
Monofunctional Platinum(II) Anticancer Agents


Abstract
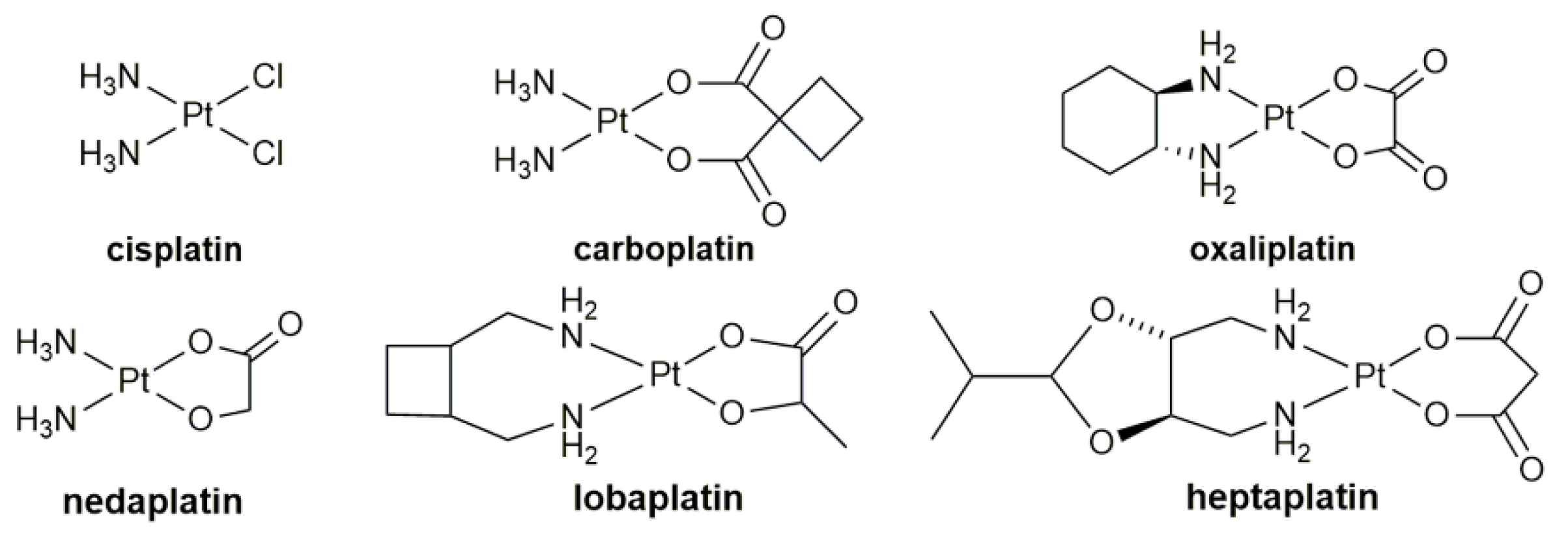
1. Introduction
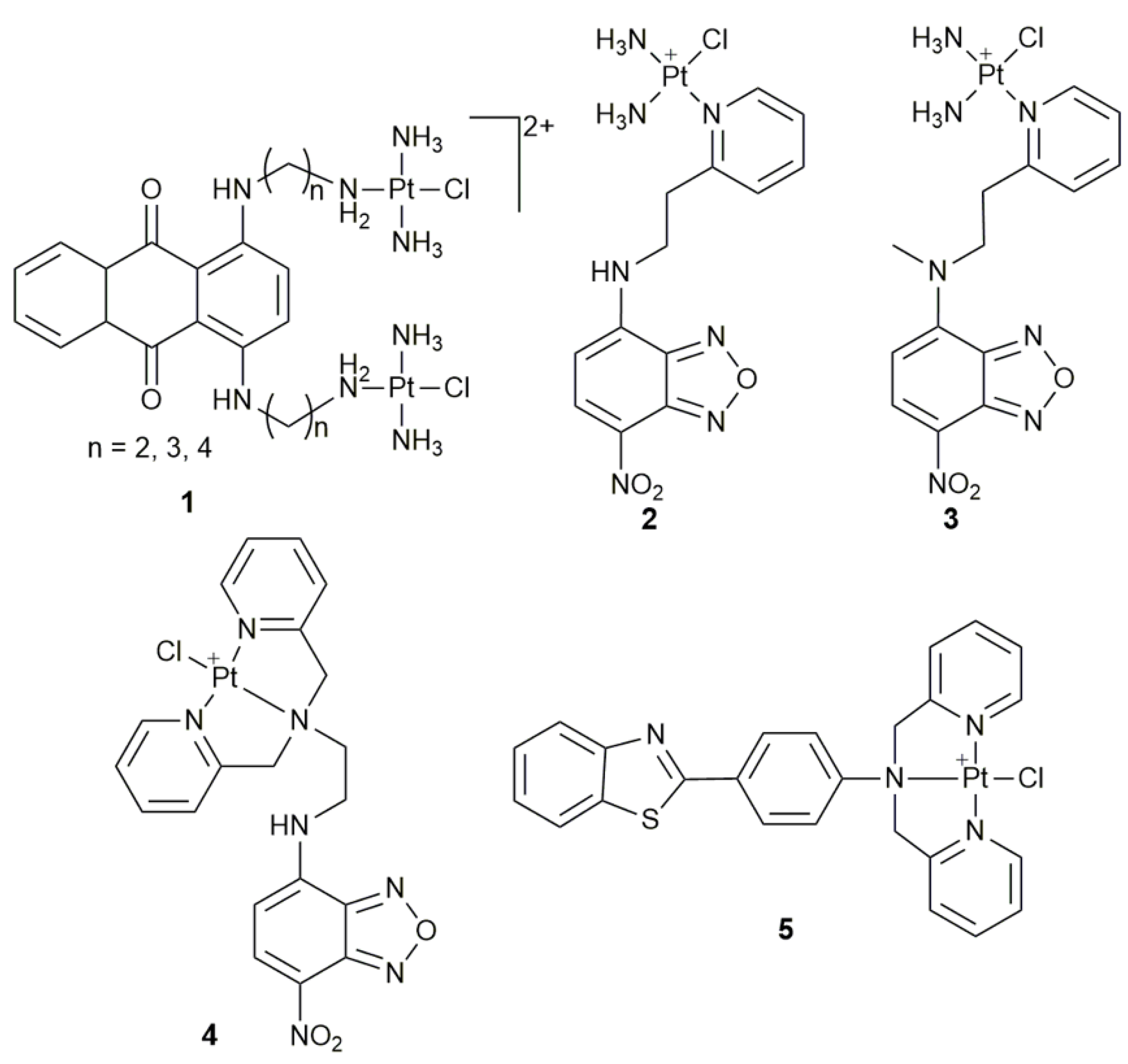
2. Fluorescent Monofunctional PtII Complexes
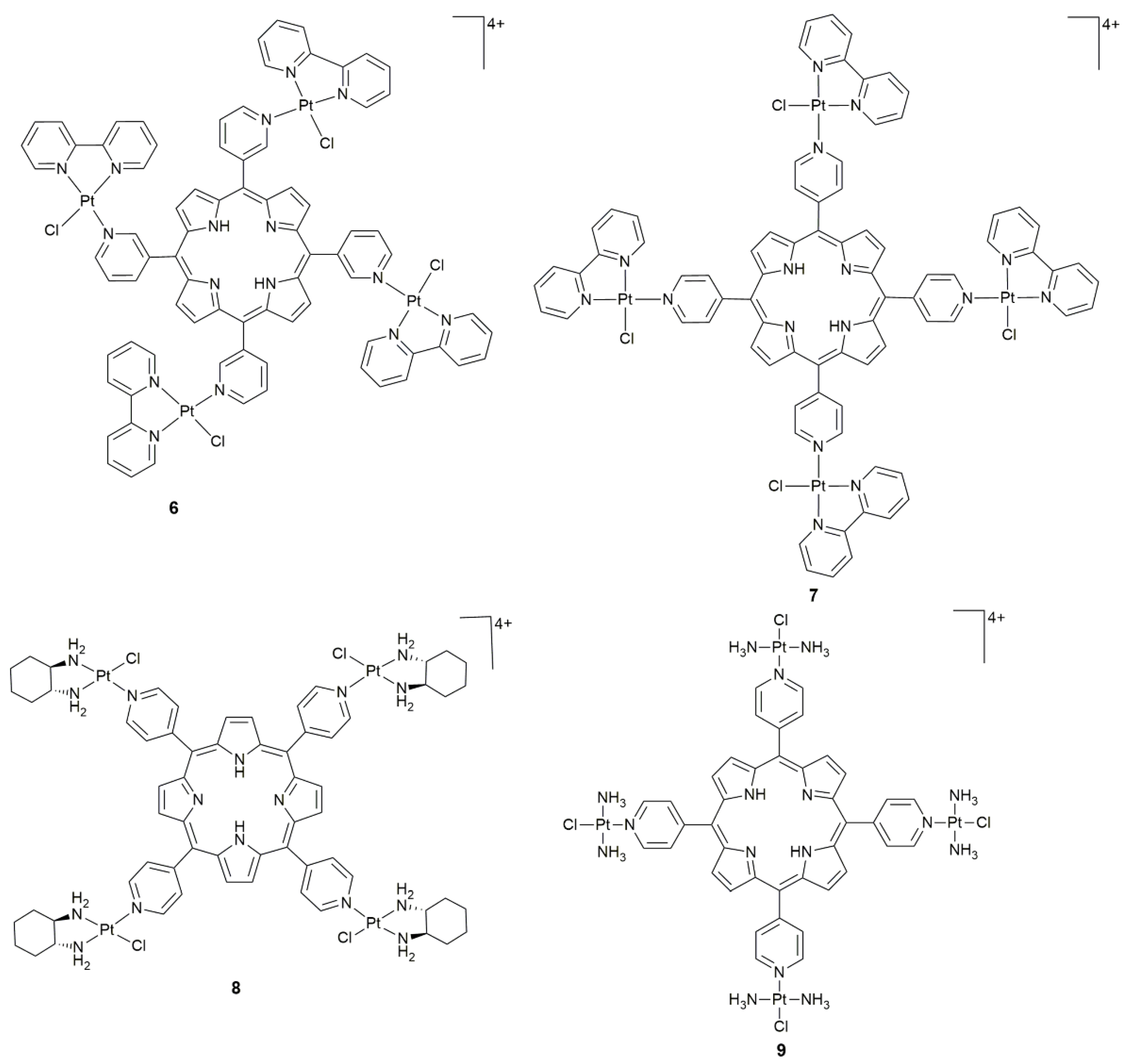
3. Photoactive Monofunctional PtII Complexes
4. Targeted Monofunctional PtII Complexes
5. Miscellaneous Monofunctinoal PtII Complexes
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Codes | Designated Cell Lines |
|---|---|
| A2780; CP70 | human ovarian cancer cell |
| A2780/DDP | cisplatin-resistant human ovarian cancer cell |
| A375 | human melanoma cell |
| A549 | human lung carcinoma cell |
| A549cisR; A549R | cisplatin-resistant human lung carcinoma cell |
| B16-F10 | mouse melanoma cell |
| BEAS-2B | human normal bronchial epithelial cell |
| BEL-7402 | human hepatoma cell |
| Caov-3 | human ovarian cancer cell |
| CAPAN-1 | human pancreatic adenocarcinoma cell |
| colon26 | murine colon carcinoma cell |
| HaCaT | human keratinocyte |
| HCT-116 | human colon carcinoma cell |
| HEK293T; 293T | human embryonic kidney cell |
| HeLa | human cervical carcinoma cell |
| HepG2 | human hepatocarcinoma cell |
| HK-2 | human renal tubular epithelial cell |
| HL-7702; LO2 | human normal liver cell |
| HUVEC | human umbilical vein endothelial cell |
| MCF-10A | human normal breast epithelial cell |
| MCF-7 | human breast adenocarcinoma cell |
| MDA-MB-231 | human breast cancer cell |
| MG-63 | human osteosarcoma cell |
| MGC80-3 | human gastric adenocarcinoma cell |
| MRC-5 | human normal lung fibroblast cell |
| NCI-H460 | human lung carcinoma cell |
| PC-3; PC3 | human prostate cancer cell |
| sarcoma180 | mouse sarcoma cell |
| SEM-1 | human testicular seminoma cell |
| SGC-7901 | human gastric adenocarcinoma cell |
| SK-MEL-28 | human melanoma cell |
| SKOV-3 | human ovarian cancer cell |
| SKOV-3/DDP | cisplatin-resistant human ovarian cancer cell |
| SMMC | human hepatocellular carcinoma cell |
| T-24 | human bladder cancer cell |
| TCam-2 | human testicular seminoma cell |
| U2-OS | human osteosarcoma cell |
| U2-OS/Pt | cisplatin-resistant human osteosarcoma cell |
| U87M | human glioblastoma cell |
| WM1366 | human melanoma cell |
References
- Shimada, M.; Itamochi, H.; Kigawa, J. Nedaplatin: A cisplatin derivative in cancer chemotherapy. Cancer Manag. Res. 2013, 5, 67–76. [Google Scholar] [CrossRef]
- McKeage, M.J. Lobaplatin: A new antitumour platinum drug. Expert. Opin. Investig. Drugs 2001, 10, 119–128. [Google Scholar] [CrossRef]
- Lee, K.H.; Hyun, M.S.; Kim, H.K.; Jin, H.M.; Yang, J.; Song, H.S.; Do, Y.R.; Ryoo, H.M.; Chung, J.S.; Zang, D.Y.; et al. Randomized, multicenter, phase III trial of heptaplatin 1-h infusion and 5-fluorouracil combination chemotherapy comparing with cisplatin and 5-fluorouracil combination chemotherapy in patients with advanced gastric cancer. Cancer Res. Treat. 2009, 41, 12–18. [Google Scholar] [CrossRef]
- Kenny, R.G.; Chuah, S.W.; Crawford, A.; Marmion, C.J. Platinum(IV) prodrugs—A step closer to Ehrlich’s vision? Eur. J. Inorg. Chem. 2017, 2017, 1596–1612. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Dilrub, S.; Kalayd, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Wang, X.Y.; Guo, Z.J. Towards the rational design of platinum(II) and gold(III) complexes as antitumour agents. Dalton Trans. 2008, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Polychronopoulos, P.; Iconomou, G.; Chroni, E.; Kalofonos, H.P. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat. Rev. 2008, 34, 368–377. [Google Scholar] [CrossRef] [PubMed]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef]
- Min, Y.Z.; Mao, C.Q.; Chen, S.M.; Ma, G.L.; Wang, J.; Liu, Y.Z. Combating the drug resistance of cisplatin using a platinum prodrug based delivery system. Angew. Chem. Int. Ed. 2012, 51, 6742–6747. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Brabec, V.; Hrabina, O.; Kasparkova, J. Cytotoxic platinum coordination compounds. DNA binding agents. Coord. Chem. Rev. 2017, 351, 2–31. [Google Scholar] [CrossRef]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
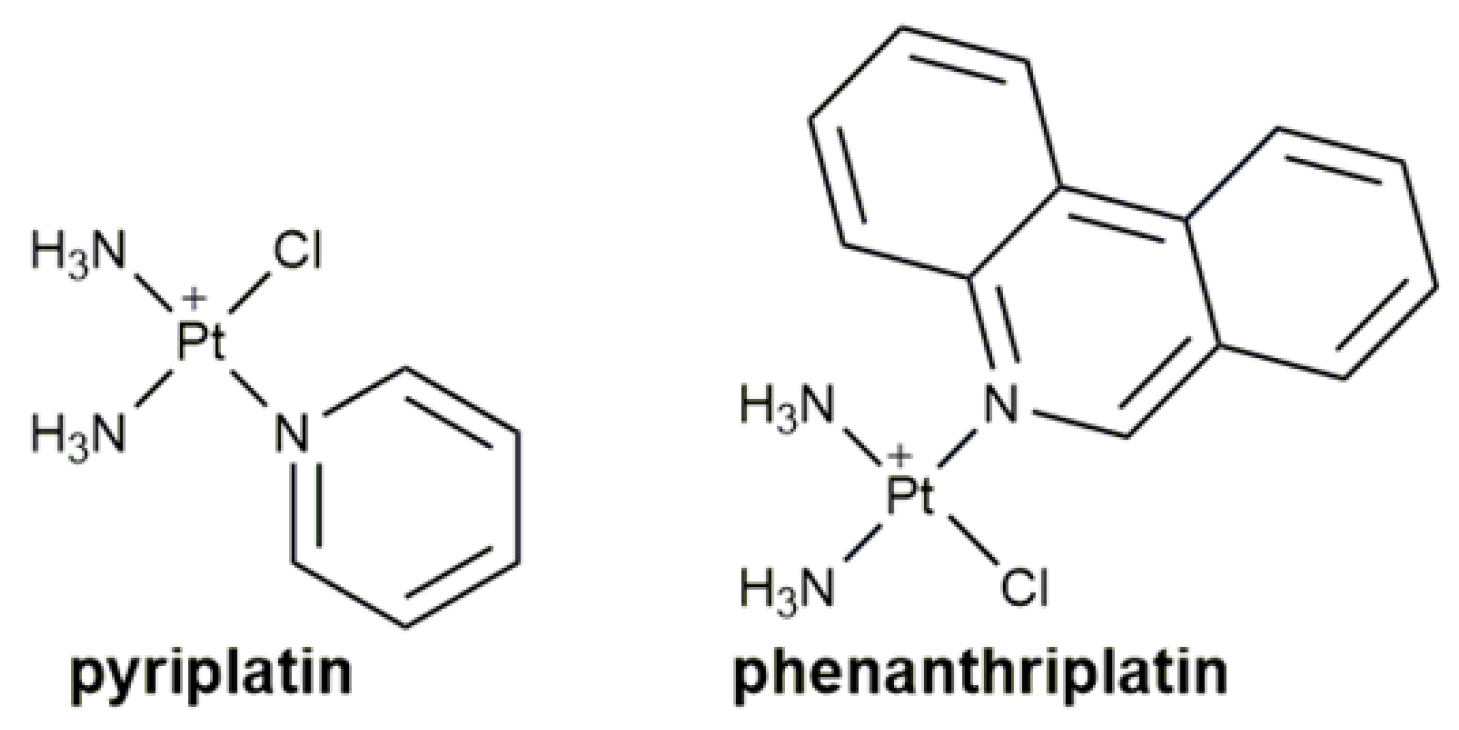
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Guo, W.J.; Zhang, Y.M.; Zhang, L.; Huang, B.; Tao, F.F.; Chen, W.; Guo, Z.J.; Xu, Q.; Sun, Y. Novel monofunctional platinum(II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013, 9, 996–1008. [Google Scholar] [CrossRef]
