4536–4541. [Google Scholar] [CrossRef]
- Obreja, O.; Rathee, P.K.; Lips, K.S.; Distler, C.; Kress, M. IL-1 potentiates heat-activated currents in rat sensory neurons: Involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002, 16, 1497–1503. [Google Scholar] [CrossRef]
- Jin, X.; Gereau, R.W., 4th. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 2006, 4, 246–255. [Google Scholar] [CrossRef]
- Schaible, H.G. Nociceptive neurons detect cytokines in arthritis. Arthritis Res. Ther. 2014, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Drummond, P.D.; Dawson, L.F.; Finch, P.M.; Drummond, E.S.; Wood, F.M.; Fear, M.W. Up-regulation of cutaneous alpha1-adrenoceptors after a burn. Burns 2015, 41, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.F.; Phillips, J.K.; Finch, P.M.; Inglis, J.J.; Drummond, P.D. Expression of alpha1-adrenoceptors on peripheral nociceptive neurons. Neuroscience 2011, 175, 300314. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Chabot, J.G.; Vercauteren, F.; Quirion, R. Injured nerve-derived COX2/PGE2 contributes to the maintenance of neuropathic pain in aged rats. Neurobiol. Aging. 2010, 31, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef]
- Thacker, M.A.; Clark, A.K.; Marchand, F.; McMahon, S.B. Pathophysiology of peripheral neuropathic pain: Immune cells and molecules. Anesth. Analg. 2007, 105, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef]
- Eisenstein, T.K. The Role of Opioid Receptors in Immune System Function. Front. Immunol. 2019, 10, 2904. [Google Scholar] [CrossRef]
- Smith, M.T.; Wyse, B.D.; Edwards, S.R.; El-Tamimy, M.; Gaetano, G.; Gavin, P. Topical application of a novel oxycodone gel formulation (tocopheryl phosphate mixture) in a rat model of peripheral inflammatory pain produces localized pain relief without significant systemic exposure. J. Pharm. Sci. 2015, 104, 2388–2396. [Google Scholar] [CrossRef]
- D’Amico, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. ALIAmides Update: Palmitoylethanolamide and Its Formulations on Management of Peripheral Neuropathic Pain. Int. J. Mol. Sci. 2020, 27, 5330. [Google Scholar] [CrossRef]
- Sehgal, N.; Smith, H.S.; Manchikanti, L. Peripherally acting opioids and clinical implications for pain control. Pain Phys. 2011, 14, 249–258. [Google Scholar]
- Nigam, R.; El-Nour, H.; Amatya, B.; Nordlind, K. GABA and GABA(A) receptor expression on immune cells in psoriasis: A pathophysiological role. Arch. Dermatol. Res. 2010, 302, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Cevikbas, F.; Braz, J.M.; Wang, X.; Solorzano, C.; Sulk, M.; Buhl, T.; Steinhoff, M.; Basbaum, A.I. Synergistic antipruritic effects of gamma aminobutyric acid A and B agonists in a mouse model of atopic dermatitis. J. Allergy Clin. Immunol. 2017, 140, 454–464.e2. [Google Scholar] [CrossRef] [PubMed]
- Misery, L. Le système neuro-immuno-cutané (SNIC) [Neuro-immuno-cutaneous system (NICS)]. Pathol. Biol. (Paris) 1996, 44, 867–874. [Google Scholar] [PubMed]
- Vidal Yucha, S.E.; Tamamoto, K.A.; Kaplan, D.L. The importance of the neuro-immuno-cutaneous system on human skin equivalent design. Cell Prolif. 2019, 52, e12677. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Sakamoto, T.; Tiwari, V. Selective keratinocyte stimulation is sufficient to evoke nociception in mice. Pain 2015, 156, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Radtke, C.; Vogt, P.M.; Devor, M.; Kocsis, J.D. Keratinocytes acting on injured afferents induce extreme neuronal hyperexcitability and chronic pain. Pain 2010, 148, 94–102. [Google Scholar] [CrossRef]
