
New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-benzothiazolones

 , , , , and
, , , , and 
Abstract
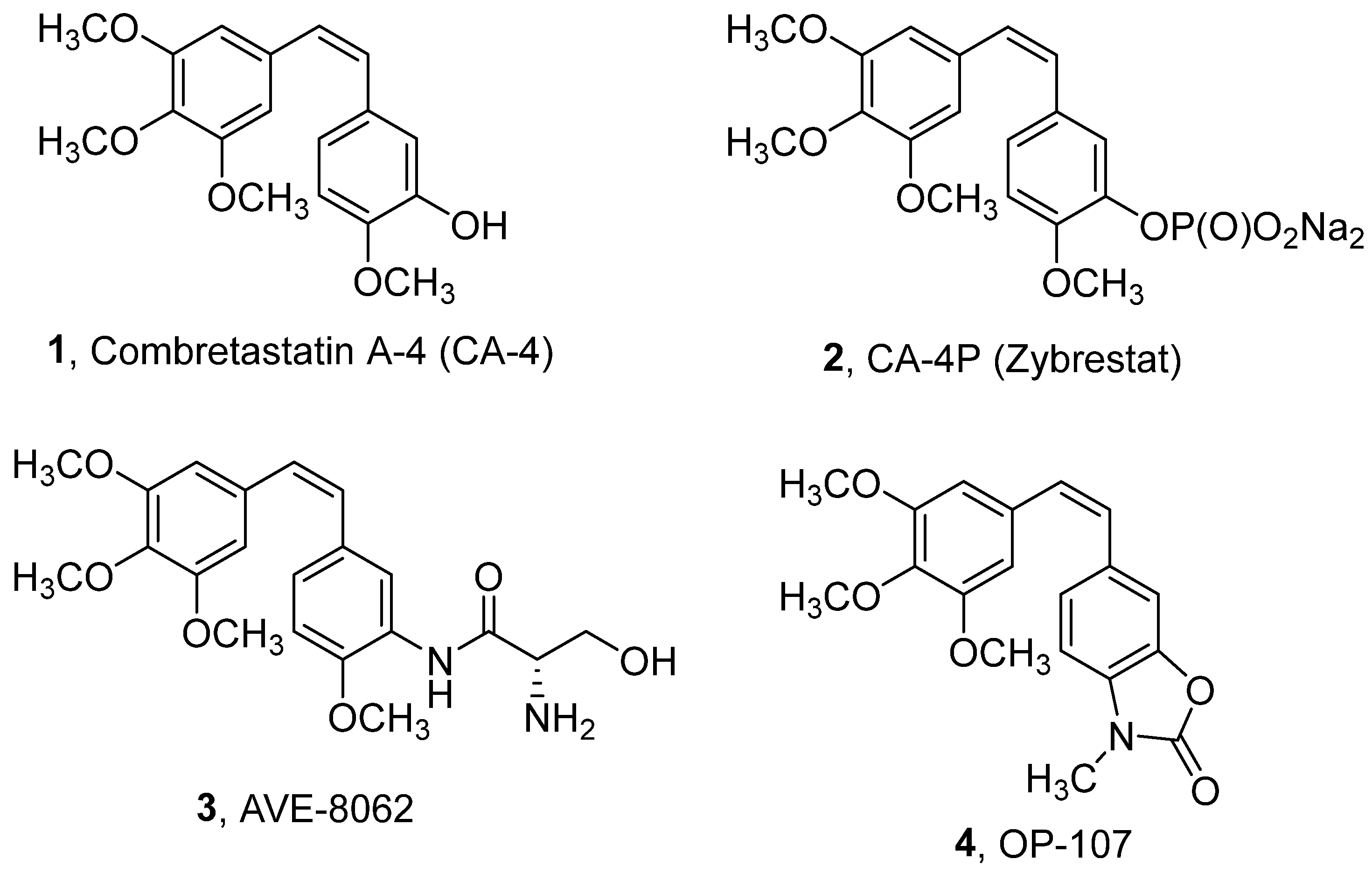
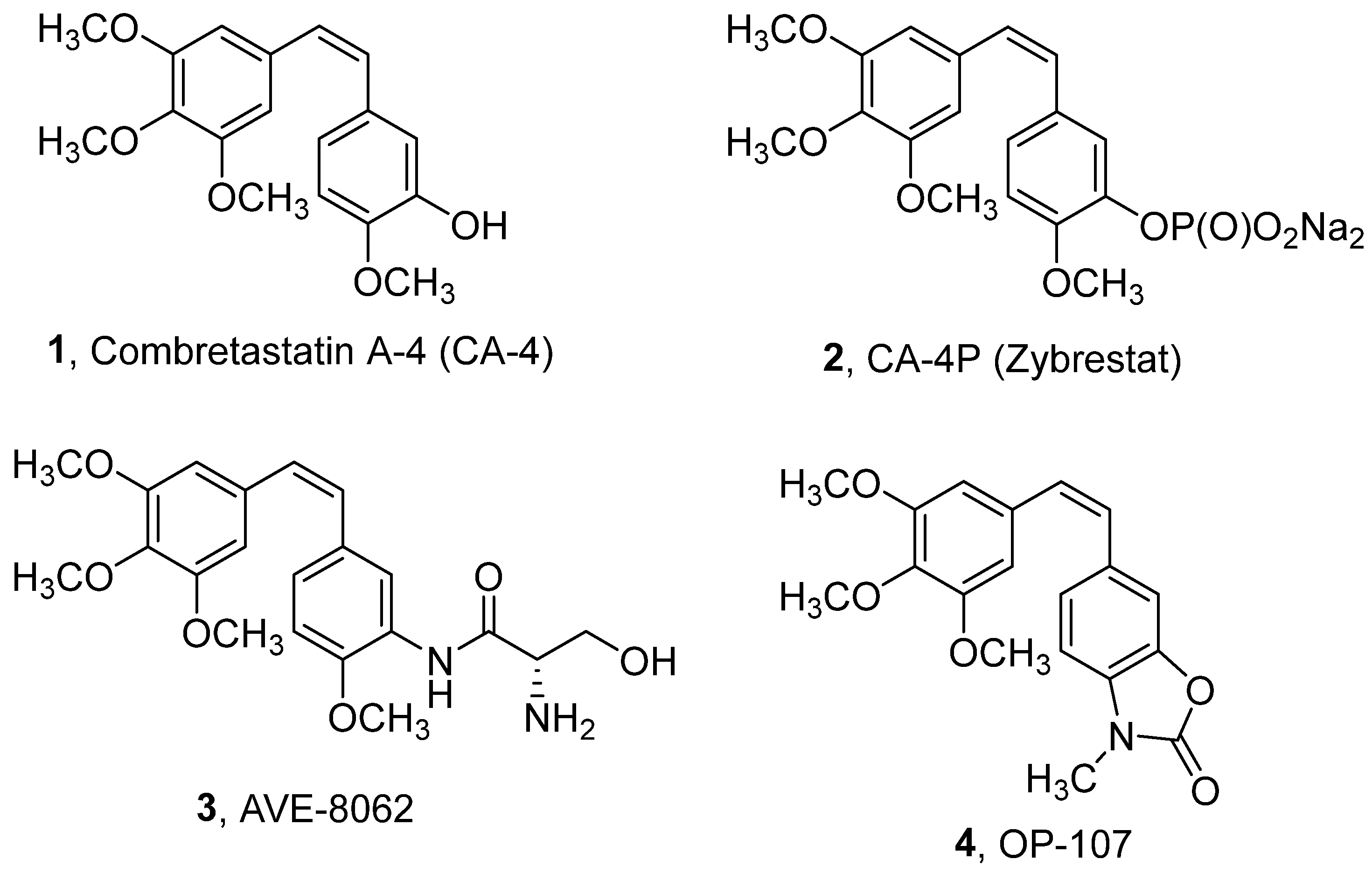
:1. Introduction
2. Results and Discussion
