
Small EVs-Associated DNA as Complementary Biomarker to Circulating Tumor DNA in Plasma of Metastatic Colorectal Cancer Patients


 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
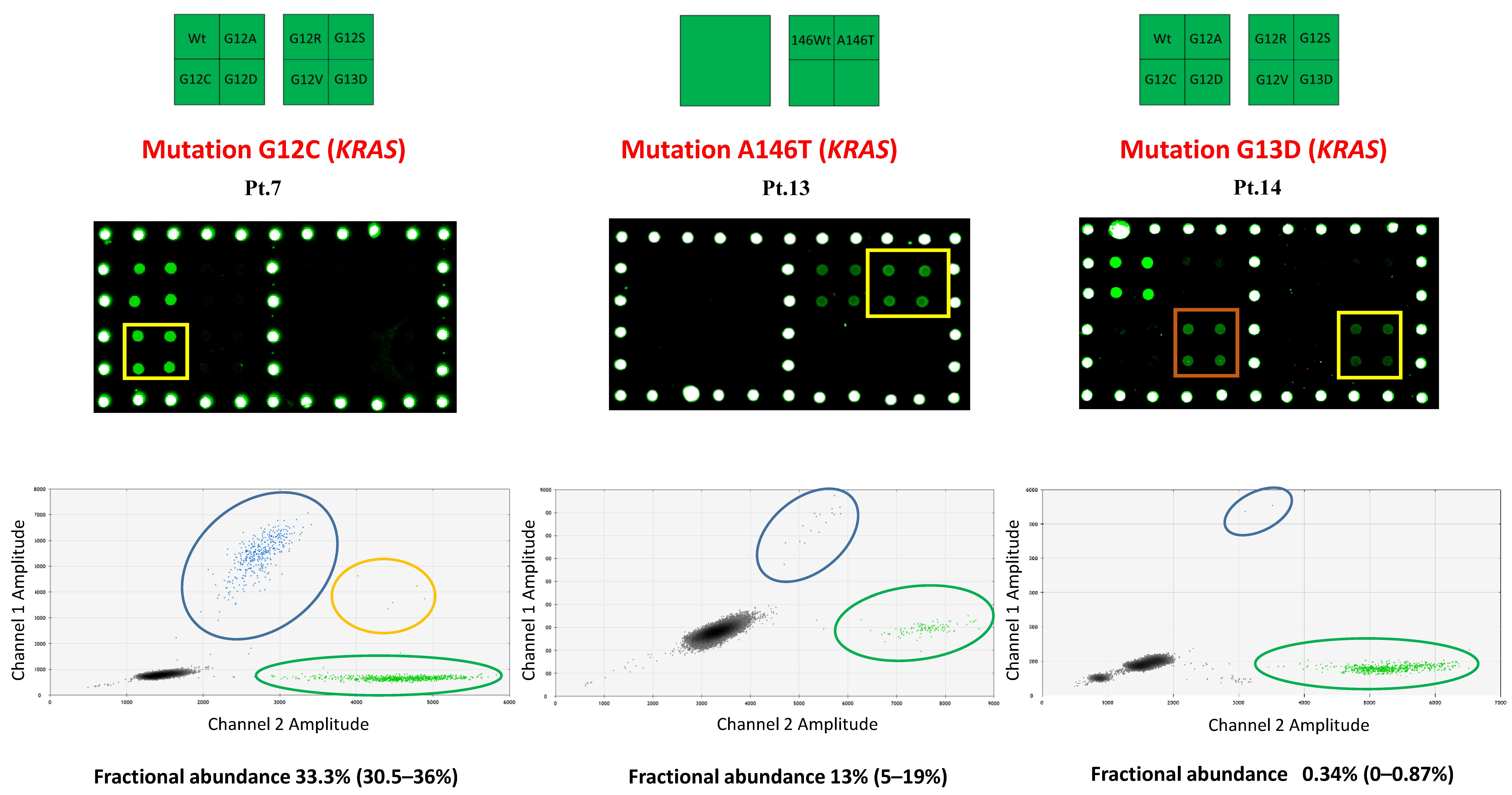
2.1. sEV-Associated DNA Analysis in mCRC Patients
2.2. sEV-Associated DNA Analysis after DNase I Treatment in mCRC Patients
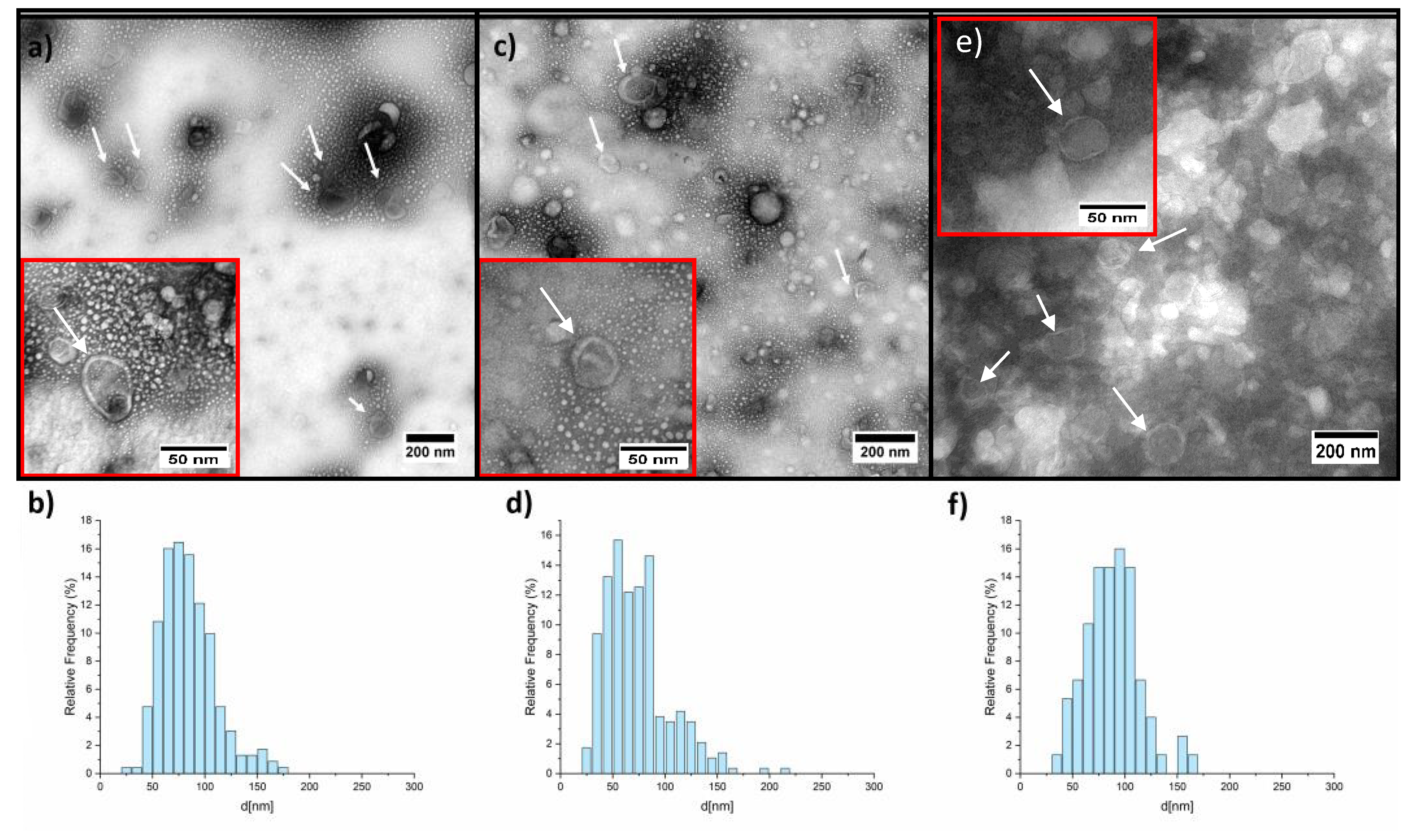
2.3. TEM Analysis on HEK Cells’ sEVs
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Isolation of sEVs from Plasma
4.2.1. Ultracentrifugation
4.2.2. Exosome Precipitation Kit
4.2.3. Immunoprecipitation of sEVs on Magnetic Beads Decorated with DNA-Directed AntiCD63
Magnetic Beads Functionalization
Capture and Release of sEVs from Magnetic Beads
4.3. Extraction of DNA from sEVs and DNase I Treatment
4.4. sEV-Associated DNA Analysis
4.4.1. Microarray Analysis
4.4.2. ddPCR Analysis
4.5. Cell Culture
Exosome Isolation and Treatment with and without DNase I
4.6. sEVs Analysis
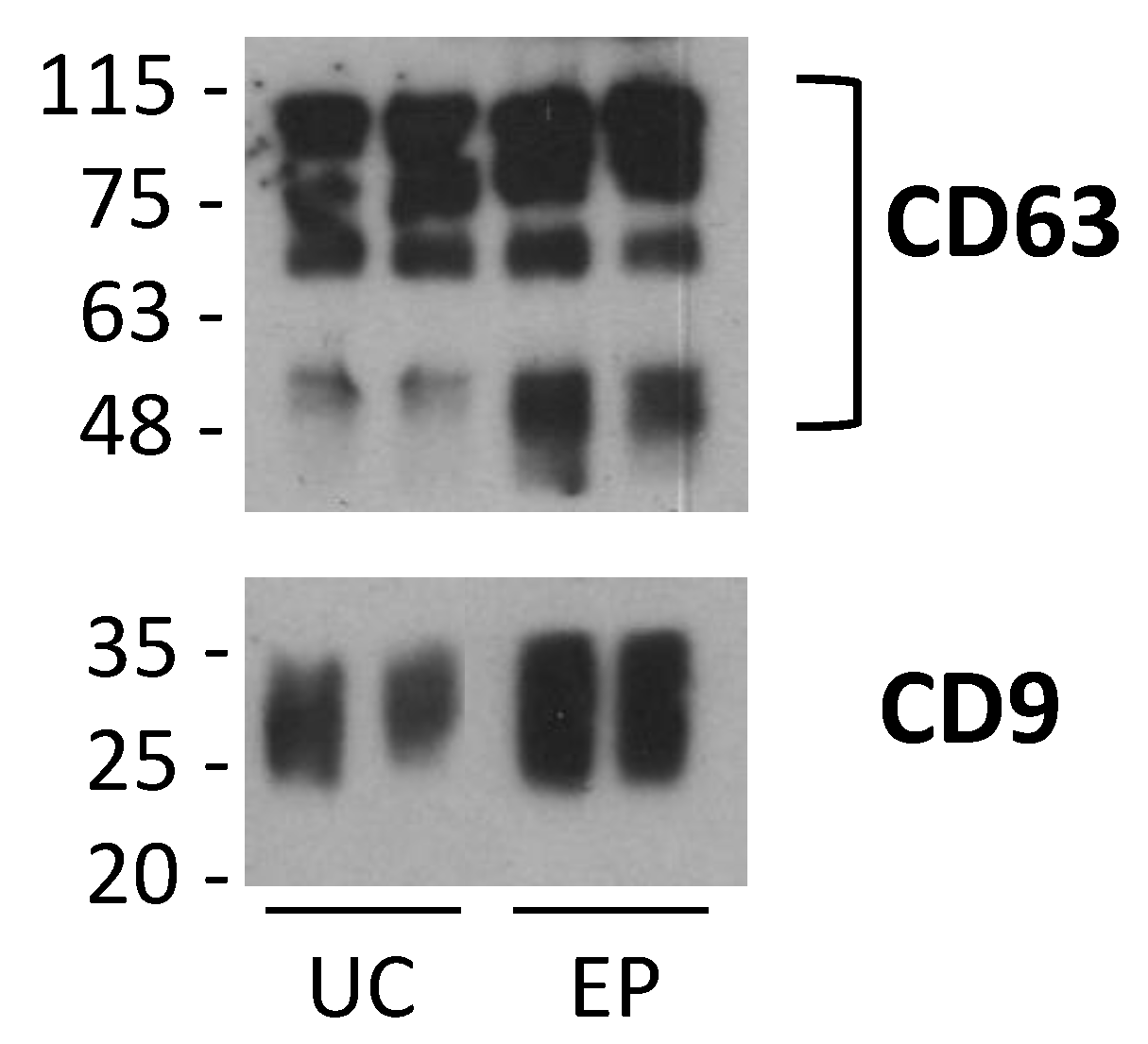
4.6.1. Western Blotting Analysis
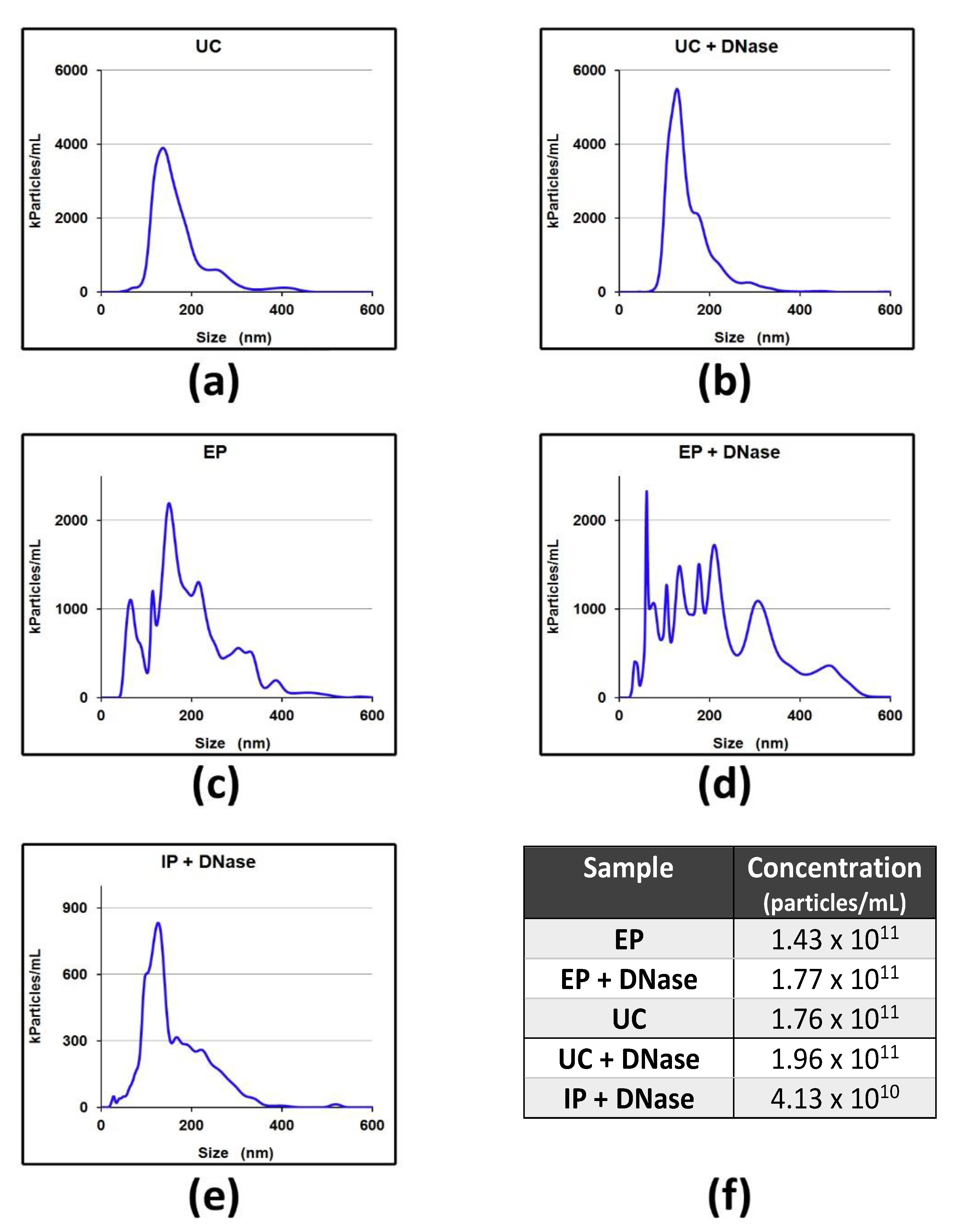
4.6.2. Nanoparticle Tracking Analysis (NTA)
4.6.3. Transmission Electron Microscope (TEM)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scholl, J.N.; Dias, C.K.; Muller, L.; Battastini, A.M.O.; Figueirò, F. Extracellular vesicles in cancer progression: Are they part of the problem or part of the solution? Nanomedicine 2020, 26, 2625–2641. [Google Scholar] [CrossRef]