- Riddell, I.A.; Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Nucleotide binding preference of the monofunctional platinum anticancer-agent phenanthriplatin. Chem. Eur. J. 2016, 22, 7574–7581. [Google Scholar] [CrossRef]
- Malina, J.; Farrell, N.P.; Brabec, V. DNA condensing effects and sequence selectivity of dna binding of antitumor noncovalent polynuclear platinum complexes. Inorg. Chem. 2014, 53, 1662–1671. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Lippard, S.J. The chiral potential of phenanthriplatin and its influence on guanine binding. J. Am. Chem. Soc. 2014, 136, 2126–2134. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Alexander, S.M.; Lin, W.; Lippard, S.J. Effects of monofunctional platinum agents on bacterial growth: A retrospective study. J. Am. Chem. Soc. 2014, 136, 116–118. [Google Scholar] [CrossRef]
- Lovejoy, K.S.; Serova, M.; Bieche, I.; Emami, S.; D’Incalci, M.; Broggini, M.; Erba, E.; Gespach, C.; Cvitkovic, E.; Faivre, S.; et al. Spectrum of cellular responses to pyriplatin, a monofunctional cationic antineoplastic platinum(II) compound, in human cancer cells. Mol. Cancer Ther. 2011, 10, 1709–1719. [Google Scholar] [CrossRef]
- Zhou, W.; Almeqdadi, M.; Xifaras, M.E.; Riddell, I.A.; Yilmaz, Ö.H.; Lippard, S.J. The effect of geometric isomerism on the anticancer activity of the monofunctional platinum complex trans-[Pt(NH3)2(phenanthridine)Cl]NO3. Chem. Commun. 2018, 54, 2788–2791. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Wilson, J.J.; Lippard, S.J. Monofunctional and higher-valent platinum anticancer agents. Inorg. Chem. 2013, 52, 12234–12249. [Google Scholar] [CrossRef] [PubMed]
- Macquet, J.P.; Butour, J.L. Platinum-amine compounds: Importance of the labile and inert ligands for their pharmacological activities toward L1210 leukemia cells. J. Natl. Cancer Inst. 1983, 70, 899–905. [Google Scholar] [PubMed]
- Cleare, M.J.; Hoeschele, J.D. Antitumor activity of group VIII transition metal complexes. I. Platinum(II) complexes. Bioinorg. Chem. 1973, 2, 187–210. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Bancroft, D.P.; Lippard, S.J. Synthesis, characterization, and biological activity of cis-diammineplatinum(II) complexes of the DNA intercalators 9-aminoacridine and chloroquine. J. Am. Chem. Soc. 1990, 112, 1590–1596. [Google Scholar] [CrossRef]
- Hollis, L.S.; Sundquist, W.I.; Burstyn, J.N.; Heiger-Bernays, W.J.; Bellon, S.F.; Ahmed, K.J.; Amundsen, A.R.; Stern, E.W.; Lippard, S.J. Mechanistic studies of a novel class of trisubstituted platinum(II) antitumor agents. Cancer Res. 1991, 51, 1866–1875. [Google Scholar]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef]
- Lovejoy, K.S.; Todd, R.C.; Zhang, S.; McCormick, M.S.; D’Aquino, J.A.; Reardon, J.T.; Sancar, A.; Giacomini, K.M.; Lippard, S.J. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proc. Natl. Acad. Sci. USA 2008, 105, 8902–8907. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, G.Y.; Huang, X.H.; Lippard, S.J. X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc. Natl. Acad. Sci. USA 2010, 107, 9584–9589. [Google Scholar] [CrossRef]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Reedijk, J. Interactions of anticancer Pt compounds with proteins: An overlooked topic in medicinal inorganic chemistry. Chem. Sci. 2012, 3, 3135–3144. [Google Scholar] [CrossRef]
- Kellinger, M.W.; Park, G.Y.; Chong, J.; Lippard, S.J.; Wang, D. Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J. Am. Chem. Soc. 2013, 135, 13054–13061. [Google Scholar] [CrossRef] [PubMed]
- Almaqwashi, A.A.; Zhou, W.; Naufer, M.N.; Riddell, I.A.; Yilmaz, Ö.H.; Lippard, S.J.; Williams, M.C. DNA intercalation facilitates efficient DNA-targeted covalent binding of phenanthriplatin. J. Am. Chem. Soc. 2019, 141, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.T.; Park, G.Y.; Johnstone, T.C.; Lee, Y.S.; Yang, W.; Lippard, S.J. Structural and mechanistic studies of polymerase η bypass of phenanthriplatin DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 9133–9138. [Google Scholar] [CrossRef] [PubMed]
- Baruah, H.; Rector, C.L.; Monnier, S.M.; Bierbach, U. Mechanism of action of non-cisplatin type DNA-targeted platinum anticancer agents: DNA interactions of novel acridinylthioureas and their platinum conjugates. Biochem. Pharm. 2002, 64, 191–200. [Google Scholar] [CrossRef]