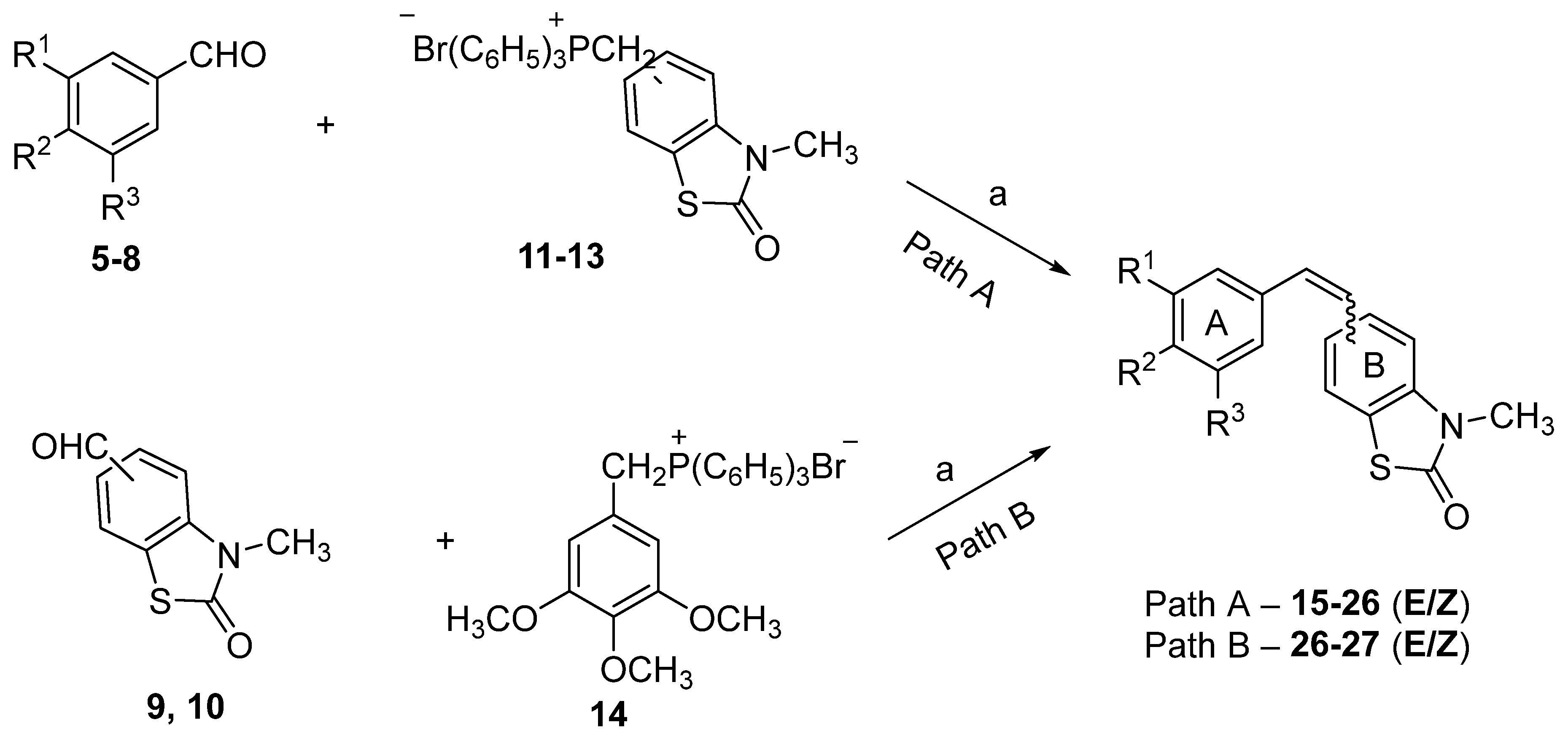
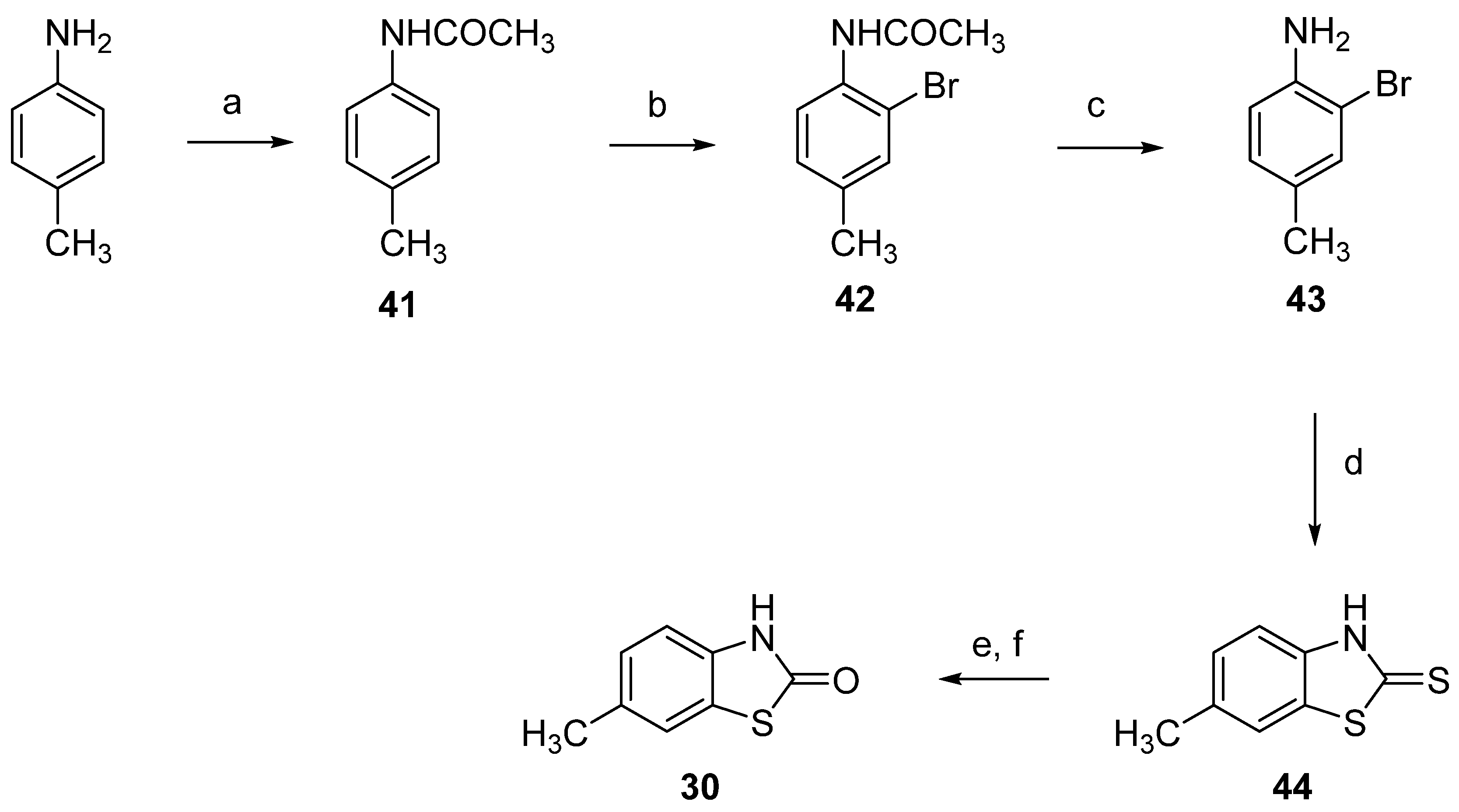
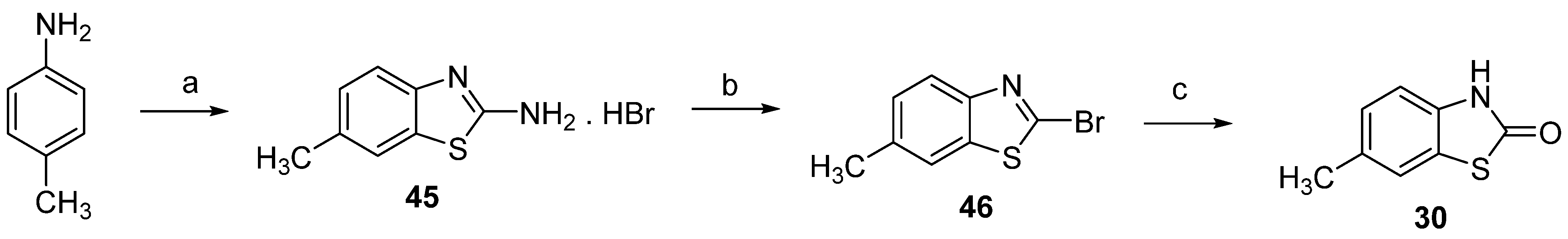
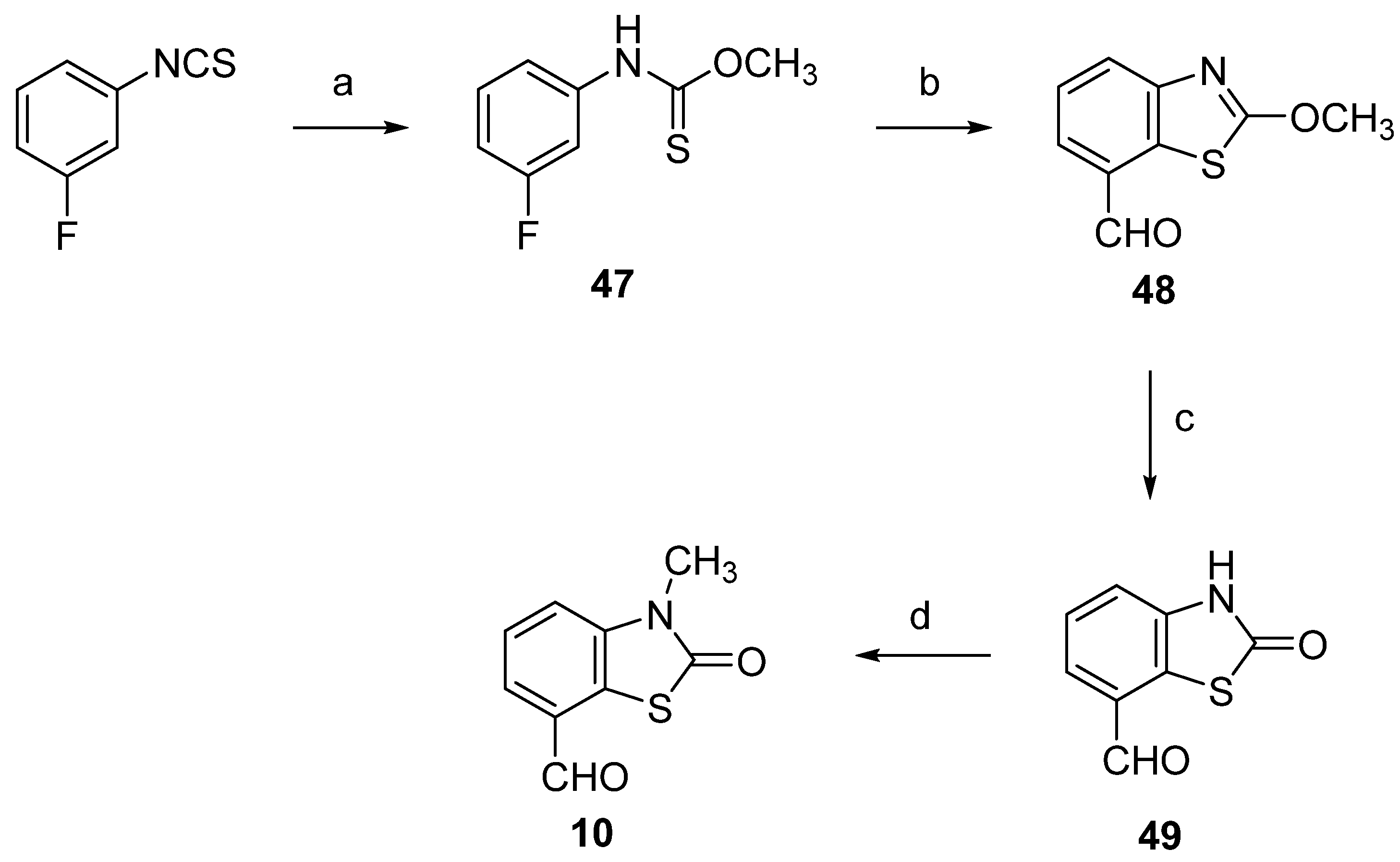
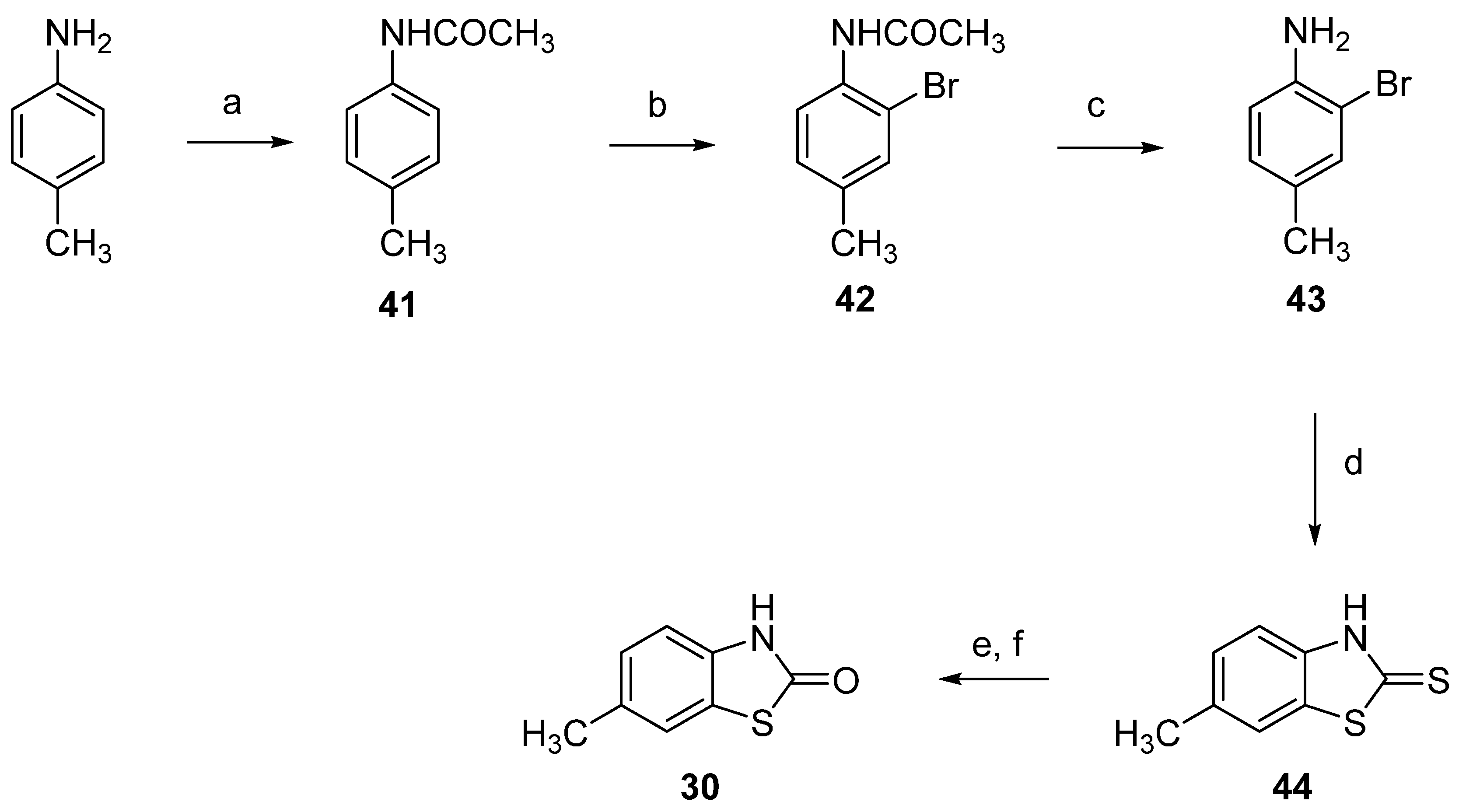
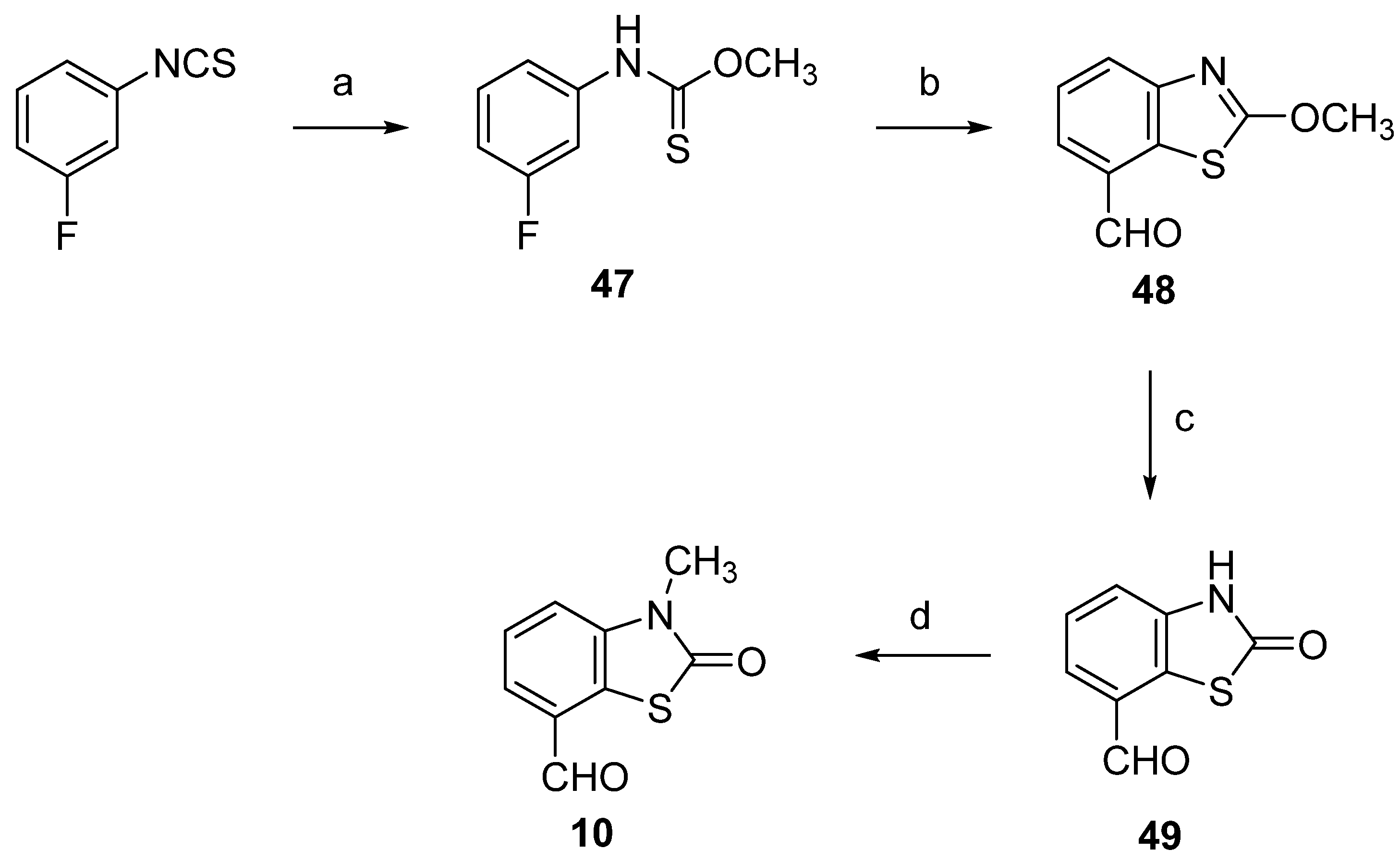
2.1. Chemistry