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome mediated communication within the tumor microenvironment. J. Control. Release 2015, 219, 278–294. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xian, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta 2019, 1871, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef]
- Yang, S.; Che, S.P.Y.; Kurywchak, P.; Tavormina, J.L.; Gansmo, L.B.; Correa de Sampaio, P.; Tachezy, M.; Bockhorn, M.; Gebauer, F.; Haltom, A.R.; et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 2017, 18, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Lazaro-Ibanez, E.; Sanz-Garcia, A.; Visakorpi, T.; Escobedo-Lucea, C.; Siljander, P.; Ayuso-Sacido, A.; Yliperttula, M. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: Apoptotic bodies, microvesicles, and exosomes. Prostate 2014, 74, 1379–1390. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Zocco, D.; Bernardi, S.; Novelli, M.; Astrua, C.; Fava, P.; Zarovni, N.; Carpi, F.M.; Bianciardi, L.; Malavenda, O.; Quaglino, P.; et al. Isolation of extracellular vesicles improves the detection of mutant DNA from plasma of metastatic melanoma patients. Sci. Rep. 2020, 10, 15745. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, S.; Damin, F.; Ferraro, L.; Soriani, N.; Burgio, V.; Ronzoni, M.; Gianni, L.; Ferrari, M.; Chiari, M. Microarray Approach Combined with ddPCR: An Useful Pipeline for the Detection and Quantification of Circulating Tumour DNA Mutations. Cells 2019, 8, 769. [Google Scholar] [CrossRef]