- Guddneppanavar, R.; Saluta, G.; Kucera, G.L.; Bierbach, U. Synthesis, biological activity, and DNA-damage profile of platinum threading intercalator conjugates designed to target adenine. J. Med. Chem. 2006, 49, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Guddneppanavar, R.; Choudhury, J.R.; Kheradi, A.R.; Steen, B.D.; Saluta, G.; Kucera, G.L.; Day, C.S.; Bierbach, U. Effect of the diamine nonleaving group in platinum-acridinylthiourea conjugates on DNA damage and cytotoxicity. J. Med. Chem. 2007, 50, 2259–2263. [Google Scholar] [CrossRef]
- Smyre, C.L.; Saluta, G.; Kute, T.E.; Kucera, G.L.; Bierbach, U. Inhibition of DNA synthesis by a platinum−acridine hybrid agent leads to potent cell kill in nonsmall cell lung cancer. ACS Med. Chem. Lett. 2011, 2, 870–874. [Google Scholar] [CrossRef]
- Kalayda, G.V.; Jansen, B.A.J.; Wielaard, P.; Tanke, H.J.; Reedijk, J. Dinuclear platinum anticancer complexes with fluorescent N,N’-bis(aminoalkyl)-1,4-diaminoanthraquinones: Cellular processing in two cisplatin-resistant cell lines reflects the differences in their resistance profiles. J. Biol. Inorg. Chem. 2005, 10, 305–315. [Google Scholar] [CrossRef]
- Kalayda, G.V.; Jansen, B.A.J.; Molenaar, C.; Wielaard, P.; Tanke, H.J.; Reedijk, J. Dinuclear platinum complexes with N,N’-bis(aminoalkyl)-1,4-diaminoanthraquinones as linking ligands. Part II. Cellular processing in A2780 cisplatin-resistant human ovarian carcinoma cells: New insights into the mechanism of resistance. J. Biol. Inorg. Chem. 2004, 9, 414–422. [Google Scholar] [CrossRef]
- Jansen, B.A.J.; Wielaard, P.; Kalayda, G.V.; Ferrari, M.; Molenaar, C.; Tanke, H.J.; Brouwer, J.; Reedijk, J. Dinuclear platinum complexes with N,N’-bis(aminoalkyl)-1,4-diaminoanthraquinones as linking ligands. Part I. Synthesis, cytotoxicity, and cellular studies in gA2780 human ovarian carcinoma cells. J. Biol. Inorg. Chem. 2004, 9, 403–413. [Google Scholar] [CrossRef]
- Wu, S.D.; Wang, X.Y.; Zhu, C.C.; Song, Y.J.; Wang, J.; Li, Y.Z.; Guo, Z.J. Monofunctional platinum complexes containing a 4-nitrobenzo-2-oxa-1,3-diazole fluorophore: Distribution in tumour cells. Dalton Trans. 2011, 40, 10376–10382. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.D.; Zhu, C.C.; Zhang, C.L.; Yu, Z.; He, W.J.; He, Y.F.; Li, Y.Z.; Wang, J.; Guo, Z.J. In vitro and in vivo fluorescent imaging of a monofunctional chelated platinum complex excitable using visible light. Inorg. Chem. 2011, 50, 11847–11849. [Google Scholar] [CrossRef]
- Chen, Z.F.; Zhang, S.P.; Shen, L.; Zhu, Z.Z.; Zhang, J. Fluorescence imaging of a new monofunctional platinum(II) complex containing a thioflavin-T (ThT)-based fluorophore. New J. Chem. 2015, 39, 1592–1596. [Google Scholar] [CrossRef]
- Zhang, C.; Guan, R.L.; Liao, X.X.; Ouyang, C.; Rees, T.W.; Liu, J.P.; Chen, Y.; Ji, L.N.; Chao, H. A mitochondria-targeting dinuclear Ir-Ru complex as a synergistic photoactivated chemotherapy and photodynamic therapy agent against cisplatin-resistant tumour cells. Chem. Commun. 2019, 55, 12547–12550. [Google Scholar] [CrossRef]
- Bonnet, S. Why develop photoactivated chemotherapy? Dalton Trans. 2018, 47, 10330–10343. [Google Scholar] [CrossRef] [PubMed]
- Dabids, L.M.; Kleemann, B. Combating melanoma: The use of photodynamic therapy as a novel, adjuvant therapeutic tool. Cancer Treat Rev. 2011, 37, 465–475. [Google Scholar]
- Zhou, Z.J.; Song, J.B.; Nie, L.M.; Chen, X.Y. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 2016, 45, 6597–6626. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.H.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Rubbiani, R.; Gasser, G.; Spingler, B. Visible-light-induced annihilation of tumor cells with platinum-porphyrin conjugates. Angew. Chem. Int. Ed. 2014, 53, 6938–6941. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.C.; Yu, S.; Saha, M.L.; Zhou, J.; Cook, T.R.; Yung, B.C.; Chen, J.; Mao, Z.W.; Zhang, F.W.; Zhou, Z.J.; et al. A discrete organoplatinum(II) metallacage as a multimodality theranostic platform for cancer photochemotherapy. Nat. Commun. 2018, 9, 4335. [Google Scholar] [CrossRef]
- Santoro, A.M.; Lo Giudice, M.C.; D’Urso, A.; Lauceri, R.; Purrello, R.; Milardi, D. Cationic porphyrins are reversible proteasome inhibitors. J. Am. Chem. Soc. 2012, 134, 10451–10457. [Google Scholar] [CrossRef]
- Bacellar, I.O.; Tsubone, T.M.; Pavani, C.; Baptista, M.S. Photodynamic efficiency: From molecular photochemistry to cell death. Int. J. Mol. Sci. 2015, 16, 20523–20559. [Google Scholar] [CrossRef] [PubMed]
- Tasso, T.T.; Tsubone, T.M.; Baptista, M.S.; Mattiazzi, L.M.; Acunha, T.V.; Iglesias, B.A. Isomeric effect on the properties of tetraplatinated porphyrins showing optimized phototoxicity for photodynamic therapy. Dalton Trans. 2017, 46, 11037–11045. [Google Scholar] [CrossRef]