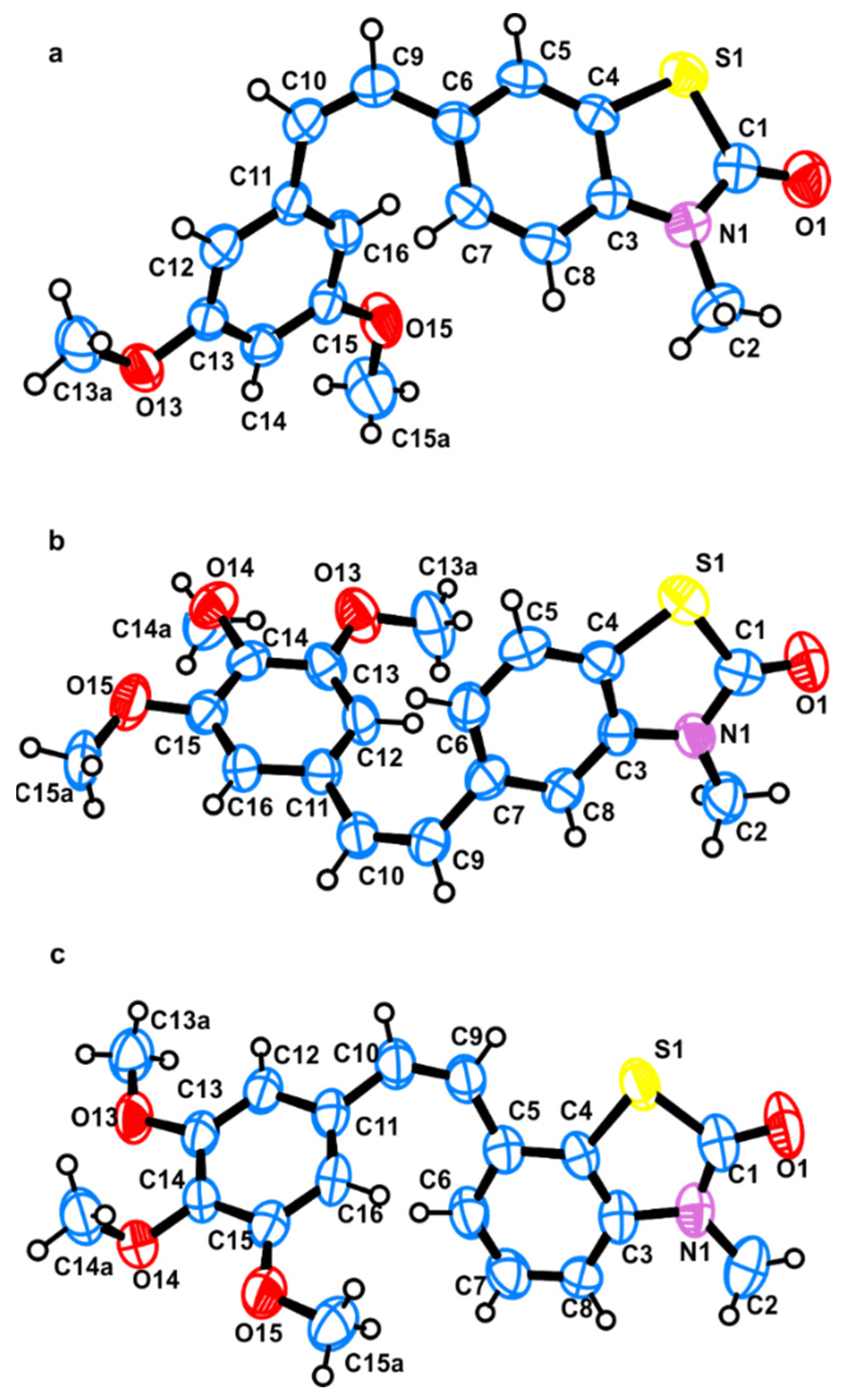
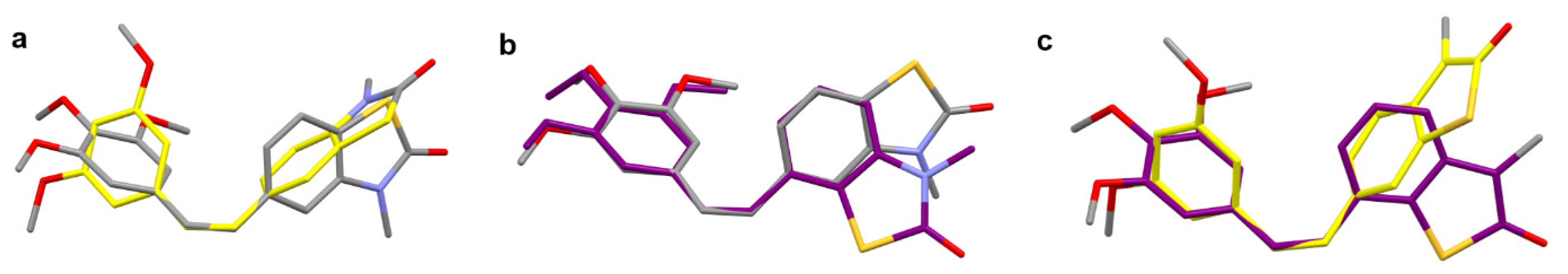
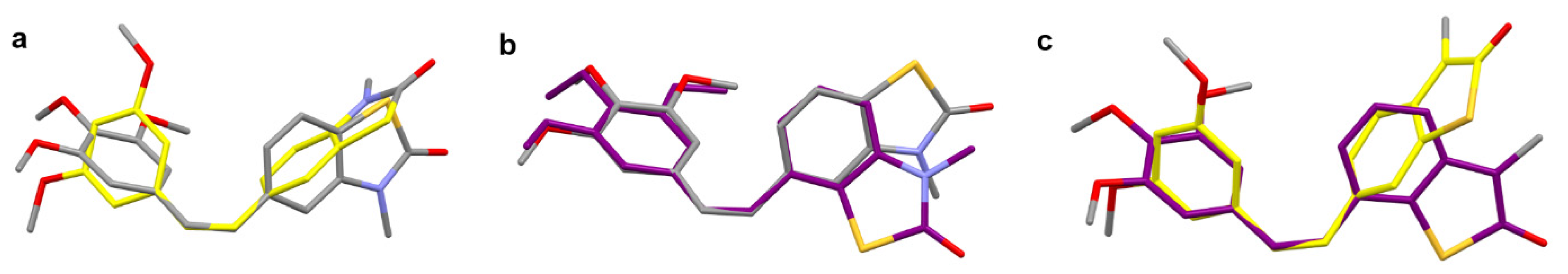
2.2. X-ray Crystallography and Docking
2.2.1. Single Crystal X-ray Diffraction
2.2.2. Molecular Docking of 26Z, 25Z, 22Z, and 27Z in the Colchicine Site of α,β-Tubulin