- Hou, Q.; Barr, T.; Gee, L.; Vickers, J.; Wymer, J.; Borsani, E.; Rodella, L.; Getsios, S.; Burdo, T.; Eisenberg, E.; et al. Keratinocyte expression of calcitonin gene-related peptide β: Implications for neuropathic and inflammatory pain mechanisms. Pain 2011, 152, 2036–2051. [Google Scholar] [CrossRef]
- Pan, B.; Schröder, W.; Jostock, R.; Schwartz, M.; Rosson, G.; Polydefkis, M. Nociceptin/orphanin FQ opioid peptide-receptor expression in pachyonychia congenita. J. Peripher. Nerv. Syst. 2018, 23, 241–248. [Google Scholar] [CrossRef]
- Zhao, P.; Barr, T.P.; Hou, Q.; Dib-Hajj, S.D.; Black, J.A.; Albrecht, P.J.; Petersen, K.; Eisenberg, E.; Wymer, J.P.; Rice, F.L.; et al. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: Evidence for a role in pain. Pain 2008, 30, 90–105. [Google Scholar] [CrossRef]
- Dussor, G.; Koerber, H.R.; Oaklander, A.L.; Rice, F.L.; Molliver, D.C. Nucleotide signaling and cutaneous mechanisms of pain transduction. Brain Res. Rev. 2009, 60, 2435. [Google Scholar] [CrossRef]
- Southall, M.D.; Li, T.; Gharibova, L.S.; Pei, Y.; Nicol, G.D.; Travers, J.B. Activation of epidermal vanilloid receptor-1 induces release of proinflammatory mediators in human keratinocytes. J. Pharmacol. Exp. Ther. 2003, 304, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sloniecka, M.; Le Roux, S.; Boman, P.; Bystrom, B.; Zhou, Q.; Danielson, P. Expression profiles of neuropeptides, neurotransmitters, and their receptors in human keratocytes in vitro and in situ. PLoS ONE 2015, 10, e0134157. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, X.; Huang, P.; Lin, F.; Liu, Q.; Xu, L.; Xu, J.; Huang, Y. N-methyl-d-aspartate receptor subunit 2B on keratinocyte mediates peripheral and central sensitization in chronic post-ischemic pain in male rats. Brain Behav. Immun. 2020, 87, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Denda, M.; Fujiwara, S.; Hibino, T. Expression of voltage-gated calcium channel subunit alpha1C in epidermal keratinocytes and effects of agonist and antagonists of the channel on skin barrier homeostasis. Exp. Dermatol. 2006, 15, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, E.P.; Ong, W.Y.; Guo, Y.; Chow, A.W. Epidermal transient receptor potential vanilloid 1 in idiopathic small nerve fibre disease, diabetic neuropathy, and healthy human subjects. Histopathology 2007, 51, 674–680. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, L.H.; Su, T.F.; Li, L.; Zhou, R.; Peng, M.; Wu, C.H.; Yuan, X.C.; Sun, N.; Meng, X.F.; et al. Signaling Mechanism of Cannabinoid Receptor-2 Activation-Induced β-Endorphin Release. Mol. Neurobiol. 2016, 53, 3616–3625. [Google Scholar] [CrossRef]
- Katsuyama, S.; Mizoguchi, H.; Kuwahata, H.; Komatsu, T.; Nagaoka, K.; Nakamura, H.; Bagetta, G.; Sakurada, T.; Sakurada, S. Involvement of peripheral cannabinoid and opioid receptors in β-caryophyllene-induced antinociception. Eur. J. Pain 2013, 17, 664–675. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Porreca, F.; Lai, J.; Albrecht, P.J.; Rice, F.L.; Khodorova, A.; Davar, G.; Makriyannis, A.; Vanderah, T.W.; Mata, H.P.; et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc. Natl. Acad. Sci. USA 2005, 22, 3093–3098. [Google Scholar] [CrossRef]
- Irving, G. The role of the skin in peripheral neuropathic pain. Eur. J. Pain Suppl. 2010, 4, 157–160. [Google Scholar] [CrossRef]