- Couto, G.K.; Pacheco, B.S.; Borba, V.M.; Junior, J.C.R.; Oliveira, T.L.; Segatto, N.V.; Seixas, F.K.; Acunha, T.V.; Iglesias, B.A.; Collares, T. Tetra-cationic platinum(II) porphyrins like a candidate photosensitizers to bind, selective and drug delivery for metastatic melanoma. J. Photoch. Photobio. B. 2020, 202, 111725. [Google Scholar] [CrossRef]
- Hu, X.J.; Ogawa, K.; Li, S.; Kiwada, T.; Odani, A. A platinum functional porphyrin conjugate: An excellent cancer killer for photodynamic therapy. Bull. Chem. Soc. Jpn. 2019, 92, 790–796. [Google Scholar] [CrossRef]
- Xu, X.L.; Lin, F.W.; Du, Y.; Zhang, X.; Wu, J.; Xu, Z.K. Graphene oxide nanofiltration membranes stabilized by cationic porphyrin for high salt rejection. Acs Appl. Mater. Interfaces 2016, 8, 12588–12593. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.J.; Ogawa, K.; Kiwada, T.; Odani, A. Water-soluble metalloporphyrinates with excellent photo-induced anticancer activity resulting from high tumor accumulation. J. Inorg. Biochem. 2017, 170, 1–7. [Google Scholar] [CrossRef]
- Li, X.; Zheng, B.D.; Peng, X.H.; Li, S.Z.; Ying, J.W.; Zhao, Y.; Huang, J.D.; Yoon, J. Phthalocyanines as medicinal photosensitizers: Developments in the last five years. Coord. Chem. Rev. 2017, 379, 147–160. [Google Scholar] [CrossRef]
- Wonga, R.C.H.; Lo, P.C.; Ng, D.K.P. Stimuli responsive phthalocyanine-based fluorescent probes and photosensitizers. Coord. Chem. Rev. 2017, 379, 30–46. [Google Scholar] [CrossRef]
- Mitra, K.; Samsó, M.; Lyonsb, C.E.; Hartman, M.C.T. Hyaluronic acid grafted nanoparticles of a platinum(II)–silicon(IV) phthalocyanine conjugate for tumor and mitochondria-targeted photodynamic therapy in red light. J. Mater. Chem. B 2018, 6, 7373–7377. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.F.; Zhang, Y.M.; Zhu, J.H.; Zhang, C.L.; Guo, Z.J. Molecular combo of photodynamic therapeutic agent silicon(IV) phthalocyanine and anticancer drug cisplatin. Chem. Commun. 2009, 908–910. [Google Scholar] [CrossRef]
- Raza, M.K.; Gautam, S.; Garai, A.; Mitra, K.; Kondaiah, P.; Chakravarty, A.R. Monofunctional BODIPY-appended imidazoplatin for cellular imaging and mitochondria-targeted photocytotoxicity. Inorg. Chem. 2017, 56, 11019–11029. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yuan, H.X.; Liu, Z.; Nie, C.Y.; Liu, L.B.; Lv, F.T.; Wang, Y.L.; Wang, S. Cationic oligo(p-phenylene vinylene) materials for combating drug resistance of cancer cells by light manipulation. Adv. Mater. 2014, 26, 5986–5990. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.L.; Zhu, C.C.; Chen, H.C.; Bai, Y.; Shi, X.C.; Jiao, Y.; Chen, Z.Y.; Miao, Y.P.; He, W.J.; Guo, Z.J. A new approach to sensitize antitumor monofunctional platinum(II) complexes via short time photo-irradiation. Inorg. Chem. 2017, 56, 3754–3762. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.J.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.V.; Hambley, T.W. Platinum drug distribution in cancer cells and tumors. Chem. Rev. 2009, 109, 4911–4920. [Google Scholar] [CrossRef]
- Xue, X.L.; Qian, C.G.; Fang, H.B.; Liu, H.K.; Yuan, H.; Guo, Z.J.; Bai, Y.; He, W.J. Photoactivated lysosomal escape of a monofunctional PtII complex Pt-BDPA for nucleus access. Angew. Chem. Int. Ed. 2019, 58, 12661–12666. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.T.; Zhong, Y.F.; Liu, L.Y.; Shen, C.T.; Zeng, W.J.; Wang, F.Y.; Yang, D.Z.; Mao, Z.W. Solution structures of multiple G-quadruplex complexes induced by a platinum(II)-based tripod reveal dynamic binding. Nat. Commun. 2018, 9, 3496. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.H.; Mu, G.; Zhong, Y.F.; Zhang, T.P.; Cao, Q.; Ji, L.N.; Zhao, Y.; Mao, Z.W. Trigeminal star-like platinum complexes induce cancer cell senescence through quadruplex-mediated telomere dysfunction. Chem. Commun. 2016, 52, 14101–14104. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.F.; Zhang, H.; Liu, W.T.; Zheng, X.H.; Zhou, Y.W.; Cao, Q.; Shen, Y.; Zhao, Y.; Qin, P.Z.; Ji, L.N.; et al. A platinum(II)-based photosensitive tripod as an effective photodynamic anticancer agent through DNA damage. Chem. Eur. J. 2017, 23, 16442–16446. [Google Scholar] [CrossRef]
- Cao, Q.; Li, Y.; Freisinger, E.; Qin, P.Z.; Sigel, R.K.O.; Mao, Z.W. G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs. Inorg. Chem. Front. 2017, 4, 10–32. [Google Scholar] [CrossRef]