2.3. Biology Experiments
2.3.1. Evaluation of Benzothiazolone CA-4 Analog’s Cytotoxic Activity in Human Endothelial and Tumor Cell Lines
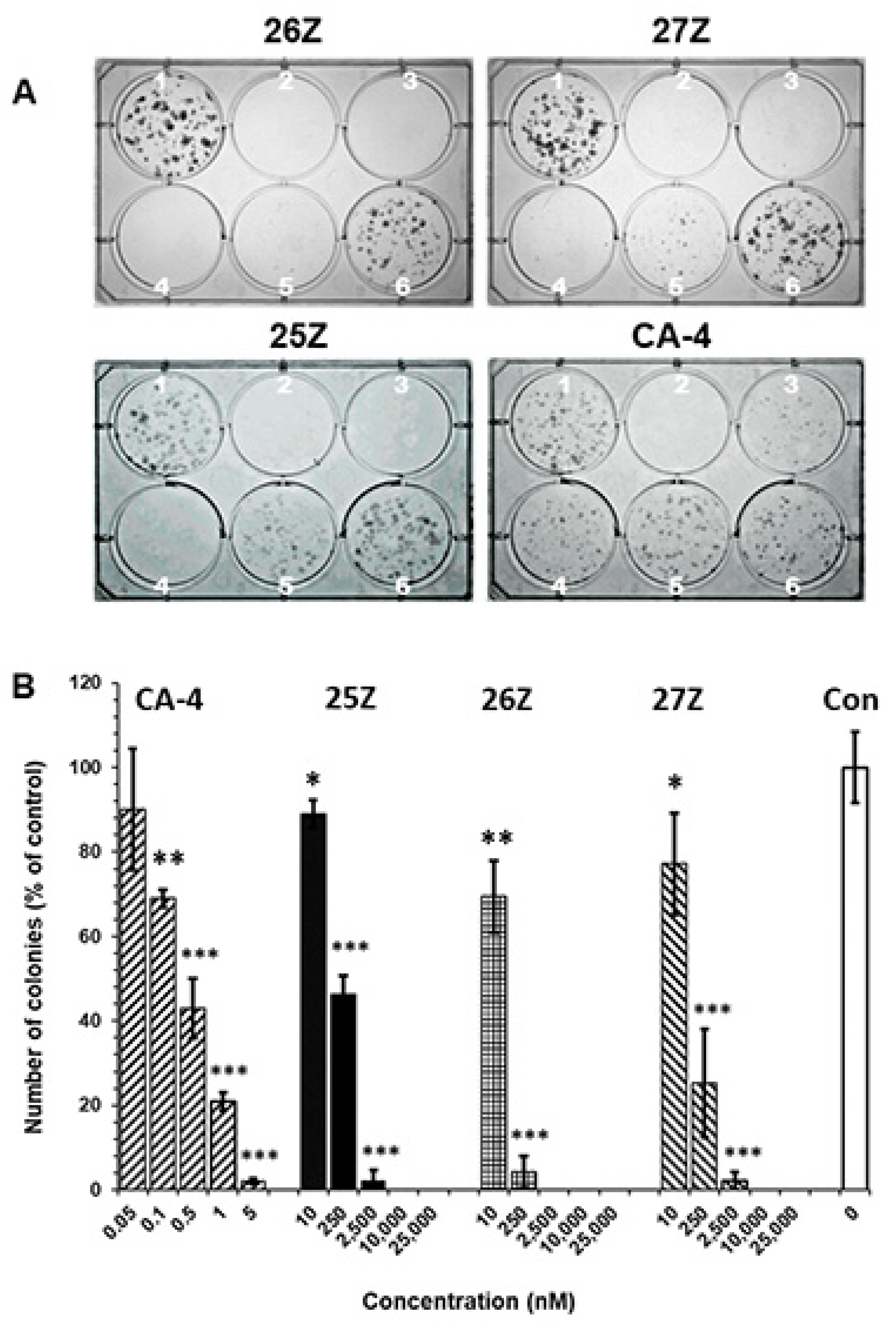
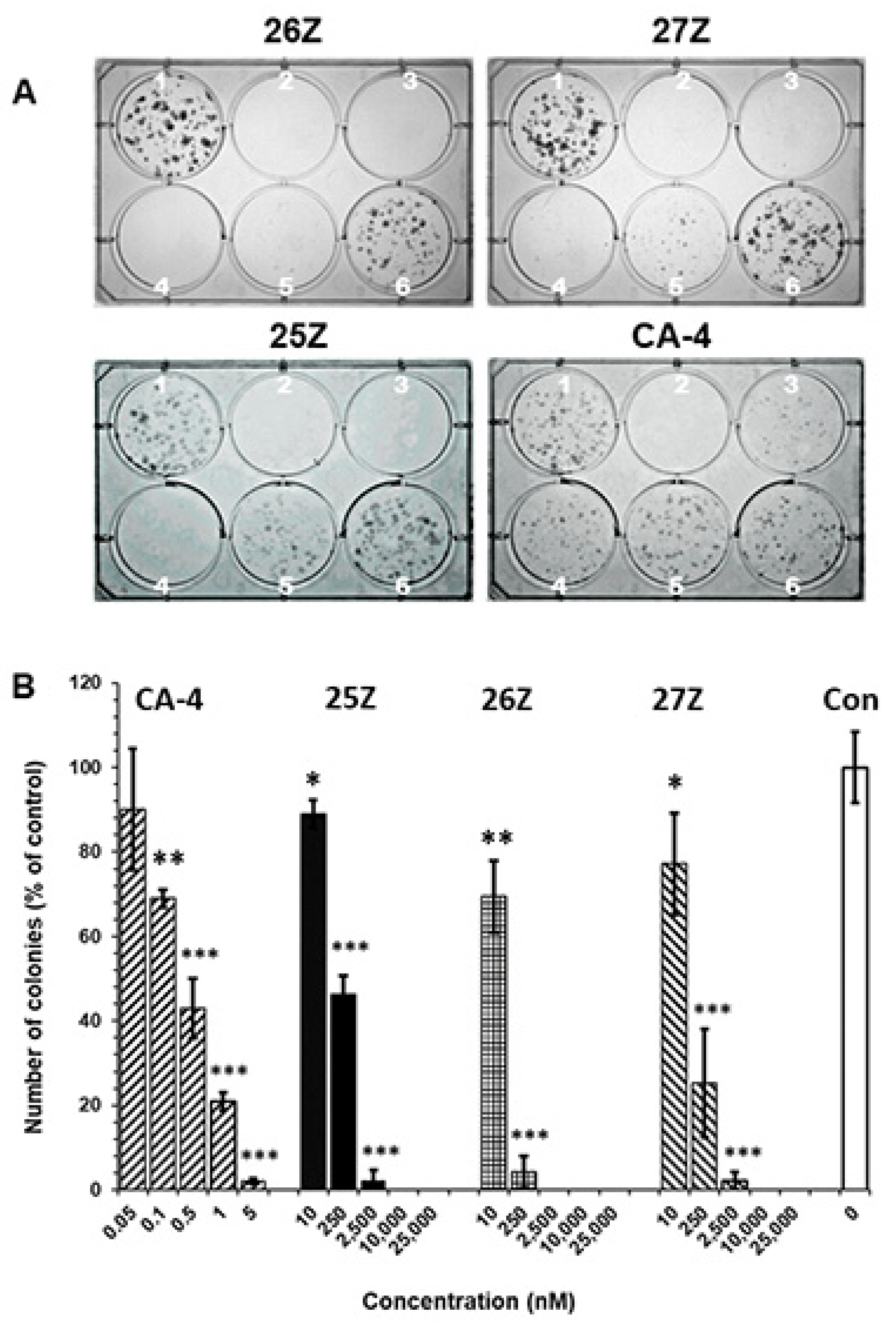