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Gracely, R.H.; Lynch, S.A.; Bennett, G.J. Painful neuropathy: Altered central processing maintained dynamically by peripheral input. PAIN 1992, 51, 175–194. [Google Scholar] [CrossRef]
- Bennett, D.L.; Woods, C.G. Painful and painless channelopathies. Lancet Neurol. 2014, 13, 587–599. [Google Scholar] [CrossRef]
- Drummond, P.D.; Morellini, N.; Finch, P.M.; Birklein, F.; Knudsen, L.F. Complex regional pain syndrome: Intradermal injection of phenylephrine evokes pain and hyperalgesia in a subgroup of patients with upregulated alpha1-adrenoceptors on dermal nerves. PAIN 2018, 159, 2296–2305. [Google Scholar] [CrossRef] [PubMed]
- Balkarli, A.; Sengül, C.; Tepeli, E.; Balkarli, H.; Cobankara, V. Synaptosomal-associated protein 25 (Snap-25) gene polymorphism frequency in fibromyalgia syndrome and relationship with clinical symptoms. BMC Musculoskelet. Disord. 2014, 15, 191. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Moisset, X.; Bouhassira, D.; Avez Couturier, J.; Alchaar, H.; Conradi, S.; Delmotte, M.H.; Lanteri-Minet, M.; Lefaucheur, J.P.; Mick, G.; Piano, V.; et al. Pharmacological and non-pharmacological treatments for neuropathic pain: Systematic review and French recommendations. Rev. Neurol. (Paris) 2020, 176, 325–352. [Google Scholar] [CrossRef]
- Fialho, M.; Brum, E.; Pegoraro, N.S.; Couto, A.; Trevisan, G.; Cruz, L.; Oliveira, S.M. Topical transient receptor potential ankyrin 1 antagonist treatment attenuates nociception and inflammation in an ultraviolet B radiation-induced burn model in mice. J. Dermatol. Sci. 2020, 97, 135–142. [Google Scholar] [CrossRef]
- Ann, J.; Kim, H.S.; Thorat, S.A.; Kim, H.; Ha, H.J.; Choi, K.; Kim, Y.H.; Kim, M.; Hwang, S.W.; Pearce, L.V.; et al. Discovery of Nonpungent Transient Receptor Potential Vanilloid 1 (TRPV1) Agonist as Strong Topical Analgesic. J. Med. Chem. 2020, 63, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.C.; Lewis, R.J. Sodium channels and pain: From toxins to therapies. Br. J. Pharmacol. 2018, 175, 2138–2157. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, M.; Inui, K.; Kakigi, R. Evaluation of nociceptive Aδ- and C-fiber dysfunction with lidocaine using intraepidermal electrical stimulation. Clin. Neurophysiol. 2014, 125, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Whirl-Carrillo, M.; Leeder, J.S.; Klein, T.E.; Altman, R.B. PharmGKB summary: Phenytoin pathway. Pharm. Genom. 2012, 22, 466–470. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Vrancken, A.F.J.E.; Keppel Hesselink, J.M.; van Eijk, R.P.A.; Notermans, N.C. Usefulness of a Double-Blind Placebo-Controlled Response Test to Demonstrate Rapid Onset Analgesia with Phenytoin 10% Cream in Polyneuropathy. J. Pain Res. 2020, 13, 877–882. [Google Scholar] [CrossRef]
- Zhu, W.; Li, T.; Silva, J.R.; Chen, J. Conservation and divergence in NaChBac and NaV1.7 pharmacology reveals novel drug interaction mechanisms. Sci. Rep. 2020, 10, 10730. [Google Scholar] [CrossRef]
- Kern, K.U.; Weiser, T. Topical ambroxol for the treatment of neuropathic pain. An initial clinical observation. Schmerz 2015, 29, S89–S96. [Google Scholar] [CrossRef]
- Pancrazio, J.J.; Kamatchi, G.L.; Roscoe, A.K.; Lynch, C., 3rd. Inhibition of neuronal Na+ channels by antidepressant drugs. J. Pharmacol. Exp. Ther. 1998, 284, 208–214. [Google Scholar]
- Obata, H. Analgesic Mechanisms of Antidepressants for Neuropathic Pain. Int. J. Mol. Sci. 2017, 18, 2483. [Google Scholar] [CrossRef]
- Thompson, D.; Brooks, K.G. Systematic review of topical amitriptyline for the treatment of neuropathic pain. J. Clin. Pharm. Ther. 2015, 40, 496–503. [Google Scholar] [CrossRef]