- Galbiati, S.; Damin, F.; Burgio, V.; Brisci, A.; Soriani, N.; Belcastro, B.; Di Resta, C.; Gianni, L.; Chiari, M.; Ronzoni, M.; et al. Evaluation of three advanced methodologies, COLD-PCR, microarray and ddPCR, for identifying the mutational status by liquid biopsies in metastatic colorectal cancer patients. Clin. Chim. Acta 2019, 489, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Damin, F.; Galbiati, S.; Soriani, N.; Burgio, V.; Ronzoni, M.; Ferrari, M.; Chiari, M. Analysis of KRAS, NRAS and BRAF mutational profile by combination of in-tube hybridization and universal tag-microarray in tumor tissue and plasma of colorectal cancer patients. PLoS ONE 2018, 13, e020787. [Google Scholar] [CrossRef]
- Brambilla, D.; Sola, L.; Chiodi, E.; Zarovni, N.; Fortunato, D.; Criscuoli, M.; Dolo, V.; Giusti, I.; Murdica, V.; Vago, R.; et al. Separation of Extracellular Vesicles by DNA-Directed Immunocapturing Followed by Enzymatic Release. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Sahebi, R.; Langari, H.; Fathinezhad, Z.; Bahari Sani, Z.; Avan, A.; Ghayour Mobarhan, M.; Rezayi, M.J. Exosomes: New insights into cancer mechanisms. Cell Biochem. 2020, 121, 7–16. [Google Scholar] [CrossRef]