2.3.2. 25Z, 26Z, and 27Z Decrease the Clonogenic Survival of Endothelial Cells
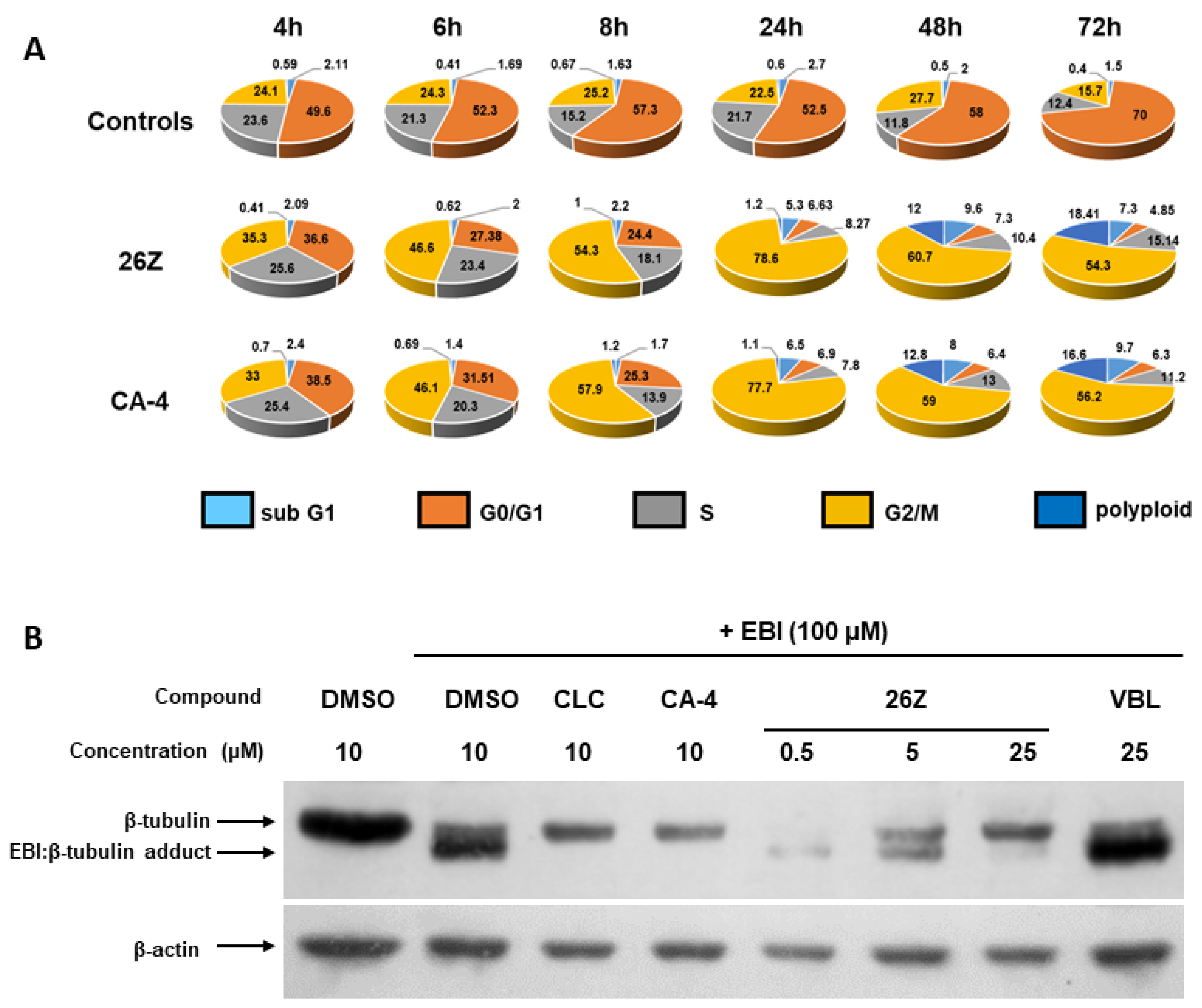
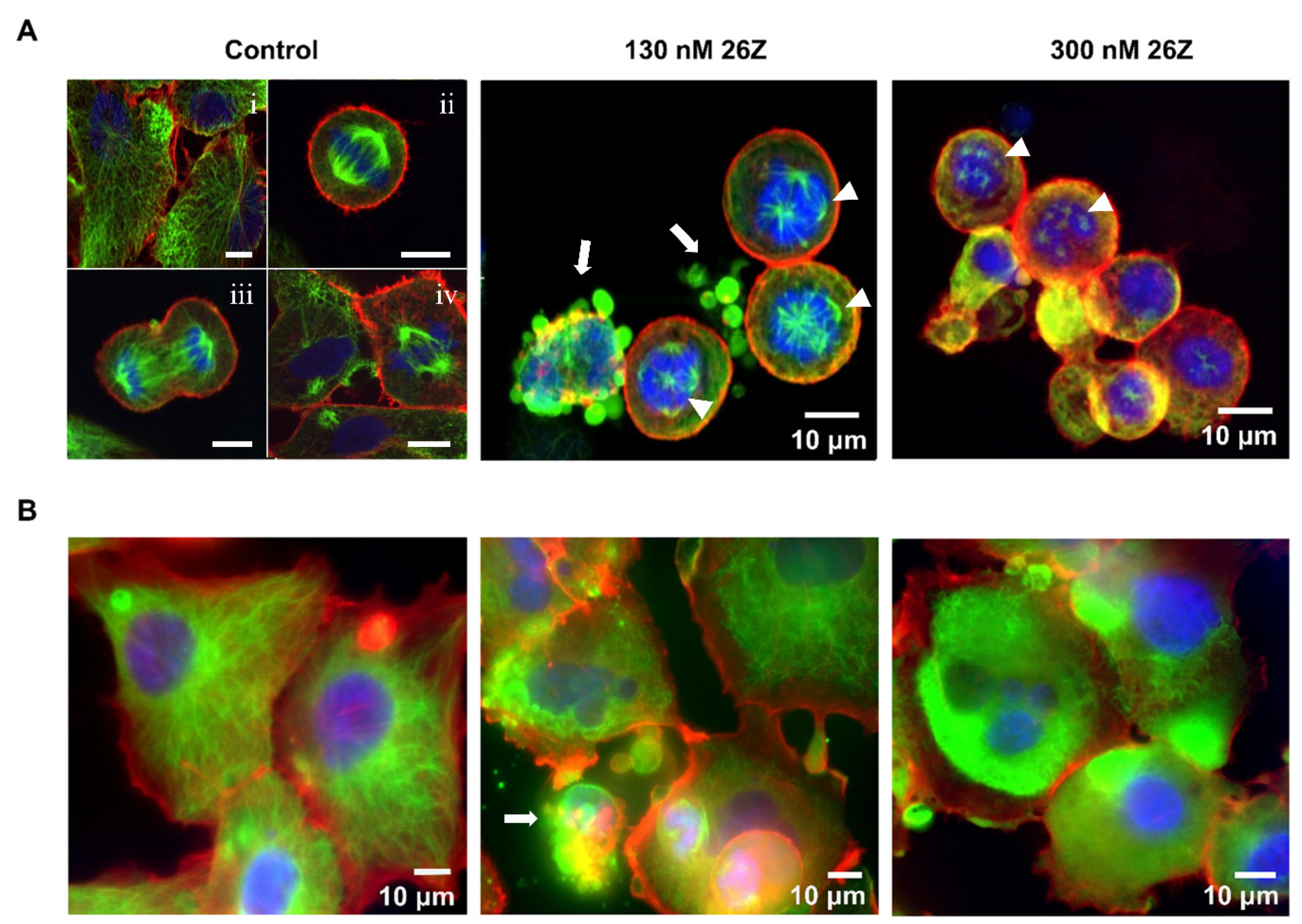
2.3.3. 26Z Causes Mitotic Arrest in Treated EA.hy926 Cells
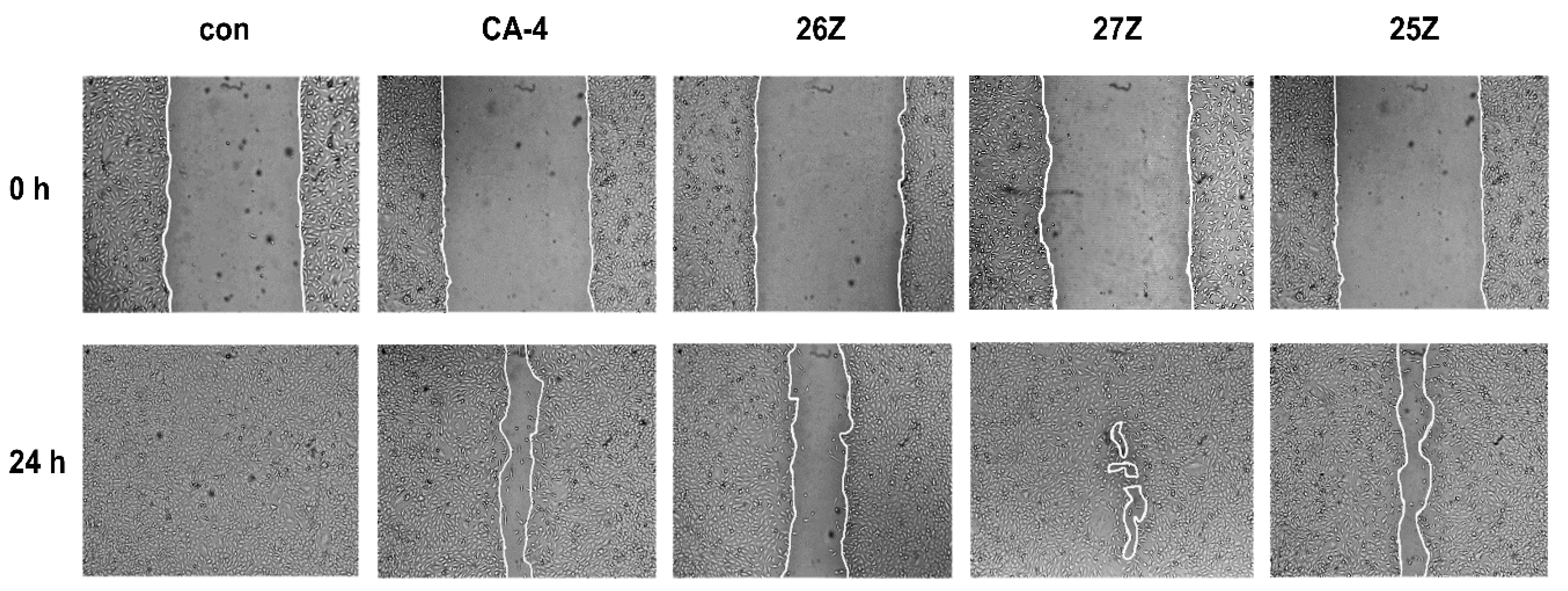