- Price, N.; Namdari, R.; Neville, J.; Proctor, K.J.; Kaber, S.; Vest, J.; Fetell, M.; Malamut, R.; Sherrington, R.P.; Pimstone, S.N.; et al. Safety and Efficacy of a Topical Sodium Channel Inhibitor (TV-45070) in Patients With Postherpetic Neuralgia (PHN): A Randomized, Controlled, Proof-of-Concept, Crossover Study, With a Subgroup Analysis of the Nav1.7 R1150W Genotype. Clin. J. Pain 2017, 33, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, E. Inhibition of Fast Nerve Conduction Produced by Analgesics and Analgesic Adjuvants-Possible Involvement in Pain Alleviation. Pharmaceuticals (Basel) 2020, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y. TRPs and pain. Semin Immunopathol. 2016, 38, 277e91. [Google Scholar] [CrossRef]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Vij, A.S.; Sharma, M. Mechanisms and clinical uses of capsaicin. Eur. J. Pharmacol. 2013, 720, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Rice, A.S.; Cole, P.; Tan, T.; Moore, R.A. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst. Rev. 2017, 13, CD007393. [Google Scholar] [CrossRef] [PubMed]
- Nozadze, I.; Tsiklauri, N.; Gurtskaia, G.; Tsagareli, M.G. NSAIDs attenuate hyperalgesia induced by TRP channel activation. Data Brief. 2016, 6, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, J.; Goto, M.; Denda, S.; Nakatani, M.; Takasugi, Y.; Tsuchiya, K.; Shimizu, Y.; Takatsuru, Y.; Denda, M. External negative electric potential accelerates exocytosis of lamellar bodies in human skin ex vivo. Exp. Dermatol. 2013, 22, 421–423. [Google Scholar] [CrossRef]
- Kawabata, A. Targeting Ca(v)3.2 T-type calcium channels as a therapeutic strategy for chemotherapy-induced neuropathic pain. Nihon Yakurigaku Zasshi. 2013, 141, 81–84. [Google Scholar] [CrossRef][Green Version]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflug. Arch. 2014, 466, 701–706. [Google Scholar] [CrossRef]
- Hiom, S.; Patel, G.K.; Newcombe, R.G.; Khot, S.; Martin, C. Severe postherpetic neuralgia and other neuropathic pain syndromes alleviated by topical gabapentin. Br. J. Dermatol. 2015, 173, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Oyama, Y.; Sadoshima, J.; Tokutomi, N.; Akaike, N. Some properties of inhibitory action of lidocaine on the Ca2þ current of single isolated frog sensory neurons. Brain Res. 1988, 442, 223e8. [Google Scholar] [CrossRef]
- Woolf, C.J.; Thompson, S.W.N. The induction and maintenance of central sensitization is dependent on N-methyl-D aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 1991, 44, 293–299. [Google Scholar] [CrossRef]
- Jang, J.H.; Nam, T.S.; Jun, J.; Jung, S.J.; Kim, D.W.; Leem, J.W. Peripheral NMDA Receptors Mediate Antidromic Nerve Stimulation-Induced Tactile Hypersensitivity in the Rat. Mediat. Inflamm. 2015, 2015, 793624. [Google Scholar] [CrossRef] [PubMed]
- Warncke, T.; Jørum, E.; Stubhaug, A. Local treatment with the N-methyl-d-aspartate receptor antagonist ketamine, inhibit development of secondary hyperalgesia in man by a peripheral action. Neurosci. Lett. 1997, 227, 1–4. [Google Scholar] [CrossRef]
- Morhenn, V.B.; Murakami, M.; O’Grady, T.; Nordberg, J.; Gallo, R.L. Characterization of the expression and function of N-methyl-D-aspartate receptor in keratinocytes. Exp. Dermatol. 2004, 13, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kopsky, D.J.; Keppel Hesselink, J.M.; Bhaskar, A.; Hariton, G.; Romanenko, V.; Casale, R. Analgesic effects of topical ketamine. Minerva Anestesiol. 2015, 81, 440–449. [Google Scholar]