- Hosseini, M.; Khatamianfar, S.; Hassanian, S.M.; Nedaeinia, R.; Shafiee, M.; Maftouh, M.; Ghayour-Mobarhan, M.; Shahid Sales, S.; Avan, A. Exosome Encapsulated microRNAs as Potential Circulating Biomarkers in Colon Cancer. Curr. Pharm. Des. 2017, 23, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Maisano, D.; Mimmi, S.; Russo, R.; Fioravanti, A.; Fiume, G.; Vecchio, E.; Nisticò, N.; Quinto, I.; Iaccino, E. Uncovering the Exosomes Diversity: A Window of Opportunity for Tumor Progression Monitoring. Pharmaceuticals 2020, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Borzi, C.; Calzolari, L.; Ferretti, A.M.; Caleca, L.; Pastorino, U.; Sozzi, G.; Fortunato, O. c-Myc shuttled by tumour-derived extracellular vesicles promotes lung bronchial cell proliferation through miR-19b and miR-92a. Cell Death Dis. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ctDNA * | sEV-Associated DNA | |||
|---|---|---|---|---|
| ID Samples | Microarray | ddPCR Mutated Allele % | Microarray | ddPCR Mutated Allele % |
| 2 | BRAF V600E | BRAF V600E = 15% | BRAF V600E | BRAF V600E = 4.5% |
| 3 | wt | wt | wt | |
| 4 | wt | wt | wt | |
| 7 | KRAS G12C | KRAS G12C = 32.3% | KRAS G12C | KRAS G12C = 33.2% |
| 13 | KRAS A146T | KRAS A146T = 20% | KRAS A146T | KRAS A146T = 13% |
| 14 | KRAS G13D | KRAS G13D = 1.3% | KRAS G13D KRAS G12D | KRAS G13D = 0.35% KRAS G12D = na |
| 16 | KRAS A146T | KRAS A146T = 77.5% | KRAS A146T | KRAS A146T = 77.2% |
| 18 | KRAS G12D | KRAS G12D = 65.3% | KRAS G12D | KRAS G12D = 64.2% |
| 19 | KRAS G12S | KRAS G12S = 17.7% | KRAS G12S | KRAS G12S = 16.5% |
| 20 | KRAS G12D | KRAS G12D = 3.7% | KRAS G12D | KRAS G12D = 4.4% |