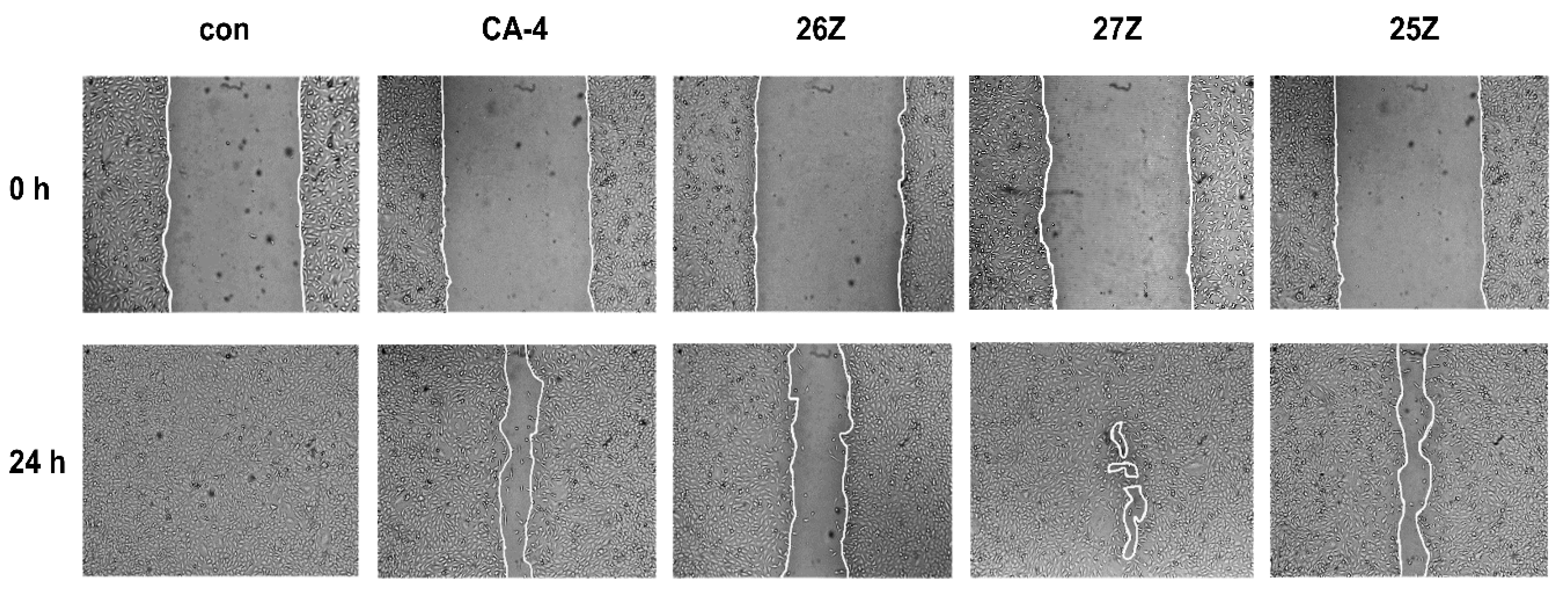
2.3.4. In Vitro Cell Migration Assay
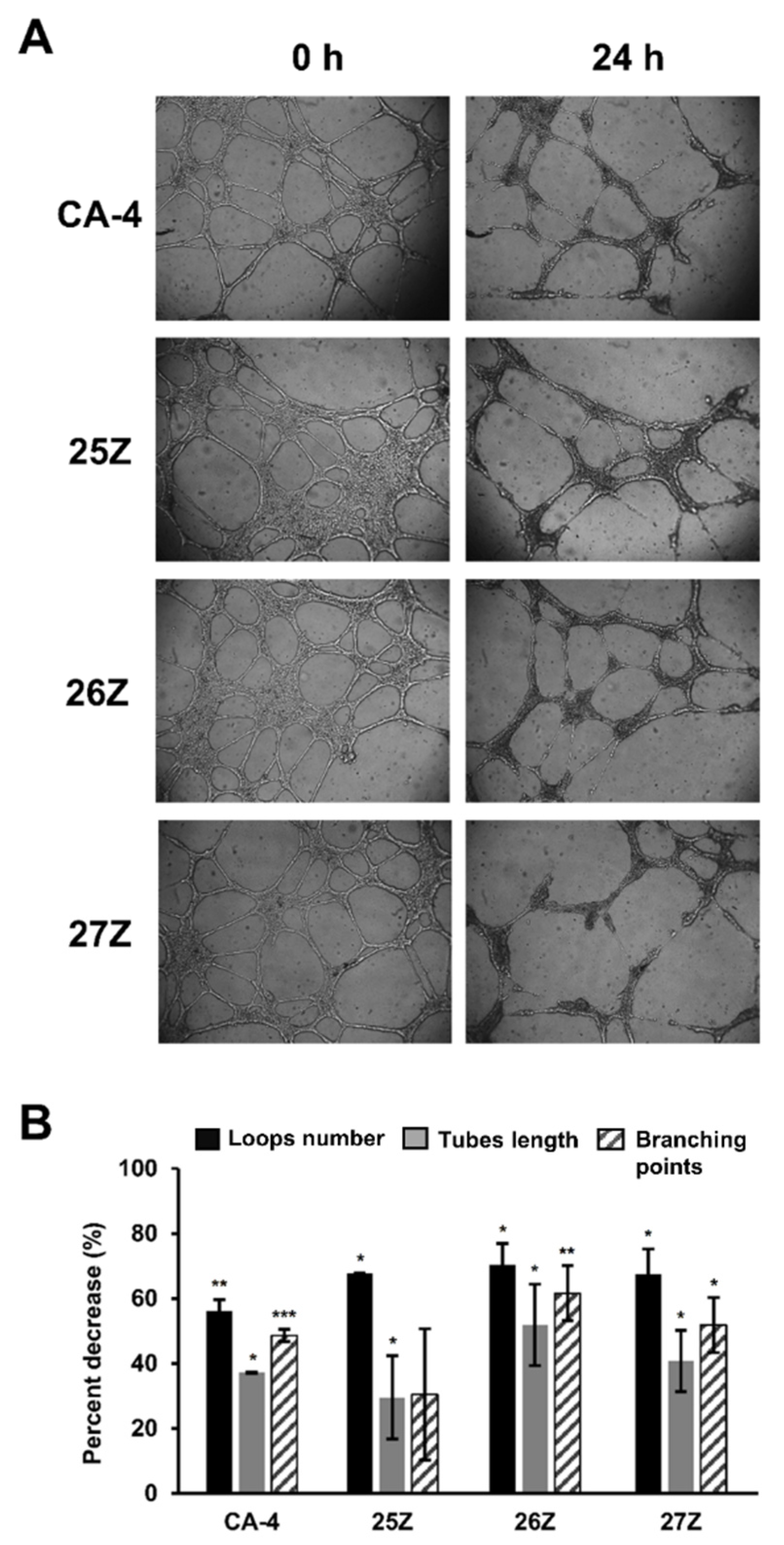
2.3.5. In Vitro Anti-Vascular Activity of Benzothiazolone CA-4 Analogs
2.3.6. Aberrant Formation of Mitotic Spindles and Microtubule Network Alterations