- Dumat, B.; Bordeau, G.; Faurel-Paul, E.; Mahuteau-Betzer, F.; Saettel, N.; Metge, G.; Fiorini-Debuisschert, C.; Charra, F.; Teulade-Fichou, M.P. DNA switches on the two-photon efficiency of an ultrabright triphenylamine fluorescent probe specific of AT regions. J. Am. Chem. Soc. 2013, 135, 12697–12706. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.F.; Zhang, H.; Mu, G.; Liu, W.T.; Cao, Q.; Tan, C.P.; Ji, L.N.; Mao, Z.W. Nucleus-localized platinum(II)–triphenylamine complexes as potent photodynamic anticancer agents. Inorg. Chem. Front. 2019, 6, 2817–2823. [Google Scholar] [CrossRef]
- Wang, X.Y.; Guo, Z.J. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef]
- Basu, U.; Banik, B.; Wen, R.; Pathak, R.K.; Dhar, S. The Platin-X series: Activation, targeting, and delivery. Dalton Trans. 2016, 45, 12992–13004. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.T.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef]
- Alpaslan, E.; Yazici, H.; Golshan, N.H.; Ziemer, K.S.; Webster, T.J. pH-dependent activity of dextran-coated cerium oxide nanoparticles on prohibiting osteosarcoma cell proliferation. ACS Biomater. Sci. Eng. 2015, 1, 1096–1103. [Google Scholar] [CrossRef]
- Reed, D.E.; Shokat, K.M. Targeting osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 18100–18101. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.S.; Davis, L.E. Osteosarcoma, chondrosarcoma, and chordoma. J. Clin. Oncol. 2018, 36, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Makris, G.; Tseligka, E.D.; Pirmettis, I.; Papadopoulos, M.S.; Vizirianakis, I.S.; Papagiannopoulou, D. Development and pharmacological evaluation of new bone-targeted 99mTc-radiolabeled bisphosphonates. Mol. Pharm. 2016, 13, 2301–2317. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Wang, X.Y.; Luo, C.; Zhu, C.C.; Wang, K.; Zhang, C.L.; Guo, Z.J. Dinuclear platinum(II) complexes with bone-targeting groups as potential anti-osteosarcoma agents. Chem. Asian J. 2017, 12, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Zhu, Z.Z.; Luo, C.; Zhu, C.C.; Zhang, S.R.; Zhang, C.L.; Guo, Z.J.; Wang, X.Y. A potential bone-targeting hypotoxic platinum(II) complex with an unusual cytostatic mechanism toward osteosarcoma cells. Inorg. Chem. 2018, 57, 3315–3322. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-targeted platinum and ruthenium drugs-a new paradigm in cancer drug treatment regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Thysiadis, S.; Katsamakas, S.; Dalezis, P.; Sigala, I.; Lazarides, T.; Nikolakaki, E.; Trafalis, D.; Gederaas, O.A.; Lindgren, M.; et al. Synthesis and biological evaluation of a platinum(II)-c(RGDyK) conjugate for integrin-targeted photodynamic therapy. Eur. J. Med. Chem. 2017, 141, 221–231. [Google Scholar] [CrossRef]
- Zamora, A.; Gandioso, A.; Massaguer, A.; Buenestado, S.; Calvis, C.; Hernández, J.L.; Mitjans, F.; Rodríguez, V.; Ruiz, J.; Marchán, V. Toward angiogenesis inhibitors based on the conjugation of organometallic platinum(II) complexes to RGD peptides. ChemMedChem 2018, 13, 1755–1762. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Franich, A.A.; Živković, M.D.; Ilić-Tomić, T.; Đorđević, I.S.; Nikodinović-Runić, J.; Pavić, A.; Janjić, G.V.; Rajković, S. New minor groove covering DNA binding mode of dinuclear Pt(II) complexes with various pyridine-linked bridging ligands and dual anticancer-antiangiogenic activities. J. Biol. Inorg. Chem. 2020, 25, 395–409. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Yang, Y.H.; Karakhanova, S.; Hartwig, W.; D’haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ros in cancer: Novel targets for anticancer therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer. 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Marrachea, S.; Dhar, S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem. Sci. 2015, 6, 1832–1845. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Lomeli, N.; Di, K.J.; Czerniawski, J.; Guzowski, J.F.; Bota, D.A. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic. Biol. Med. 2017, 102, 274–286. [Google Scholar] [CrossRef]
- Guo, Y.; He, Y.F.; Wu, S.D.; Zhang, S.R.; Song, D.F.; Zhu, Z.Z.; Guo, Z.J.; Wang, X.Y. Enhancing cytotoxicity of a monofunctional platinum complex via a dual-DNA-damage approach. Inorg. Chem. 2019, 58, 13150–13160. [Google Scholar] [CrossRef]
- Zhang, C.; Guan, R.L.; Liao, X.X.; Ouyang, C.; Liu, J.P.; Ji, L.N.; Chao, H. Mitochondrial DNA targeting and impairment by a dinuclear Ir–Pt complex that overcomes cisplatin resistance. Inorg. Chem. Front. 2020, 7, 1864–1871. [Google Scholar] [CrossRef]