| 22 | KRAS G12D | KRAS G12D = 79.8% | KRAS G12D | KRAS G12D = 80% |
| Patient n. 22 | ddPCR Copies/20 µL | Fractional Abundance (Range) | Plasma Volume |
|---|---|---|---|
| ctDNA | mut = 1876 wt = 472 tot = 2348 | 80% (79–81%) | 500 µL |
| Isolation of EV | |||
| Ultracentrifugation | mut = 224 wt = 70 tot = 294 | 76% (73–79%) | 250 µL |
| Exosome Precipitation Kit | mut = 452 wt = 122 tot = 574 | 79% (77–81%) | 250 µL |
| DNA-directed Immunoprecipitation * | mut = 7 wt = 14 tot = 22 | 33% (14–46%) | 250 µL |
| Patient n. 2 | ddPCR Copies/20 µL | Fractional Abundance (Range) | Plasma Volume |
|---|---|---|---|
| ctDNA | mut = 42 wt = 240 tot = 282 | 15% (8–21%) | 500 µL |
| Isolation of EV | |||
| Ultracentrifugation | mut = 2 wt = 21 tot = 23 | 8% (2–15%) | 250 µL |
| Ultracentrifugation + DNase I treatment | mut = 0 wt = 2 tot = 2 | 0 | 250 µL |
| Supernatant 1 | mut = 15 wt = 92 tot = 107 | 14% (10–18%) | 250 µL |
| Supernatant 2 | mut = 134 wt = 74 tot = 878 | 15% (10–20%) | 250 µL |
| Exosome Precipitation Kit | mut = 10 wt = 45 tot = 55 | 19% (12–25%) | 250 µL |
| Exosome Precipitation Kit + DNase I treatment | mut = 0 wt = 0 tot = 0 | 0 | 250 µL |
| DNA-directed Immunoprecipitation * | mut = 0 wt = 3 tot = 3 | 0 | 250 µL |
| Sample | d * Mean [nm] | Std Dev [nm] | Minimum [nm] | Maximum [nm] |
|---|---|---|---|---|
| sEVs untreated | 84.5 | 28.2 | 28.6 | 173.7 |
| sEVs thermal treated | 74 | 34.2 | 24.4 | 214.7 |