- Sawynok, J.; Zinger, C. Topical amitriptyline and ketamine for post-herpetic neuralgia and other forms of neuropathic pain. Expert Opin. Pharmacother. 2016, 17, 601–609. [Google Scholar] [CrossRef]
- Mahoney, J.M.; Vardaxis, V.; Moore, J.L.; Hall, A.M.; Haffner, K.E.; Peterson, M.C. Topical ketamine cream in the treatment of painful diabetic neuropathy: A randomized, placebo-controlled, double blind initial study. J. Am. Pediatr. Med. Assoc. 2012, 102, 178–183. [Google Scholar] [CrossRef]
- Barygin, O.I.; Nagaeva, E.I.; Tikhonov, D.B.; Belinskaya, D.A.; Vanchakova, N.P.; Shestakova, N.N. Inhibition of the NMDA and AMPA receptor channels by antidepressants and antipsychotics. Brain Res. 2017, 1660, 58–66. [Google Scholar] [CrossRef]
- Dong, X.D.; Svensson, P.; Cairns, B.E. The analgesic action of topical diclofenac may be mediated through peripheral NMDA receptor antagonism. Pain 2009, 147, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, C.J.; Rouppe van der Voort, C.; van de Pol, M.; Kavelaars, A. Cytokines regulate alpha(1)-adrenergic receptor mRNA expression in human monocytic cells and endothelial cells. J. Neuroimmunol. 2002, 125, 66–72. [Google Scholar] [CrossRef]
- Drummond, E.S.; Maker, G.; Birklein, F.; Finch, P.M.; Drummond, P.D. Topical prazosin attenuates sensitivity to tactile stimuli in patients with complex regional pain syndrome. Eur. J. Pain 2016, 20, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, F.; Kiuchi, Y.; Ishikawa, Y.; Otsuka, N.; Masuda, Y.; Oguchi, K.; Hosoyamada, A. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth. Analg. 2002, 95, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.U.; Zhang, Y.; Chen, L.; Cohen, A.; Hillary, K.S.; Vo, T.; Houghton, M.; Mao, J. Effect of 1.5% topical diclofenac on clinical neuropathic pain. Anaesthesiology 2015, 123, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Wiffen, P.J.; Kalso, E.A.; Bell, R.F.; Aldington, D.; Phillips, T.; Gaskell, H.; Moore, R.A. Topical analgesics for acute and chronic pain in adults-an overview of Cochrane Reviews. Cochrane Database Syst. Rev. 2017, 5, CD008609. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef]
- Mika, J.; Rojewska, E.; Makuch, W.; Korostynski, M.; Luvisetto, S.; Marinelli, S.; Pavone, F.; Przewlocka, B. The effect of botulinum neurotoxin A on sciatic nerve injury-induced neuroimmunological changes in rat dorsal root ganglia and spinal cord. Neuroscience 2011, 175, 358–366. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Lucchetti, F.; Marchi, F.; Cobianchi, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Botulinum neurotoxins and formalin-induced pain: Central vs. peripheral effects in mice. Brain Res. 2006, 1082, 124–131. [Google Scholar] [CrossRef]
- Ngo, D.H.; Vo, T.S. An Updated Review on Pharmaceutical Properties of Gamma-Aminobutyric Acid. Molecules 2019, 24, 2678. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qin, X.; Du, H.; Li, N.; Ren, W.; Peng, Y. The immunological function of GABAergic system. Front. Biosci. (Landmark Ed). 2017, 1, 1162–1172. [Google Scholar]