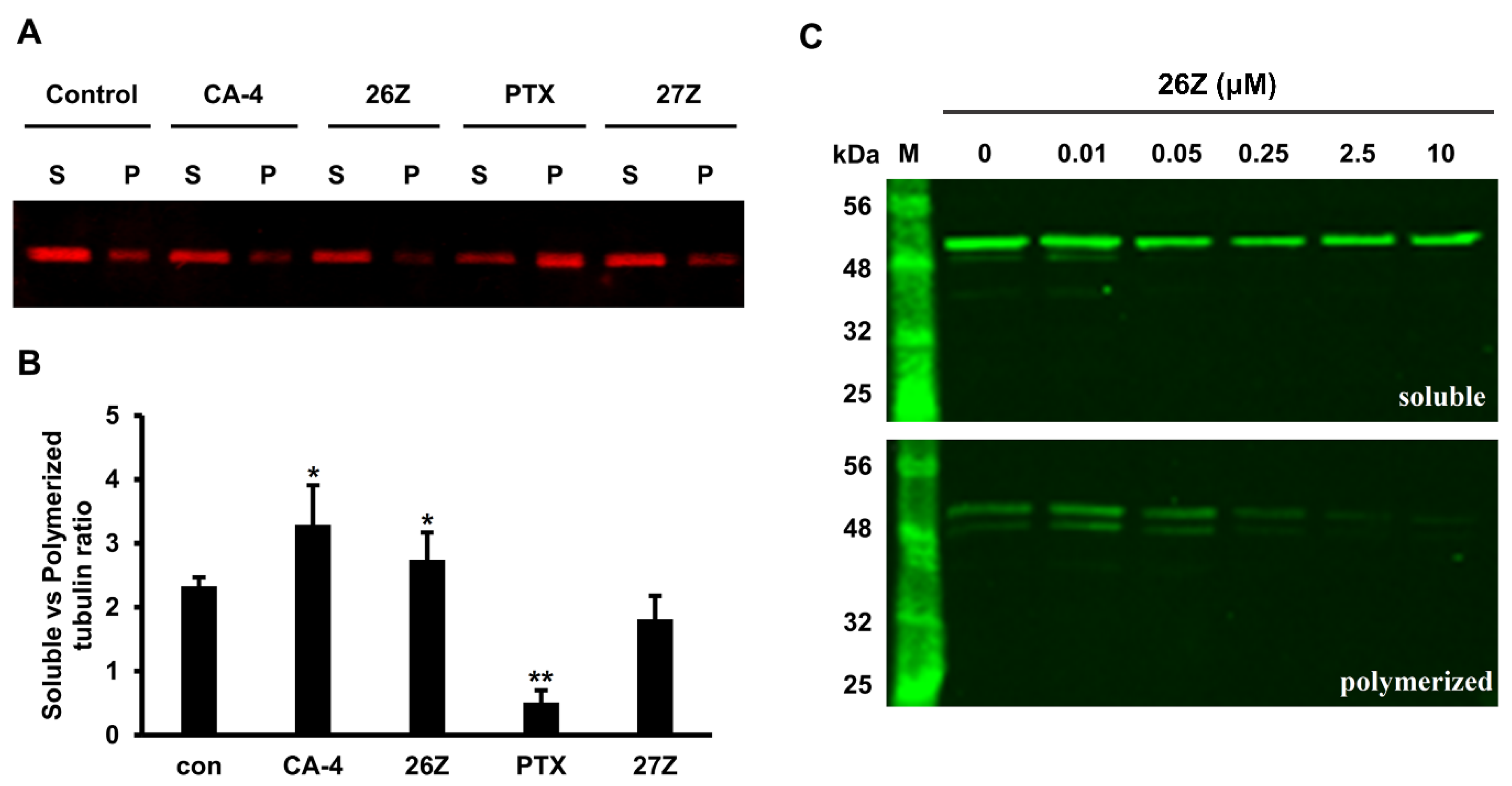
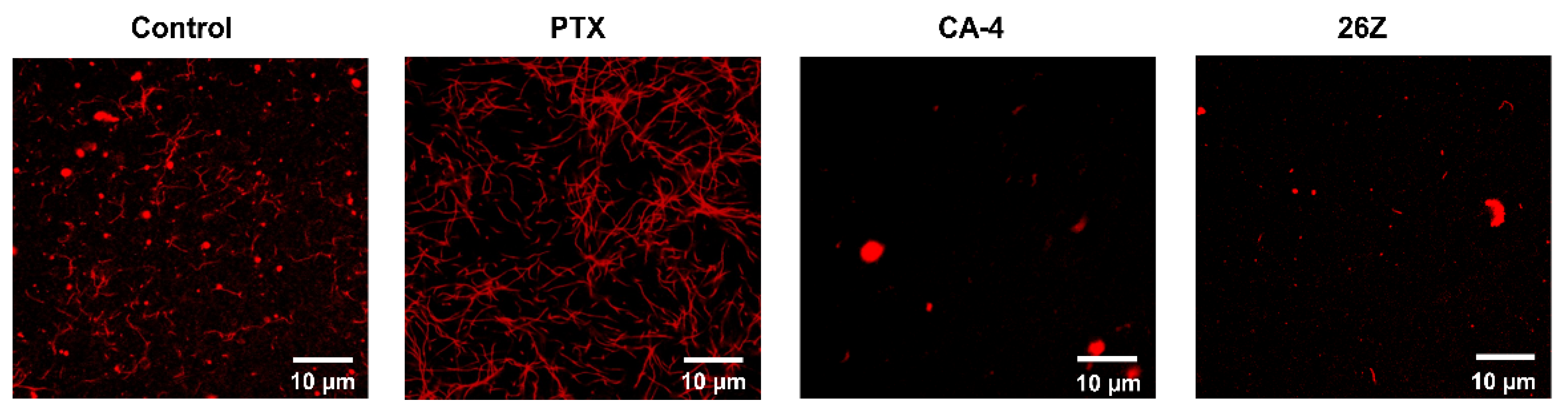
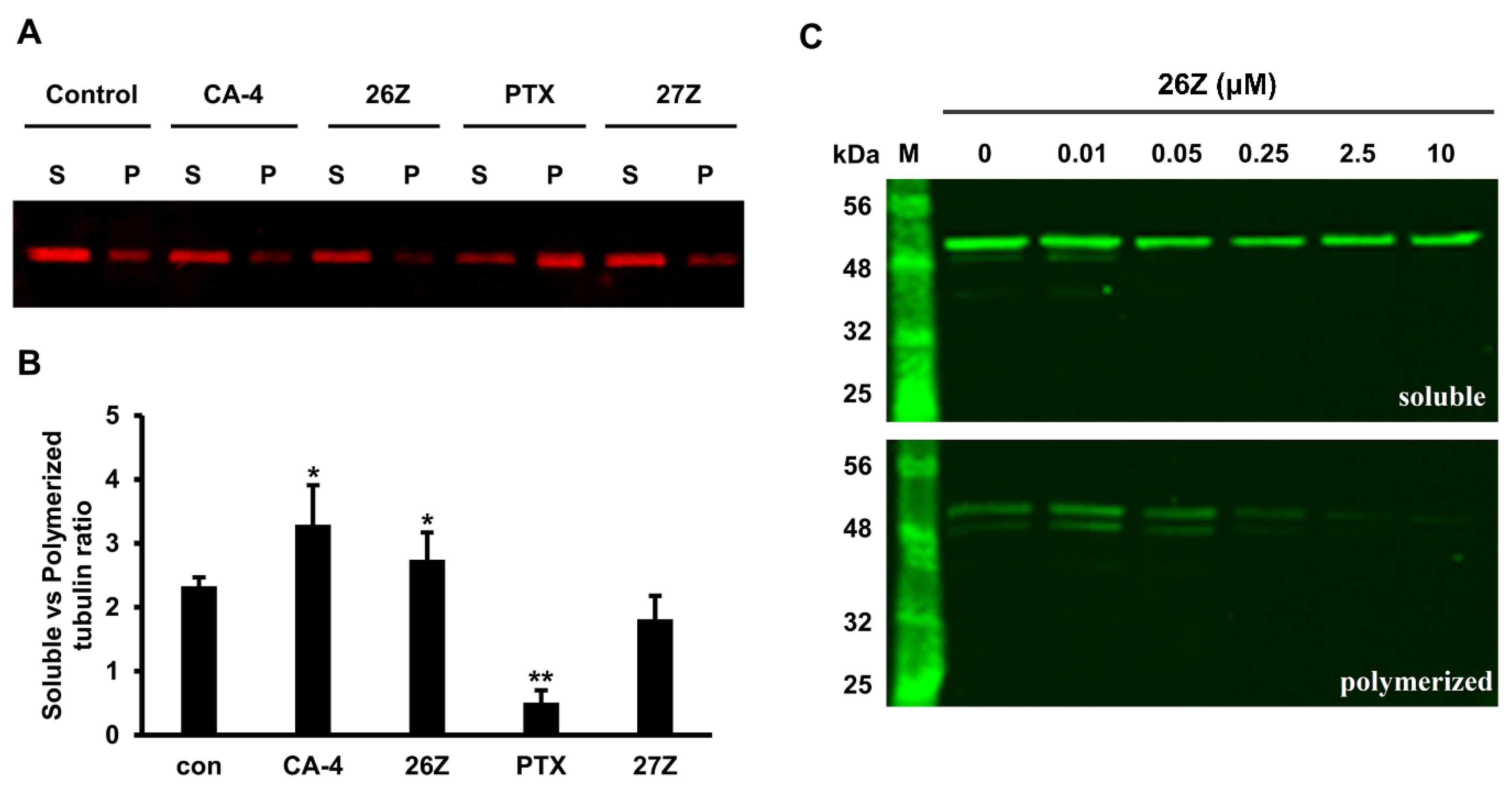
2.3.7. 26Z Acts as Polymerization Inhibitor in Ex Vivo Conditions