- Zhu, Z.Z.; Wang, Z.H.; Zhang, C.L.; Wang, Y.J.; Zhang, H.M.; Gan, Z.J.; Guo, Z.J.; Wang, X.Y. Mitochondrion-targeted platinum complexes suppressing lung cancer through multiple pathways involving energy metabolism. Chem. Sci. 2019, 10, 3089–3095. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, C.C.; He, Y.F.; Zhang, Z.Q.; Zhou, W.; Muhammad, N.; Guo, Y.; Wang, X.Y.; Guo, Z.J. Restraining cancer cells by dual metabolic inhibition with a mitochondrion-targeted platinum(II) complex. Angew. Chem. Int. Ed. 2019, 58, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, Their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.; Tipton, K. Assessment of enzyme inhibition: A review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 2017, 22, 1192. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Lu, L.P.; Zhu, M.L. Protein tyrosine phosphatase inhibition by metals and metal complexes. Antioxid. Redox Sign. 2014, 20, 2210–2224. [Google Scholar] [CrossRef]
- Yuan, C.X.; Wang, W.R.; Wang, J.W.; Li, X.H.; Wu, Y.B.; Li, S.D.; Lu, L.P.; Zhu, M.L.; Xing, S.; Fu, X.Q. Potent and selective PTP1B inhibition by a platinum(II) complex: Possible implications for a new antitumor strategy. Chem. Commun. 2020, 56, 102–105. [Google Scholar] [CrossRef]
- Zhang, D.W.; Yip, Y.M.; Li, L.B. In silico construction of HK2-VDAC1 complex and investigating the HK2 binding-induced molecular gating mechanism of VDAC1. Mitochondrion 2016, 30, 222–228. [Google Scholar] [CrossRef]
- Li, S.N.; Li, J.J.; Dai, W.Q.; Zhang, Q.H.; Feng, J.; Wu, L.W.; Liu, T.; Yu, Q.; Xu, S.Z.; Wang, W.W.; et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer. 2017, 117, 1518–1528. [Google Scholar] [CrossRef]
- Muhammad, N.; Tan, C.P.; Muhammad, K.; Wang, J.; Sadia, N.; Pan, Z.Y.; Ji, L.N.; Mao, Z.W. Mitochondria-targeting monofunctional platinum(II)–lonidamine conjugates for cancer cell de-energization. Inorg. Chem. Front. 2020, 7, 4010–4019. [Google Scholar] [CrossRef]
- Waghorn, P.A.; Jackson, M.R.; Gouverneur, V.; Vallis, K.A. Targeting telomerase with radiolabeled inhibitors. Eur. J. Med. Chem. 2017, 125, 117–129. [Google Scholar] [CrossRef]
- Paul, A.; Maji, B.; Misra, S.K.; Jain, A.K.; Muniyappa, K.; Bhattacharya, S. Stabilization and structural alteration of the G-quadruplex DNA made from the human telomeric repeat mediated by Troger’s base based novel benzimidazole derivatives. J. Med. Chem. 2012, 55, 7460–7471. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.B.; Chen, S.; Qin, Q.P.; Luo, J.R.; Tan, M.X.; Wang, Z.F.; Zou, B.Q.; Liang, H. Preparation of platinum(II) complexes with naphthalene imide derivatives and exploration of their in vitro cytotoxic activities. Inorg. Chem. Commun. 2019, 104, 124–128. [Google Scholar] [CrossRef]
- Huang, G.B.; Chen, S.; Qin, Q.P.; Luo, J.R.; Tan, M.X.; Wang, Z.F.; Zou, B.Q.; Liang, H. In vitro and in vivo activity of novel platinum(II) complexes with naphthalene imide derivatives inhibiting human non-small cell lung cancer cells. New J. Chem. 2019, 43, 8146–8152. [Google Scholar] [CrossRef]
- Qin, Q.P.; Zou, B.Q.; Wang, Z.F.; Huang, X.L.; Zhang, Y.; Tan, M.X.; Wang, S.L.; Liang, H. High in vitro and in vivo antitumor activities of luminescent platinum(II) complexes with jatrorrhizine derivatives. Eur. J. Med. Chem. 2019, 183, 111727. [Google Scholar] [CrossRef]
- Zou, H.H.; Wang, L.; Long, Z.X.; Qin, Q.P.; Song, Z.K.; Xie, T.; Zhang, S.H.; Liu, Y.C.; Lin, B.; Chen, Z.F. Preparation of 4-([2,2′:6′,2′′-terpyridin]-4′-yl)-N,N-diethylaniline NiII and PtII complexes and exploration of their in vitro cytotoxic activities. Eur. J. Med. Chem. 2016, 108, 1–12. [Google Scholar] [CrossRef]
- Sabbatini, M.; Zanellato, I.; Ravera, M.; Gabano, E.; Perin, E.; Rangone, B.; Osella, D. Pt(IV) bifunctional prodrug containing 2-(2-propynyl)octanoato axial ligand: Induction of immunogenic cell death on colon cancer. J. Med. Chem. 2019, 62, 3395–3406. [Google Scholar] [CrossRef]
- Wong, D.Y.Q.; Ong, W.W.F.; Ang, W.H. Induction of immunogenic cell death by chemotherapeutic platinum complexes. Angew. Chem. Int. Ed. 2015, 54, 6483–6487. [Google Scholar] [CrossRef]
- Huang, K.B.; Wang, F.Y.; Feng, H.W.; Luo, H.J.; Long, Y.; Zou, T.T.; Chan, A.S.C.; Liu, R.; Zou, H.H.; Chen, Z.F.; et al. An aminophosphonate ester ligand-containing platinum(II) complex induces potent immunogenic cell death in vitro and elicits effective anti-tumor immune responses in vivo. Chem. Commun. 2019, 55, 13066–13069. [Google Scholar] [CrossRef]