- Whitehead, R.A.; Puil, E.; Ries, C.R.; Schwarz, S.K.; Wall, R.A.; Cooke, J.E.; Putrenko, I.; Sallam, N.A.; MacLeod, B.A. GABA(B) receptor-mediated selective peripheral analgesia by the non-proteinogenic amino acid, isovaline. Neuroscience 2012, 213, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Kopsky, D.J.; Hesselink, J.M.K. Neuropathic pain as a result of acromegaly, treated with topical baclofen cream. J. Pain Symptom. Manag. 2013, 46, e4–e5. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.L.; Wos, E.J.; Qin, R.; Mattar, B.I.; Green, N.B.; Lanier, K.S.; Bearden, J.D., 3rd; Kugler, J.W.; Hoff, K.L.; Reddy, P.S.; et al. A double-blind, placebo-controlled trial of a topical treatment for chemotherapy-induced peripheral neuropathy: NCCTG trial N06CA. Support Care Cancer 2011, 19, 833–841. [Google Scholar] [CrossRef]
- McCarson, K.E.; Duric, V.; Reisman, S.A.; Winter, M.; Enna, S.J. GABA(B) receptor function and subunit expression in the rat spinal cord as indicators of stress and the antinociceptive response to antidepressants. Brain Res. 2006, 1068, 109–117. [Google Scholar] [CrossRef]
- Irifune, M.; Sato, T.; Kamata, Y.; Nishikawa, T.; Dohi, T.; Kawahara, M. Evidence for GABA(A) receptor agonistic properties of ketamine: Convulsive and anesthetic behavioral models in mice. Anesth. Analg. 2000, 91, 230–236. [Google Scholar]
- Granger, P.; Biton, B.; Faure, C.; Vige, X.; Depoortere, H.; Graham, D.; Langer, S.Z.; Scatton, B.; Avenet, P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995, 47, 1189–1196. [Google Scholar]
- Buerkle, H. Peripheral antinociceptive action of alpha2- adrenoceptor agonist. Baillières Clin. Anaesthesiol. 2000, 2, 411–418. [Google Scholar]
- Riedl, M.S.; Schnell, S.A.; Overland, A.C.; Chabot-Doré, A.J.; Taylor, A.M.; Ribeiro-da-Silva, A.; Elde, R.P.; Wilcox, G.L.; Stone, L.S. Coexpression of alpha 2A-adrenergic and delta-opioid receptors in substance P-containing terminals in rat dorsal horn. J. Comp. Neurol. 2009, 513, 385–398. [Google Scholar] [CrossRef]
- Shi, T.S.; Winzer-Serhan, U.; Leslie, F.; Hokfelt, T. Distribution and regulation of alpha(2)-adrenoceptors in rat dorsal root ganglia. Pain 2000, 84, 319–330. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawasaki, C.; Ueki, M.; Hamada, K.; Habe, K.; Sata, T. Dexmedetomidine suppresses proinflammatory mediator production in human whole blood in vitro. J. Trauma Acute Care Surg. 2013, 74, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, A.; Woron, J.; Dobrogowski, J.; Jakowicka-Wordliczek, J.; Wordliczek, J. Topical clonidine for neuropathic pain (review). Cochrane Database Syst. Rev. 2015, 8, CD010967. [Google Scholar] [PubMed]
- Obara, I.; Parkitna, J.R.; Korostynski, M.; Makuch, W.; Kaminska, D.; Przewlocka, B.; Przewlocki, R. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 2009, 141, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Machelska, H.; Celik, M.Ö. Opioid Receptors in Immune and Glial Cells-Implications for Pain Control. Front Immunol. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Bigliardi-Qi, M.; Sumanovski, L.T.; Büchner, S.; Rufli, T.; Bigliardi, P.L. Mu-opiate receptor and Beta-endorphin expression in nerve endings and keratinocytes in human skin. Dermatology 2004, 209, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Anderson, M.; Yang, F.; Tiwari, V.; Zheng, Q.; He, S.Q.; Zhang, T.; Shu, B.; Chen, X.; Grenald, S.A.; et al. Peripherally acting m-opioid receptor agonists attenuate ongoing pain-associated behavior and spontaneous neuronal activity after nerve injury in rats. Anesthesiology 2018, 128, 1220–1236. [Google Scholar] [CrossRef]
- Kopsky, D.J.; Bhaskar, A.K.; Zonneveldt, H.J.; Keppel Hesselink, J.M. Topical loperamide for the treatment of localized neuropathic pain: A case report and literature review. J. Pain Res. 2019, 12, 1189–1192. [Google Scholar] [CrossRef]
