
Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters



Abstract
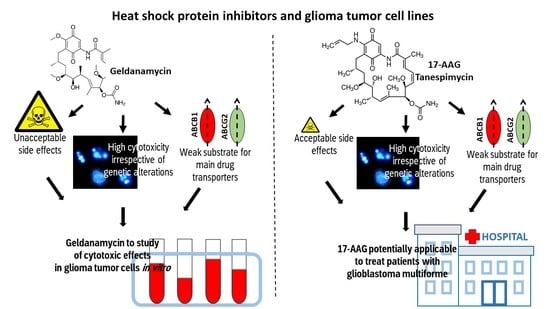
1. Introduction
2. Results
2.1. Cytotoxic Effects of HSP90 Inhibitors in Glioma Tumor Cells
2.2. Interactions of Geldanamycin and Tanespimycin with the Main Drug Transporters
3. Discussion
4. Material and Methods
4.1. Chemicals and Cell Treatment
4.2. Cell Culture
4.3. Cell Viability Assessment
4.4. Cytotoxicity Assay
4.5. Western Blot Analysis of Drug Transporter Expression
4.6. Functional Assay of ABCB1 and ABCG2
4.7. Analysis of Cell Cycle and Apoptotic Cells
4.8. Morphological Analysis of Apoptosis
4.9. Measurement of Caspase3/7 Enzymatic Activity
4.10. Cell Extracts Preparation
4.11. LC/MS/MS Analysis of GDN
4.12. LC/MS/MS Analysis of 17-AAG
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Soo, E.T.L.; Yip, G.W.; Lwin, Z.M.; Kumar, S.D.; Bay, B.-H. Heat shock proteins as novel therapeutic targets in cancer. Vivo 2008, 22, 311–315. [Google Scholar]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Wick, W.; Osswald, M.; Wick, A.; Winkler, F. Treatment of glioblastoma in adults. Ther. Adv. Neurol. Disord. 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Bai, R.-Y.; Staedtke, V.; Riggins, G.J. Molecular targeting of glioblastoma: Drug discovery and therapies. Trends Mol. Med. 2011, 17, 301–312. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro-Oncology 2011, 13, 566–579. [Google Scholar] [CrossRef]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus 2015, 38, E4. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Guan, J.; Cohen, A.L.; Jensen, R.L.; Colman, H. New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr. Neurol. Neurosci. Rep. 2017, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J., Jr.; Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017, 37, 21–33. [Google Scholar] [CrossRef]
- Litak, J.; Grochowski, C.; Litak, J.; Osuchowska, I.; Gosik, K.; Radzikowska, E.; Kamieniak, P.; Roliński, J. TLR-4 Signaling vs. Immune Checkpoints, miRNAs Molecules, Cancer Stem Cells, and Wingless-Signaling Interplay in Glioblastoma Multiforme—Future Perspectives. Int. J. Mol. Sci. 2020, 21, 3114. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; Bent, M.V.D.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Iglesia, R.P.; Fernandes, C.F.D.L.; Coelho, B.P.; Prado, M.B.; Escobar, M.I.M.; Almeida, G.H.D.R.; Lopes, M.H. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? Int. J. Mol. Sci. 2019, 20, 5794. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Yang, J.-M.; Iannone, M.; Shih, W.J.; Lin, Y.; Hait, W.N. Disruption of the EF-2 kinase/Hsp90 protein complex: A possible mechanism to inhibit glioblastoma by geldanamycin. Cancer Res. 2001, 61, 4010–4016. [Google Scholar]
- García-Morales, P.; Carrasco-García, E.; Ruiz-Rico, P.; Martínez-Mira, R.; Menéndez-Gutiérrez, M.P.; Ferragut, J.A.; Saceda, M.; Martínez-Lacaci, I. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G2/M arrest in glioblastoma cell lines. Oncogene 2007, 26, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Sauvageot, C.M.-E.; Weatherbee, J.L.; Kesari, S.; Winters, S.E.; Barnes, J.; DellaGatta, J.; Ramakrishna, N.R.; Stiles, C.D.; Kung, A.L.-J.; Kieran, M.W.; et al. Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro-Oncology 2009, 11, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Hirose, Y.; Yoshida, K.; Yazaki, T.; Kawase, T. Inhibition of 90-kD heat shock protein potentiates the cytotoxicity of chemotherapeutic agents in human glioma cells. J. Neurosurg. 2010, 112, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A Novel Extracellular Hsp90 Mediated Co-Receptor Function for LRP1 Regulates EphA2 Dependent Glioblastoma Cell Invasion. PLoS ONE 2011, 6, e17649. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Blower, P.E.; Liu, R.; Dai, Z.; Pham, A.-N.; Moon, H.; Fang, J.; Sadee, W. Chemogenomic Analysis Identifies Geldanamycins as Substrates and Inhibitors of ABCB. Pharm. Res. 2007, 24, 1702–1712. [Google Scholar] [CrossRef]