| sEVs treated with DNase I | 88.7 | 26.1 | 36.2 | 169.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbiati, S.; Damin, F.; Brambilla, D.; Ferraro, L.; Soriani, N.; Ferretti, A.M.; Burgio, V.; Ronzoni, M.; Vago, R.; Sola, L.; et al. Small EVs-Associated DNA as Complementary Biomarker to Circulating Tumor DNA in Plasma of Metastatic Colorectal Cancer Patients. Pharmaceuticals 2021, 14, 128. https://doi.org/10.3390/ph14020128
Galbiati S, Damin F, Brambilla D, Ferraro L, Soriani N, Ferretti AM, Burgio V, Ronzoni M, Vago R, Sola L, et al. Small EVs-Associated DNA as Complementary Biomarker to Circulating Tumor DNA in Plasma of Metastatic Colorectal Cancer Patients. Pharmaceuticals. 2021; 14(2):128. https://doi.org/10.3390/ph14020128
Chicago/Turabian StyleGalbiati, Silvia, Francesco Damin, Dario Brambilla, Lucia Ferraro, Nadia Soriani, Anna M. Ferretti, Valentina Burgio, Monica Ronzoni, Riccardo Vago, Laura Sola, and et al. 2021. "Small EVs-Associated DNA as Complementary Biomarker to Circulating Tumor DNA in Plasma of Metastatic Colorectal Cancer Patients" Pharmaceuticals 14, no. 2: 128. https://doi.org/10.3390/ph14020128
APA StyleGalbiati, S., Damin, F., Brambilla, D., Ferraro, L., Soriani, N., Ferretti, A. M., Burgio, V., Ronzoni, M., Vago, R., Sola, L., & Chiari, M. (2021). Small EVs-Associated DNA as Complementary Biomarker to Circulating Tumor DNA in Plasma of Metastatic Colorectal Cancer Patients. Pharmaceuticals, 14(2), 128. https://doi.org/10.3390/ph14020128