- Ciałkowska-Rysz, A.; Dzierżanowski, T. Topical morphine for treatment of cancer-related painful mucosal and cutaneous lesions: A double-blind, placebo-controlled cross-over clinical trial. Arch. Med. Sci. 2019, 15, 146–151. [Google Scholar] [CrossRef]
- Maldonado, R.; Banos, J.E.; Cabanero, D. The endocannabinoid system and neuropathic pain. Pain 2016, 157, S23–S32. [Google Scholar] [CrossRef]
- Lötsch, J.; Weyer-Menkhoff, I.; Tegeder, I. Current evidence of cannabinoid-based analgesia obtained in preclinical and human experimental settings. Eur. J. Pain 2018, 22, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid Delivery Systems for Pain and Inflammation Treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef] [PubMed]
- Keppel Hesselink, J.M.; Kopsky, D.J.; Sajben, N. New topical treatment of vulvodynia based on the pathogenetic role of cross talk between nociceptors, immunocompetent cells, and epithelial cells. J. Pain Res. 2016, 9, 757–762. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Ion Channel Enzyme | Topical Agent Utilized in Clinical Practice | Possible Site of Action | Reference | |
|---|---|---|---|---|
| EXCITATORY | Nav | Lidocaine Antidepressants: -Amitriptyline -Doxepin Phenytoin Ambroxol TV-45070 Opioids NSAIDs Clonidine | Neurons Keratinocytes | [36,37,38,91,106,114,115,116,117,118,119,120,121,122,123] |
| TRPV1 | Capsaicin NSAIDs | Neurons Keratinocytes Immune cells | [69,70,76,93,94,97,124,125,126,127,128] | |
| VGCC | Gabapentin Lidocaine | Neurons Keratinocytes | [39,61,76,91,94,96,129,130,131,132,133] | |
| NMDAR | Ketamine Antidepressants: -amitriptyline NSAID-Diclofenac | Neurons Keratinocytes Immune cells | [95,134,135,136,137,136,137,138,139,140,141,142] | |
| α1-AR | Prazosin Antidepressants | Neurons Keratinocytes Immune cells | [31,33,43,44,45,46,67,73,143,144,145] | |
| COX-2 | NSAIDs | Neurons Immune cells Schwann cells | [75,146,147] | |
| SNAP-25 | Botulinum toxin A | Neurons Immune cells Glial cells | [148,149,150,151] |
| Receptor Ion Channel | Topical Agent Utilized in Clinical Practice | Possible Site of Action | Reference | |
|---|---|---|---|---|
| INHIBITORY | GABAR | Antidepressants: Amitriptyline | Neurons Keratinocytes Immune cells | [83,84,152,153,157] |
| GABAAR | Ketamine Phenytoin | [158,159] | ||
| GABABR | Baclofen | [154,155,156] | ||
| α2-AR | Clonidine | Neurons | [160,161,162,163,164] | |
| OR | Opioids | Neurons Keratinocytes Immune cells | [79,80,82,165,166,167,168,169,170] | |
| CB | Cannabinoids | Neurons Keratinocytes Immune cells | [78,81,98,99,100,171,172,173,174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain. Pharmaceuticals 2021, 14, 77. https://doi.org/10.3390/ph14020077
Kocot-Kępska M, Zajączkowska R, Mika J, Wordliczek J, Dobrogowski J, Przeklasa-Muszyńska A. Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain. Pharmaceuticals. 2021; 14(2):77. https://doi.org/10.3390/ph14020077
Chicago/Turabian StyleKocot-Kępska, Magdalena, Renata Zajączkowska, Joanna Mika, Jerzy Wordliczek, Jan Dobrogowski, and Anna Przeklasa-Muszyńska. 2021. "Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain" Pharmaceuticals 14, no. 2: 77. https://doi.org/10.3390/ph14020077
APA StyleKocot-Kępska, M., Zajączkowska, R., Mika, J., Wordliczek, J., Dobrogowski, J., & Przeklasa-Muszyńska, A. (2021). Peripheral Mechanisms of Neuropathic Pain—The Role of Neuronal and Non-Neuronal Interactions and Their Implications for Topical Treatment of Neuropathic Pain. Pharmaceuticals, 14(2), 77. https://doi.org/10.3390/ph14020077