- Mccollum, A.K.; TenEyck, C.J.; Stensgard, B.; Morlan, B.W.; Ballman, K.V.; Jenkins, R.B.; Toft, D.O.; Erlichman, C. P-Glycoprotein–Mediated Resistance to Hsp90-Directed Therapy Is Eclipsed by the Heat Shock Response. Cancer Res. 2008, 68, 7419–7427. [Google Scholar] [CrossRef]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat Shock Transcription Factor 1-activating Compounds Suppress Polyglutamine-induced Neurodegeneration through Induction of Multiple Molecular Chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef]
- Nomura, M.; Nomura, N.; Newcomb, E.W.; Lukyanov, Y.; Tamasdan, C.; Zagzag, D. Geldanamycin induces mitotic catastrophe and subsequent apoptosis in human glioma cells. J. Cell. Physiol. 2004, 201, 374–384. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kosztyu, P.; Dolezel, P.; Mlejnek, P. Can P-glycoprotein mediate resistance to nilotinib in human leukaemia cells? Pharmacol. Res. 2013, 67, 79–83. [Google Scholar] [CrossRef]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Bates, S.E.; Ruzickova, E. Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem. Interact. 2017, 273, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ruzickova, E.; Janska, R.; Dolezel, P.; Mlejnek, P. Clinically relevant interactions of anti-apoptotic Bcl-2 protein inhibitors with ABC transporters. Die Pharm. 2017, 72, 751–758. [Google Scholar]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Annereau, J.P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.-L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of Multidrug Resistance Gene in Human Cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- Miller, D.A.W. Regulation of ABC transporters at the blood-brain barrier. Clin. Pharmacol. Ther. 2014, 97, 395–403. [Google Scholar] [CrossRef]
- Richardson, P.G.; Chanan-Khan, A.A.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Mitsiades, C.S.; Anderson, K.C. Tanespimycin monotherapy in relapsed multiple myeloma: Results of a phase 1 dose-escalation study. Br. J. Haematol. 2010, 150, 438–445. [Google Scholar] [CrossRef]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef]
- Egorin, M.J.; Zuhowski, E.G.; Rosen, D.M.; Sentz, D.L.; Covey, J.M.; Eiseman, J.L. Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) in CD2F1 mice. Cancer Chemother. Pharmacol. 2001, 47, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Keum, G.; Pae, A.N. Discovery and development of heat shock protein 90 inhibitors as anticancer agents: A review of patented potent geldanamycin derivatives. Expert Opin. Ther. Patents 2013, 23, 919–943. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Yoon, N.G.; Lee, J.-E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.-H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef]
- Tang, R.; Faussat, A.-M.; Perrot, J.-Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.-P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8, 51. [Google Scholar] [CrossRef]
- Elkind, N.B.; Apáti, Á.; Várady, G.; Ujhelly, O.; Szabó, K.; Homolya, L.; Buday, L.; Német, K.; Sarkadi, B.; Szentpétery, Z.; et al. Multidrug Transporter ABCG2 Prevents Tumor Cell Death Induced by the Epidermal Growth Factor Receptor Inhibitor Iressa (ZD1839, Gefitinib). Cancer Res. 2005, 65, 1770–1777. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kosztyu, P.; Dolezel, P.; Vaclavikova, R.; Mlejnek, P. Can the assessment of ABCB 1 gene expression predict its function in vitro? Eur. J. Haematol. 2015, 95, 150–159. [Google Scholar] [CrossRef]