- Al-Khayal, K.; Vaali-Mohammed, M.A.; Elwatidy, M.; Bin Traiki, T.; Al-Obeed, O.; Azam, M.; Khan, Z.; Abdulla, M.; Ahmad, R. A novel coordination complex of platinum (PT) induces cell death in colorectal cancer by altering redox balance and modulating MAPK pathway. Bmc Cancer 2020, 20, 685. [Google Scholar] [CrossRef] [PubMed]
- Choroba, K.; Machura, B.; Raposo, L.R.; Małecki, J.G.; Kula, S.; Pająk, M.; Erfurt, K.; Maroń, A.M.; Fernandes, A.R. Platinum(II) complexes showing high cytotoxicity toward A2780 ovarian carcinoma cells. Dalton Trans. 2019, 48, 13081–13093. [Google Scholar] [CrossRef]
- Wang, F.Y.; Tang, X.M.; Wang, X.; Huang, K.B.; Feng, H.W.; Chen, Z.F.; Liu, Y.N.; Liang, H. Mitochondria-targeted platinum(II) complexes induce apoptosis-dependent autophagic cell death mediated by ER-stress in A549 cancer cells. Eur. J. Med. Chem. 2018, 155, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.; Marzo, T.; Infrasca, T.; Cilibrizzi, A.; Vilar, R.; Messori, L.; Merlino, A. A case of extensive protein platination: The reaction of lysozyme with a Pt(II)–terpyridine complex. Dalton Trans. 2018, 47, 8716–8723. [Google Scholar] [CrossRef]
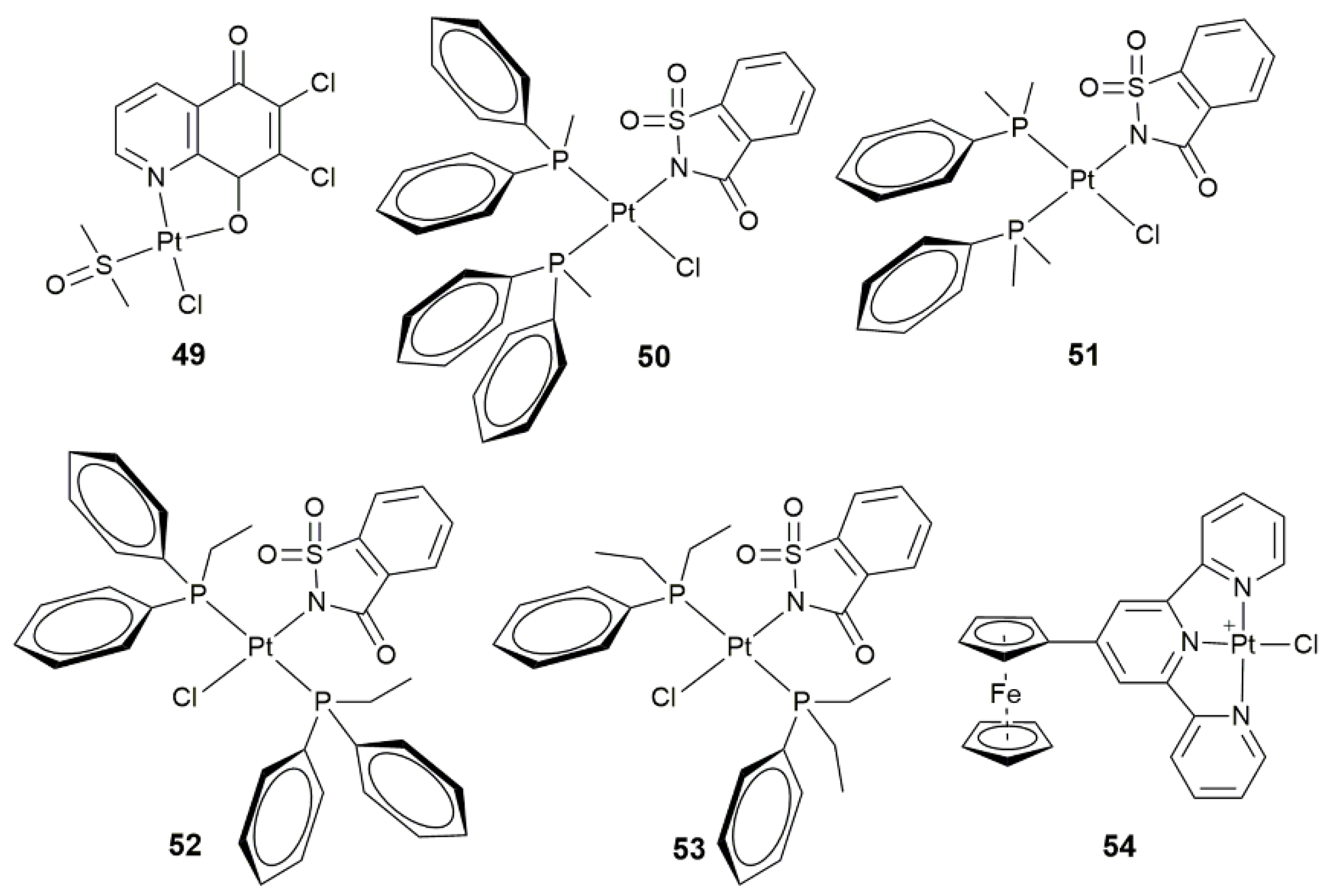
- Li, Z.; Zhou, J.; Gan, Y.; Yin, Y.H.; Zhang, W.C.; Yang, J.F.; Tang, Y.X.; Dai, Y.B. Synthesis of a novel platinum(II) complex with 6,7-dichloro-5,8-quinolinedione and the study of its antitumor mechanism in testicular seminoma. J. Inorg. Biochem. 2019, 197, 110701. [Google Scholar] [CrossRef] [PubMed]
- Icsel, C.; Yilmaz, V.T.; Cevatemre, B.; Aygun, M.; Ulukaya, E. Structures and anticancer activity of chlorido platinum(II) saccharinate complexes with mono- and dialkylphenylphosphines. J. Inorg. Biochem. 2019, 195, 39–50. [Google Scholar] [CrossRef]
- Mitra, K.; Basu, U.; Khan, I.; Maity, B.; Kondaiah, P.; Chakravarty, A.R. Remarkable anticancer activity of ferrocenyl-terpyridine platinum(II) complexes in visible light with low dark toxicity. Dalton Trans. 2014, 43, 751–763. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, X.Y.; Tu, C.; Lin, J.; Ding, J.; Lin, L.P.; Wang, Z.M.; He, C.; Yan, C.H.; You, X.Z.; et al. Monofunctional platinum complexes showing potent cytotoxicity against human liver carcinoma cell line BEL-7402. J. Med. Chem. 2003, 46, 3502–3507. [Google Scholar] [CrossRef]

















| Complex | Functional Group | Function | Tested Cells or Animals | Ref. |
|---|---|---|---|---|
| Fluorescent Complexes | ||||
| 1 | anthraquinone | monitor subcellular localization | U2-OS, U2-OS/Pt, A2780, A2780/DDP | [40] |
| 2, 3 | 4-nitrobenzo-2-oxa-1,3-diazole | track cellular distribution | HeLa | [43] |
| 4 | 4-amino-7-nitro-2,1,3- benzoxadiazole | in vitro and vivo fluorescence imaging | MCF-7, A549, 293T; zebrafish larva | [44] |
| 5 | ThT derivative | cellular imaging | HeLa | [45] |
| Photoactive Complexes | ||||
| 6, 7 | isomeric tetra-cationic(pyridyl)porphyrins | PDT on metastatic melanoma cells | WM1366 | [56] |
| 8 | 5,10,15,20-tetra-(4-pyridyl)-21H,23H-porphine | photocytotoxicity (50 W LED light, 6 J cm−2, 30 min) | colon26, sarcoma180; colon26 tumor-bearing mice | [57] |
| 9 | 5,10,15,20-tetra(4-pyridyl)porphyrin | photocytotoxicity (6.95 J cm−2, 420 nm, 15 min), DNA photocleavage | MRC-5, HeLa, A2780, CP70 | [51] |
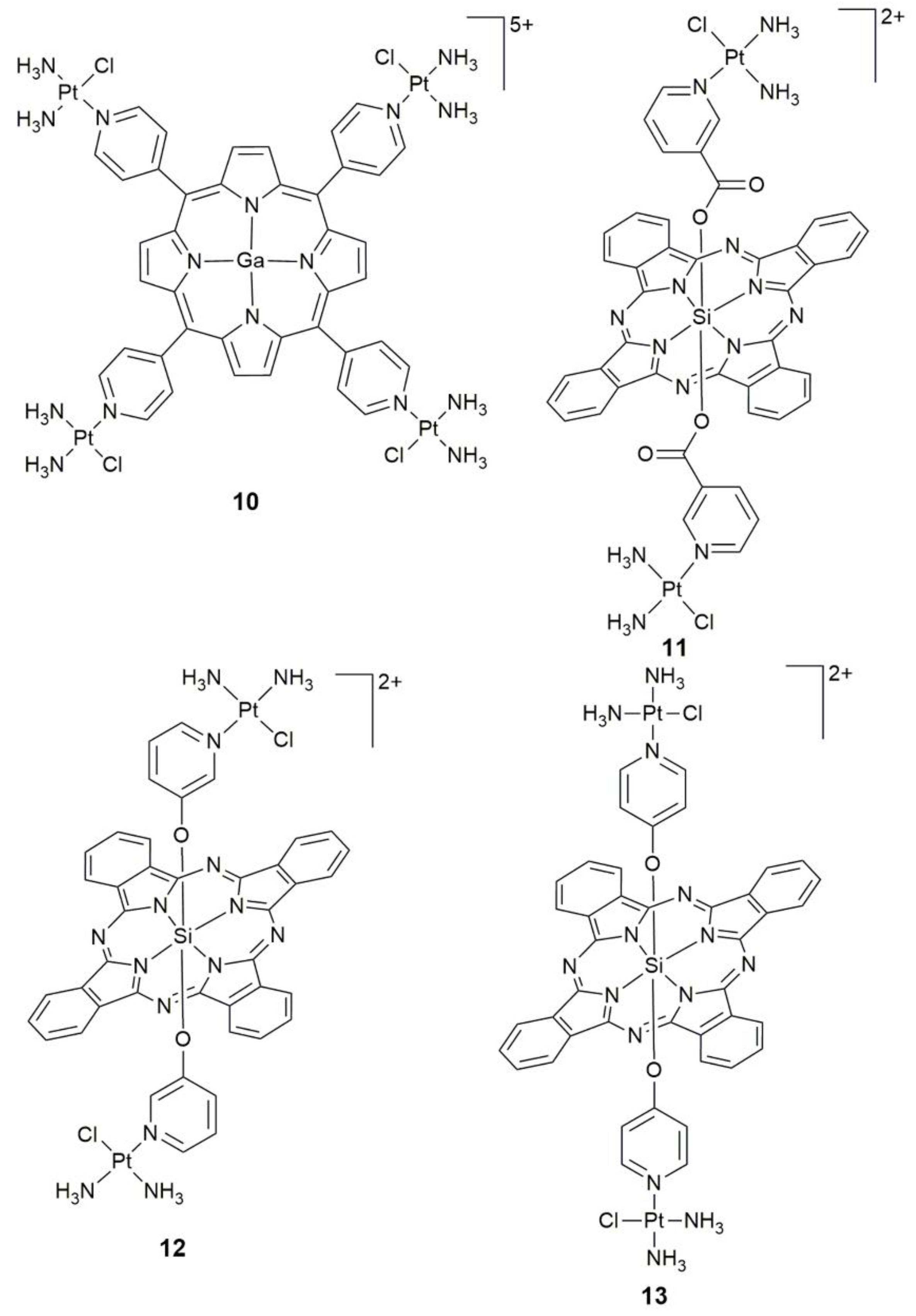
| 10 | porphyrin containing GaIII center | singlet oxygen generator, photocytotoxicity, DNA interaction | colon 26, sarcoma 180; colon26 tumor-bearing mice | [59] |
| 11 | SiIV phthalocyanine | specific cellular uptake, mitochondrial accumulation, photocytotoxicity (45 min, 660–680 nm, 5.5 ± 2.5 mW cm−2) | MDA-MB-231, HEK293T | [62] |
| 12, 13 | SiIV phthalocyanine | photocytotoxicity, DNA-targeting PDT agents | HeLa | [63] |
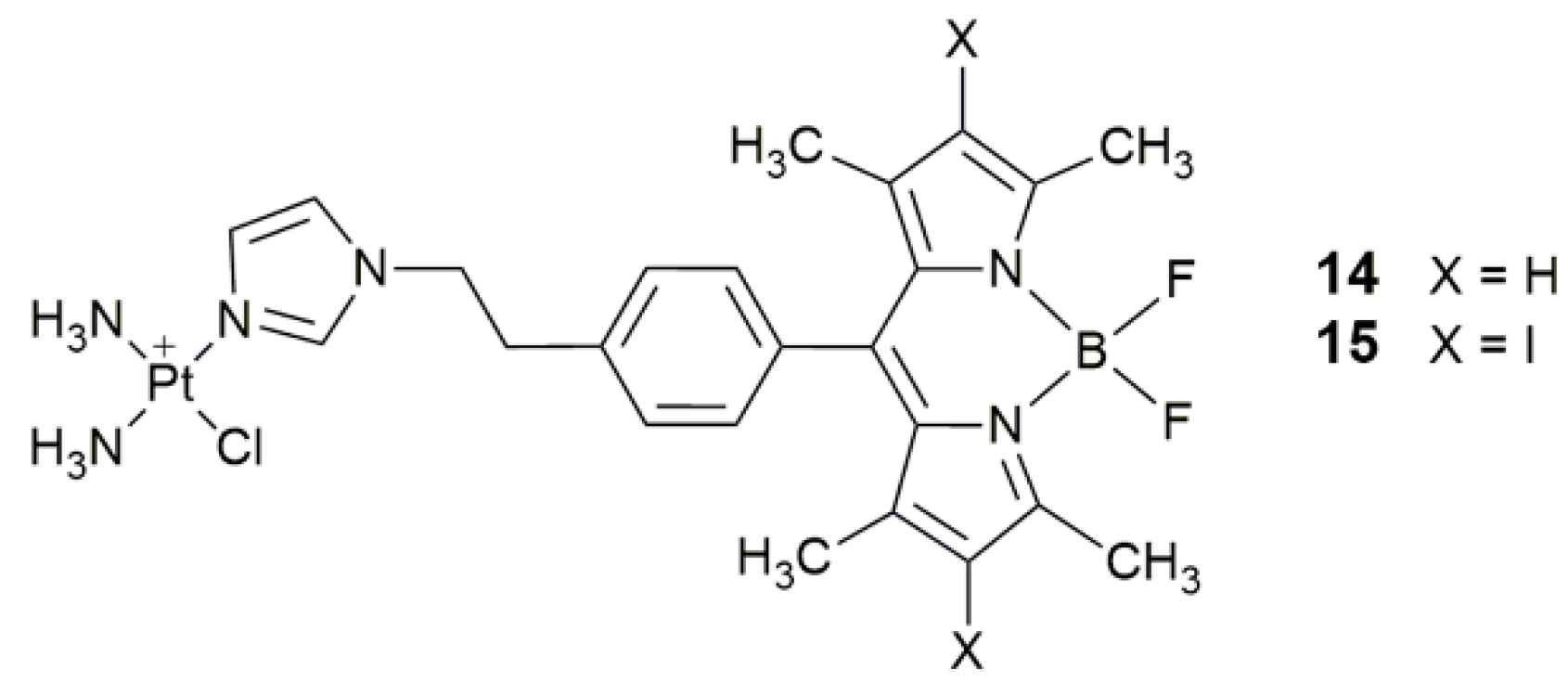
| 14, 15 | 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) and its diiodo derivative | photocytotoxicity (400−700 nm, 10 J cm−2), cellular imaging | HaCaT, MCF-7 | [64] |
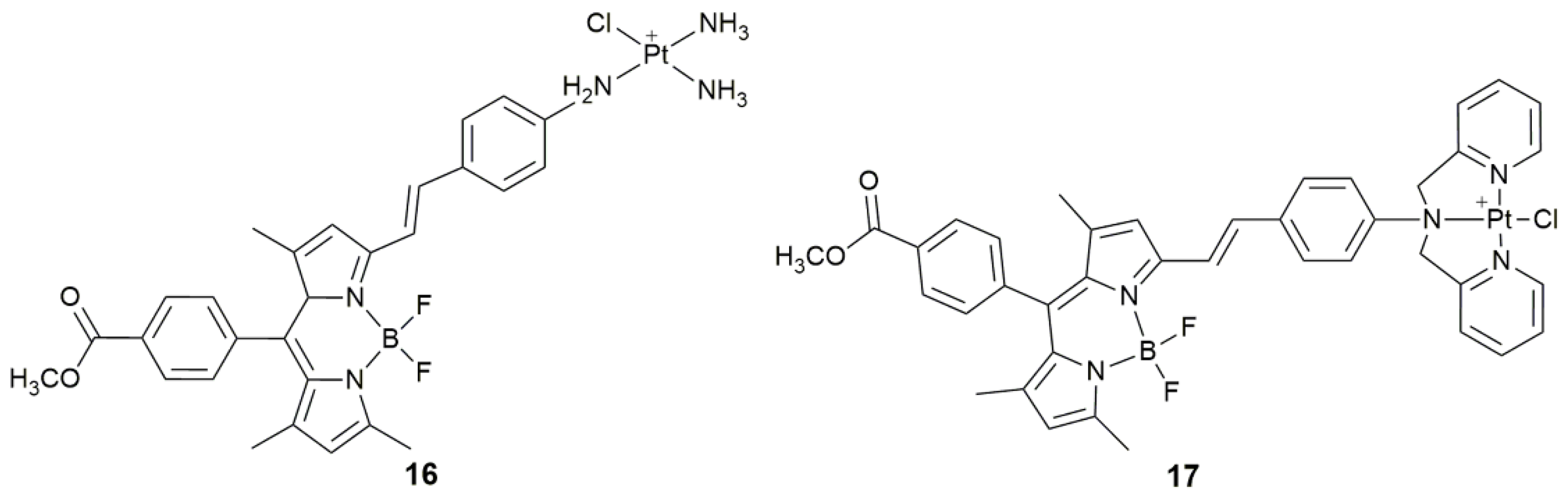
| 16 | α-(4-amino)styryl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene | photocytotoxicity (532 nm, 3.5 mW cm−2, 5 min) | MCF-7, SGC-7901, A549, HeLa | [66] |
| 17 | α-(4-amino)styryl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene | photocytotoxicity, lysosomal escape, increase accessibility to nDNA, decrease intracellular GSH | MCF-7, SGC-7901, A549, HeLa | [70] |
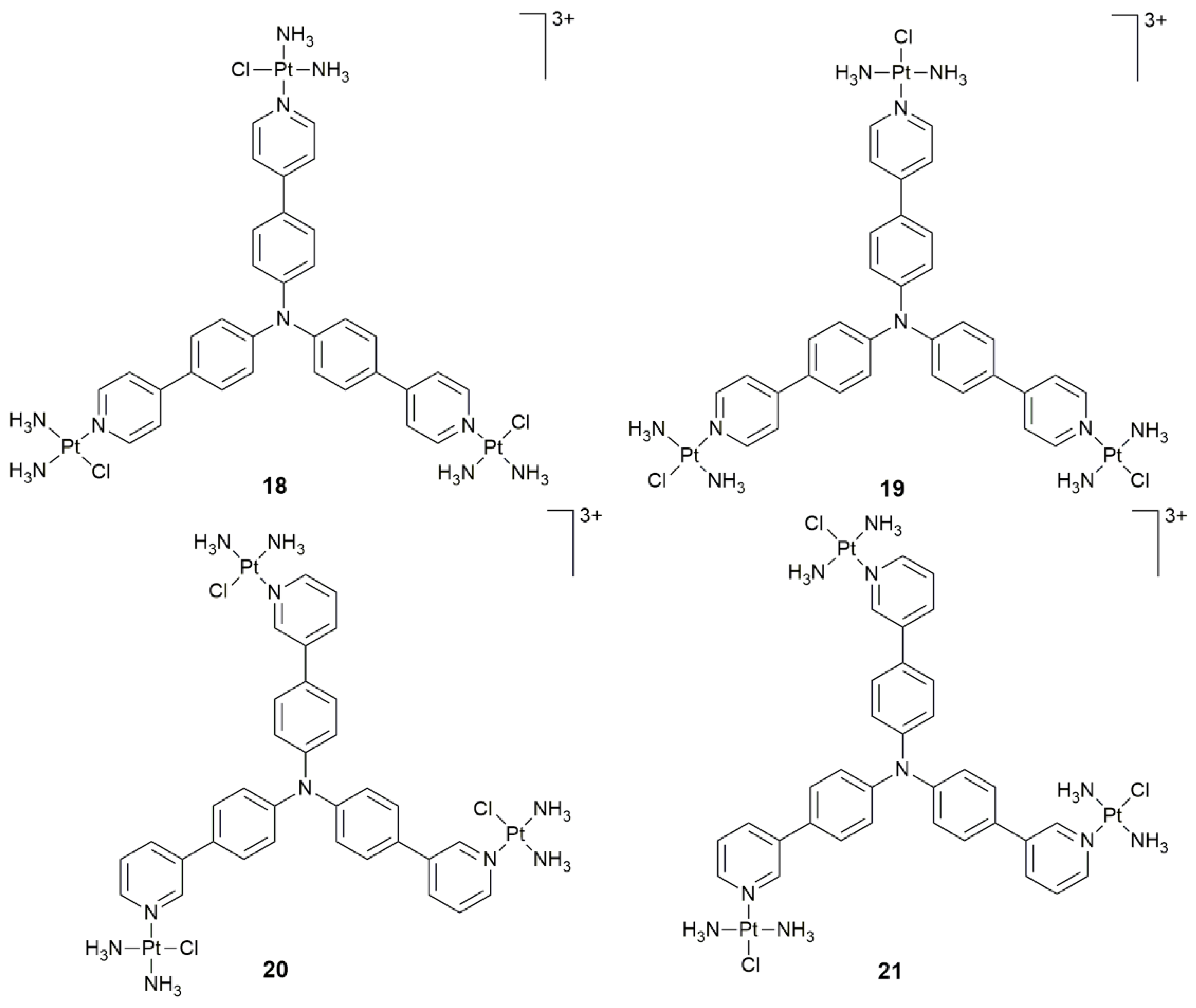
| 18–21 | triphenylamine core | PDT activity | HeLa, HepG2, A549, A549cisR, LO2; HeLa xenograft-bearing nude mice | [76] |
| Targeted Complexes | ||||
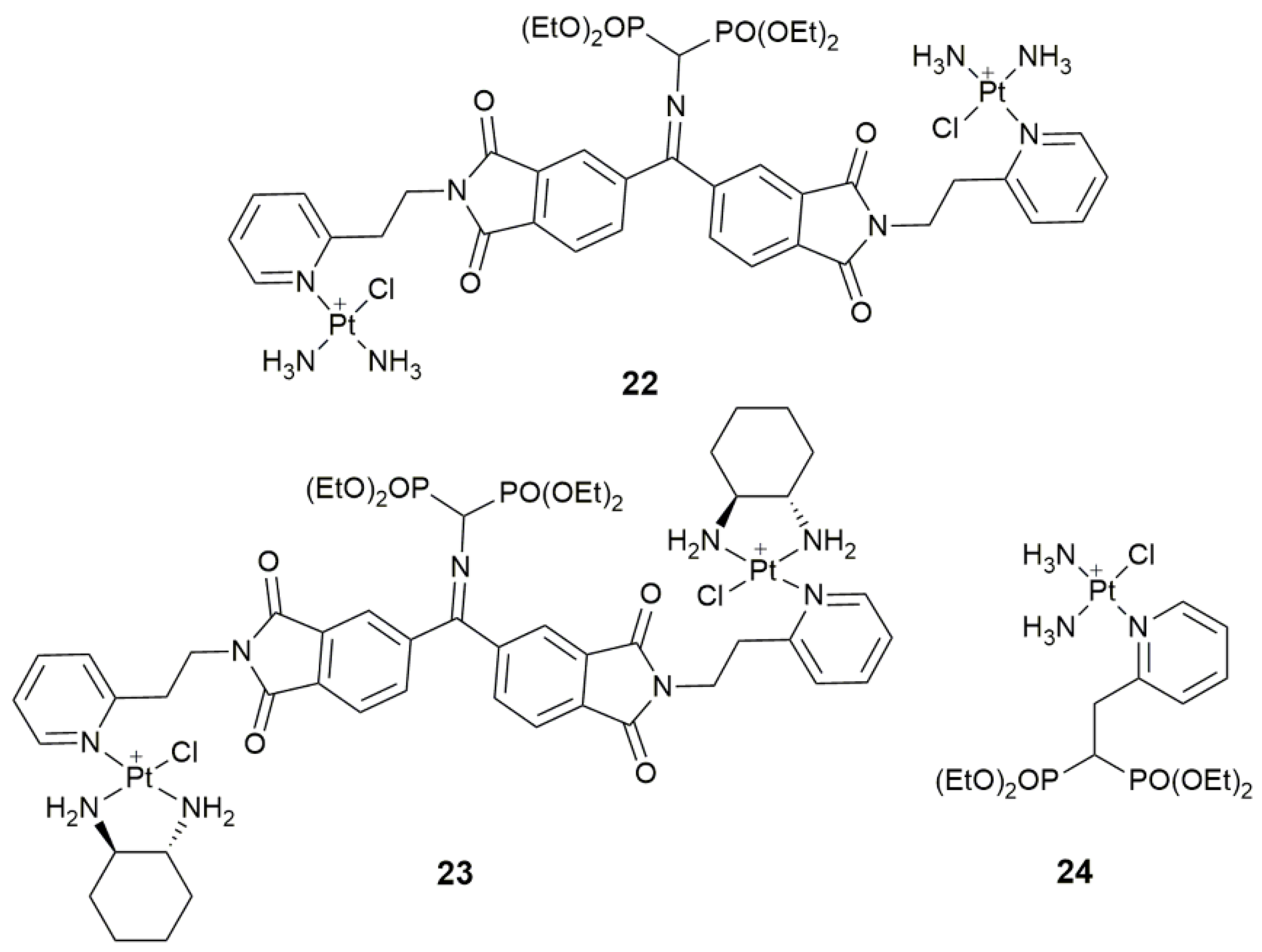
| 22–23 | bisphosphonate | bone targeting | U2-OS, MG-63; male ICR mice | [84] |
| 24 | bisphosphonate | bone targeting, decrease systemic toxicity | U2-OS, MG-63, LO2; male ICR mice | [85] |
| 25 | c(RGDyK) | tumor targeting, integrin-targeted PDT | SKOV-3, PC-3, A549, MCF-7, MDA-MB-231, U87M | [88] |
| 26 | cyclic peptide containing RGD sequence (-Arg-Gly-Asp-) | target angiogenesis, antiangiogenic and antitumor activity | SK-MEL-28, MDA-MB-231, CAPAN-1, HUVEC | [89] |
| 27–29 | 4,4’-bipyridine, 1,2-di(pyridin-4-yl)ethane, or 1,2-di(pyridin-4-yl)ethene | target angiogenesis, overcome cisplatin resistance, block tumor neovascularization and metastasis | MRC-5, A549, A375; B16-F10 melanoma-zebrafish, HCT-116-zebrafish | [91] |
| 30 | naphthalimide | target mtDNA, damage mtDNA, regulate mtDNA-encoded protein, disturb mitochondrial physiological process | MCF-7, A549, Caov-3, HK-2; MCF-7 tumor-bearing mice | [102] |
| 31 | IrIII moiety plus imidazo[4,5-f][1,10]phenanthroline derivative | target mtDNA, accumulate in mitochondria, induce mitochondrial dysfunction via mtDNA damage | HepG2, HeLa, A549, A549R | [103] |
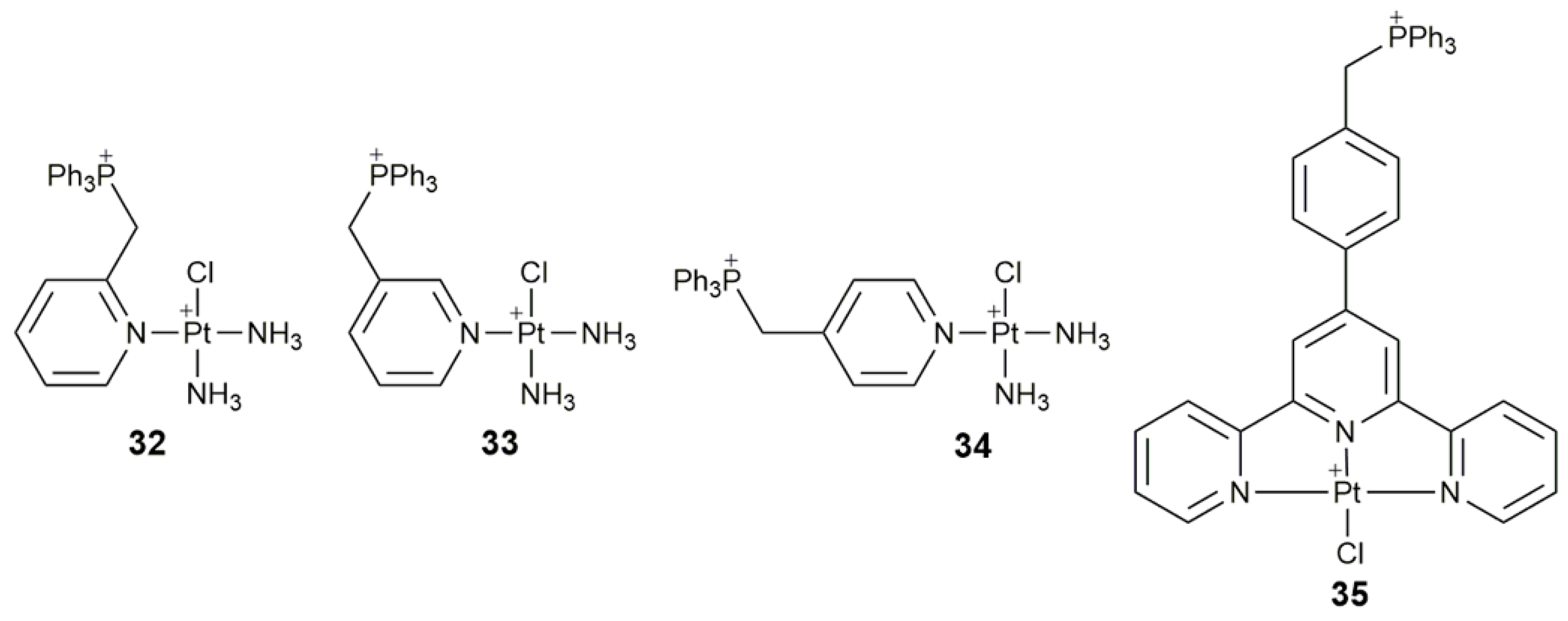
| 32–34 | triphenylphosphonium | target mtDNA, inhibit glycolysis, affect mitochondrial structure and function, damage mtDNA | A549, HeLa, SMMC, HL-7720; A549 tumor-bearing mice | [104] |
| 35 | triphenylphosphonium | target mitochondrion, inhibit mitochondrial TrxR, destroy respiratory and glycolytic metabolisms | Caov-3, A549, A549R, HK-2 | [105] |
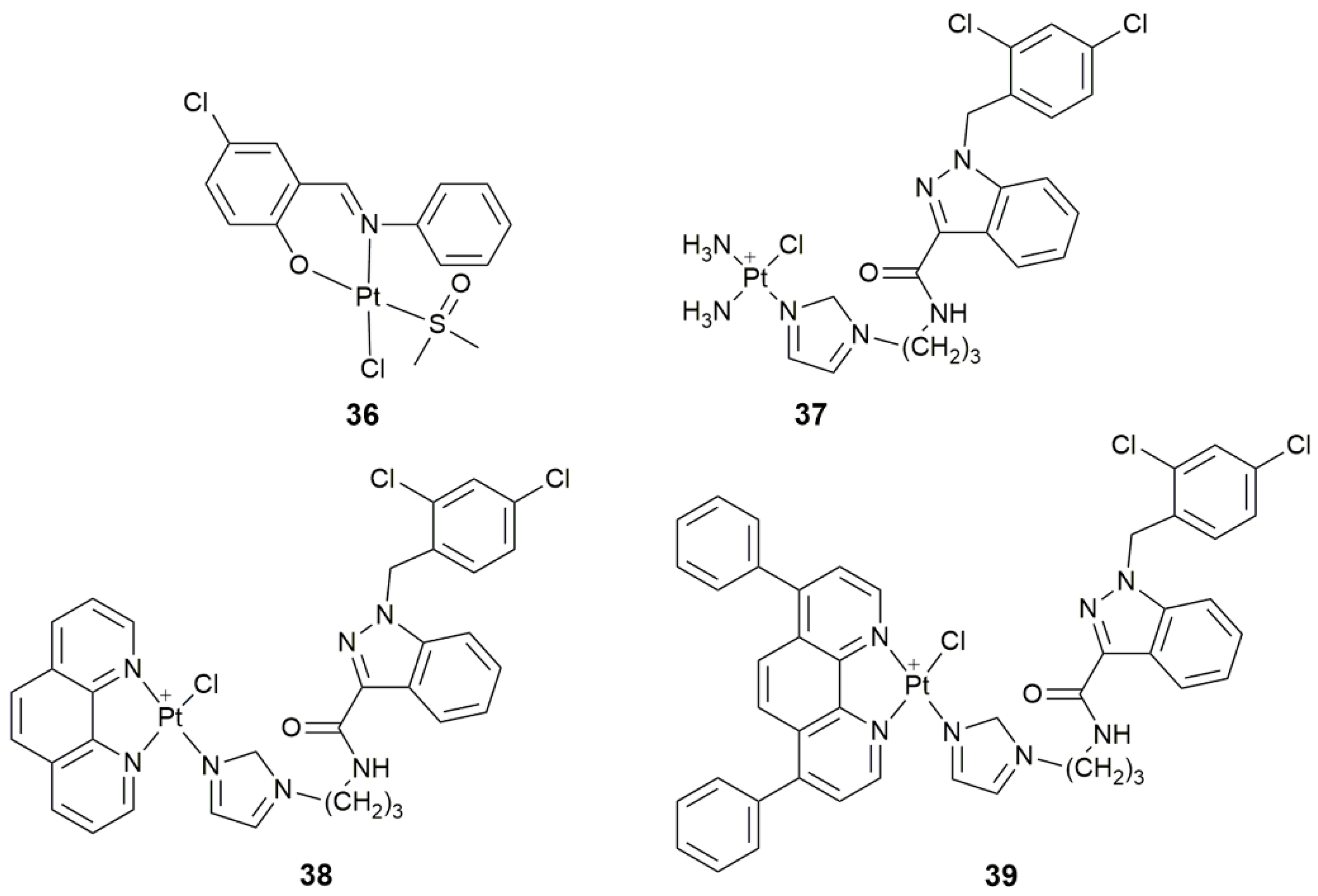
| 36 | 5-chlorosalicylideneaniline | target tyrosine phosphatases, selectively inhibit PTP1B, antiproliferative activity | MCF-7, HepG2, A549 | [110] |
| 37–39 | lonidamine | target hexokinase, disrupt mitochondrial bioenergetics, damage mtDNA | A549, PC3, Caov-3, MCF-7, MDA-MB-231, MCF-10A | [113] |
| 40, 41 | naphthalene imide derivatives | target telomerase, inhibit telomerase, disrupt mitochondrial function | SKOV-3, NCI-H460, HeLa, HL-7702, BEL-7402; NCI-H460 tumor-bearing mice | [116,117] |
| 42 | jatrorrhizine derivative | target telomerase and p53, cause mitochondrial and DNA damage, display antitumor activity and green luminescence | SKOV-3/DDP, T-24, HeLa, HL-7702, A549; HeLa tumor-bearing mice | [118] |
| 43 | 4-([2,2′:6′,2′′-terpyridin]-4′-yl)-N,N-diethylaniline | target telomerase, inhibit telomerase by interacting with c-myc quadruplex, disrupt mitochondrial function | BEL-7404, A549, MGC80-3, T-24, HL-7702 | [119] |
| Miscellaneous Complexes | ||||
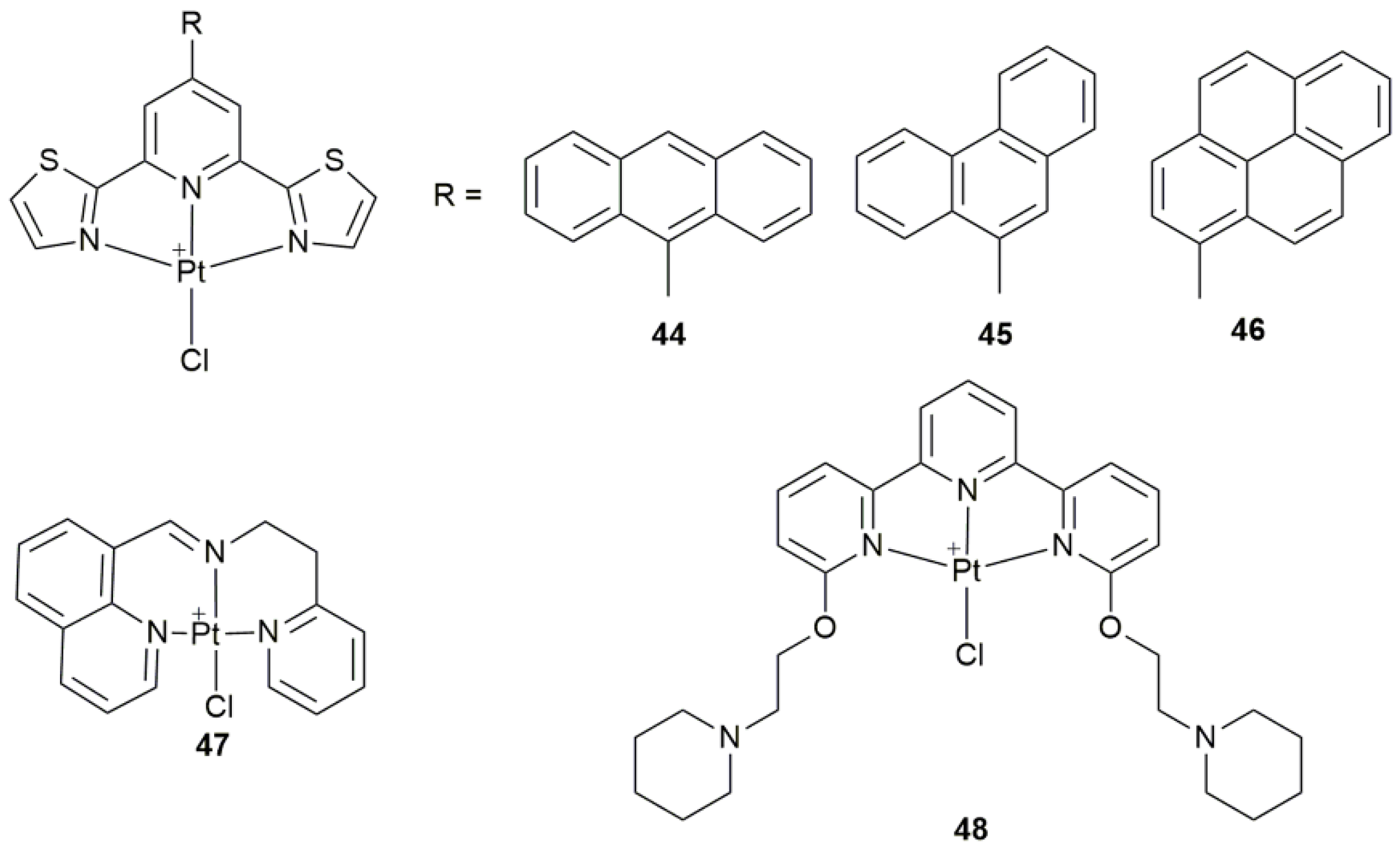
| 44–46 | 9-anthryl, 9-phenantryl, and 1-pyrenyl 2,6-bis(thiazol-2-yl)pyridines | participate in apoptotic mechanism involving mitochondrial and autophagic pathways | MCF-7, PC3, HCT-116, A2780, Fibroblasts | [124] |
| 47 | 8-substituted quinoline derivatives | induce ER stress, cause apoptosis and pro-death autophagy | BEL-7404, SKOV-3, HepG2, HCT-116, HL-7702; A549 tumor-bearing mice | [125] |
| 48 | terpyridine with two piperidine substituents | peculiar reactivity with biological macromolecules (proteins) | hen egg white lysozyme (HEWL, protein) | [126] |
| 49 | 6,7-dichloro-5,8-quinolinedione | induce apoptosis via PI3K/Akt signaling and mitochondria-mediated apoptotic pathways | TCam-2, SEM-1 | [127] |
| 50–53 | mono- and dialkylphenylphosphines | conformation-dependent biological activity | MCF-7, A549, BEAS-2B, HCT-116 | [128] |
| 54 | ferrocenyl-terpyridine | photocytotoxicity in visible light (400–700 nm) | HaCaT | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, S.; Guo, Y.; Guo, Z.; Wang, X. Monofunctional Platinum(II) Anticancer Agents. Pharmaceuticals 2021, 14, 133. https://doi.org/10.3390/ph14020133
Jin S, Guo Y, Guo Z, Wang X. Monofunctional Platinum(II) Anticancer Agents. Pharmaceuticals. 2021; 14(2):133. https://doi.org/10.3390/ph14020133
Chicago/Turabian StyleJin, Suxing, Yan Guo, Zijian Guo, and Xiaoyong Wang. 2021. "Monofunctional Platinum(II) Anticancer Agents" Pharmaceuticals 14, no. 2: 133. https://doi.org/10.3390/ph14020133
APA StyleJin, S., Guo, Y., Guo, Z., & Wang, X. (2021). Monofunctional Platinum(II) Anticancer Agents. Pharmaceuticals, 14(2), 133. https://doi.org/10.3390/ph14020133