- Nicoleti, G.; Migliorati, M.C.; Pagliacci, F.; Grignani, C.; Riccardi, A. rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bruno, S.; Del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kuglík, P. Induction of apoptosis in HL-60 cells by N(6)-benzyladenosine. J. Cell. Biochem. 2000, 77, 6–17. [Google Scholar] [CrossRef]
- Frydrych, I.; Mlejnek, P. Serine protease inhibitors N-α-tosyl-l-lysinyl-chloromethylketone (TLCK) and N-tosyl-l-phenylalaninyl-chloromethylketone (TPCK) are potent inhibitors of activated caspase proteases. J. Cell. Biochem. 2008, 103, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Novák, O.; Dolezel, P. A non-radioactive assay for precise determination of intracellular levels of imatinib and its main metabolite in Bcr-Abl positive cells. Talanta 2011, 83, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Krumpochova, P.; Kocurova, A.; Dolezel, P.; Mlejnek, P. Assay for determination of daunorubicin in cancer cells with multidrug resistance phenotype. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2011, 879, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P. N-acetylcysteine prevents the geldanamycin cytotoxicity by forming geldanamycin–N-acetylcysteine adduct. Chem. Interact. 2014, 220, 248–254. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | ATCC® | Disease | Gene Mutation/Alteration |
|---|---|---|---|
| A172 | CRL-1620 | Glioblastoma | CDKN2A |
| CDKN2B | |||
| EGFR vIII | |||
| PTEN | |||
| TP53 | |||
| H4 | HTB-148 | Neuroglioma | CDKN2A |
| CDKN2C | |||
| MGMT down | |||
| PTEN | |||
| ABCB1 up | |||
| U-87 MG | HTB-14 | Likely Glioblastoma | CDKN2A |
| CDKN2B | |||
| CDKN2C | |||
| MGMT down | |||
| PTEN | |||
| U-118 MG | HTB-15 | Glioblastoma Astrocytoma | CDKN2A |
| CDKN2B | |||
| MGMT down | |||
| PTEN | |||
| TP53 | |||
| SW1088 | HTB-12 | Astrocytoma | CDKN2A |
| CDKN2B | |||
| MGMT down | |||
| PTEN | |||
| TP53 | |||
| T98G | CRL-1690 | Glioblastoma multiforme | CDKN2A |
| CDKN2C | |||
| MGMT up | |||
| PTEN | |||
| TP53 |
| Cell Line | GDN IC50 (nM) | GDN + ZSQ IC50 (nM) | 17-AAG IC50 (nM) | 17-AAG + ZSQ IC50 (nM) |
|---|---|---|---|---|
| A-172 | 214.7 ± 39.5 | 222.5 ± 42.8 | 34.8 ± 7.1 | 33.8 ± 6.6 |
| H4 | 654.8 ± 147.5 | 183.9 ± 35.1 | 49.3 ± 8.0 | 28.2 ± 5.3 |
| U-87 MG | 104.4 ± 22.4 | 118.6 ± 27.8 | 21.4 ± 4.6 | 21.8 ± 4.3 |
| U-118 MG | 118.0 ± 20.2 | 121.5 ± 19.7 | 29.9 ± 5.2 | 28.2 ± 5.2 |
| SW1088 | 124.5 ± 24.8 | 131.2 ± 20.4 | 35.6 ± 5.9 | 34.4 ± 7.2 |
| T98G | 549.6 ± 109.0 | 579.4 ± 91.2 | 40.4 ± 6.5 | 40.9 ± 7.9 |
| Cell Line | TMZ IC50 (µM) | TMZ + LG IC50 (µM) | LG IC20 (µM) |
|---|---|---|---|
| A-172 | 54.4 ± 9.6 | 45.1 ± 9.9 | >10 |
| H4 | 78.8 ± 15.4 | 28.7 ± 6.1 | >10 |
| U-87 MG | 26.4 ± 5.1 | 23.1 ± 4.4 | >10 |
| U-118 MG | 71.3 ± 11.2 | 22.1 ± 3.7 | >10 |
| SW1088 | 51.1 ± 12.8 | 47.3 ± 8.0 | >10 |
| T98G | 51.5 ± 9.9 | 49.1 ± 11.4 | >10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastvova, N.; Dolezel, P.; Mlejnek, P. Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters. Pharmaceuticals 2021, 14, 107. https://doi.org/10.3390/ph14020107
Pastvova N, Dolezel P, Mlejnek P. Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters. Pharmaceuticals. 2021; 14(2):107. https://doi.org/10.3390/ph14020107
Chicago/Turabian StylePastvova, Nikola, Petr Dolezel, and Petr Mlejnek. 2021. "Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters" Pharmaceuticals 14, no. 2: 107. https://doi.org/10.3390/ph14020107
APA StylePastvova, N., Dolezel, P., & Mlejnek, P. (2021). Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters. Pharmaceuticals, 14(2), 107. https://doi.org/10.3390/ph14020107