2.3.8. Benzothiazolones Depolymerizing Activity in EA.hy926 Cells
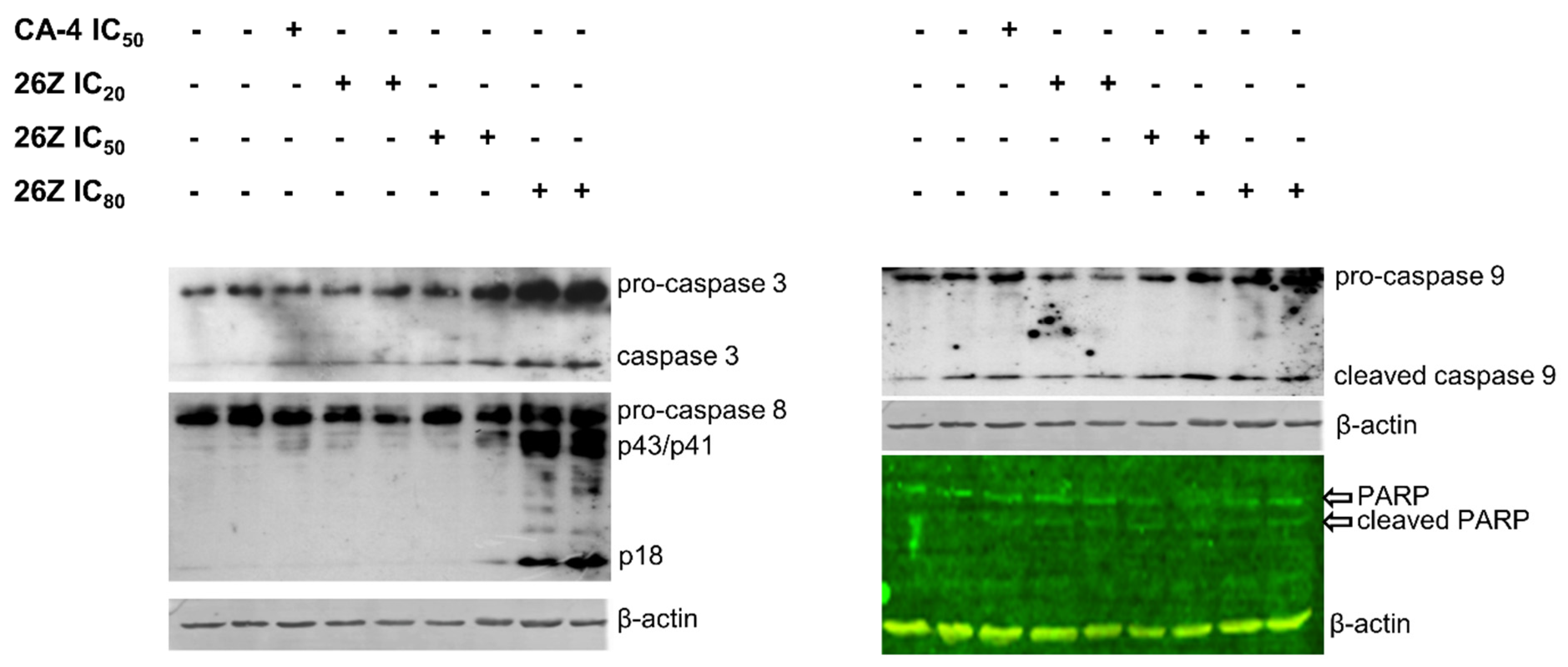
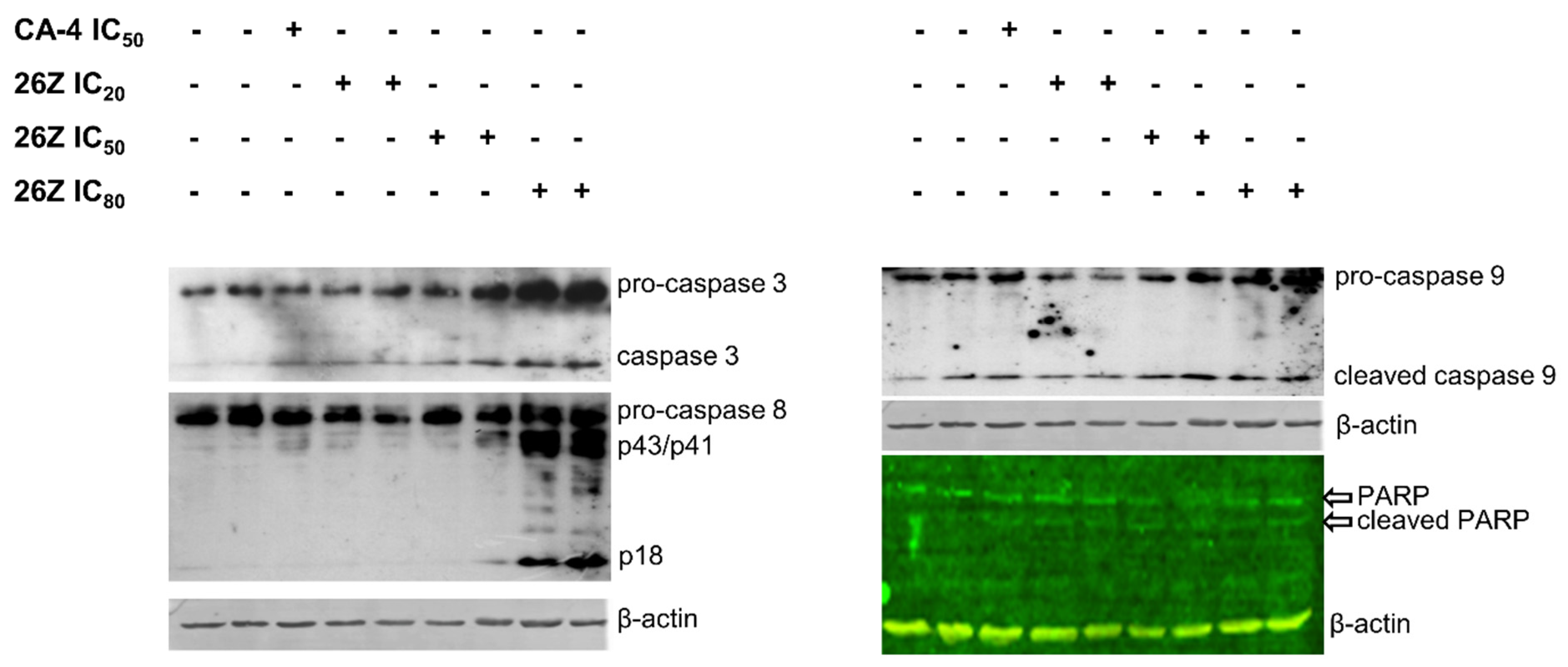
2.3.9. 26Z Activates Apoptotic Signaling Pathways in EA.hy926 Cells
3. Materials and Methods
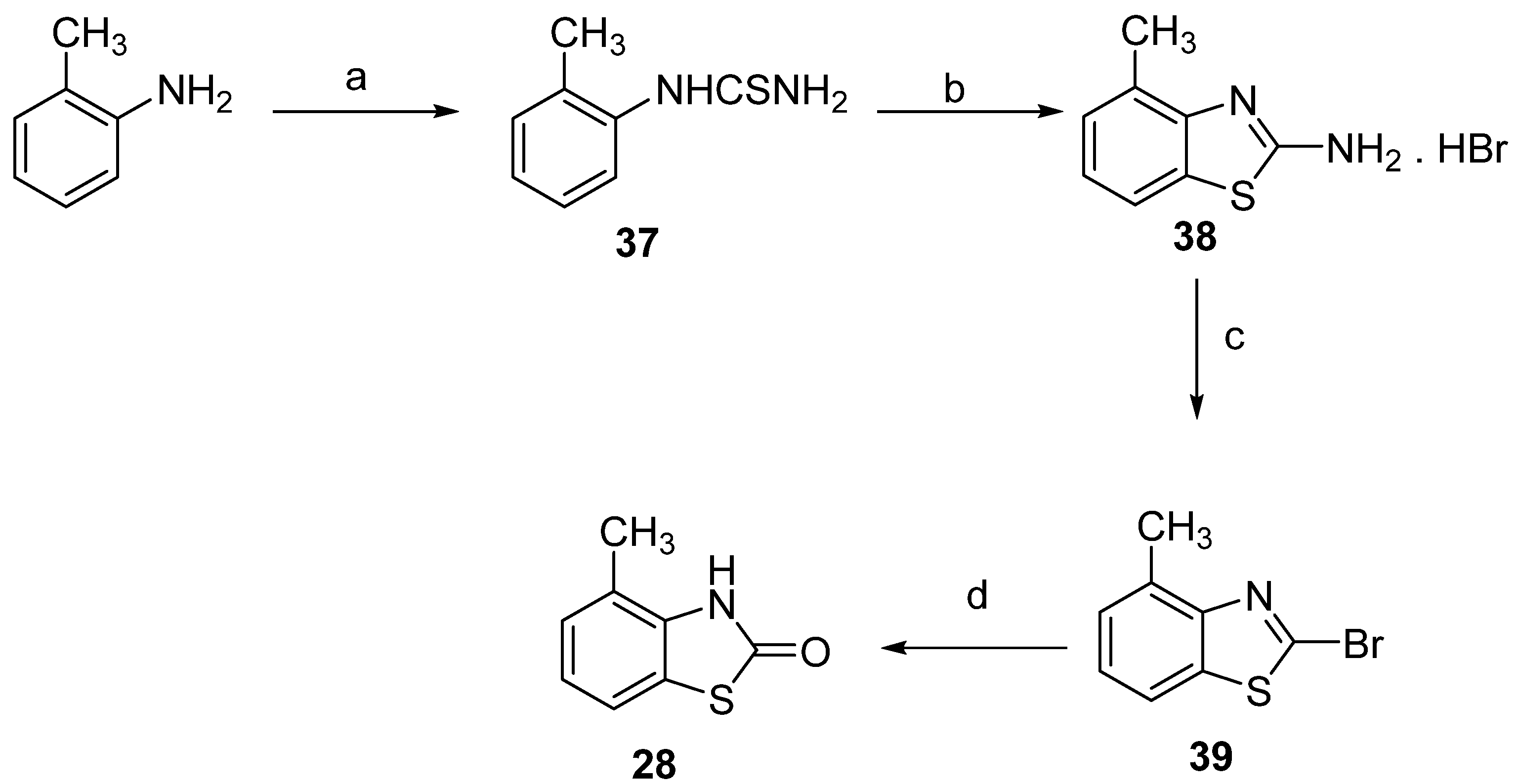
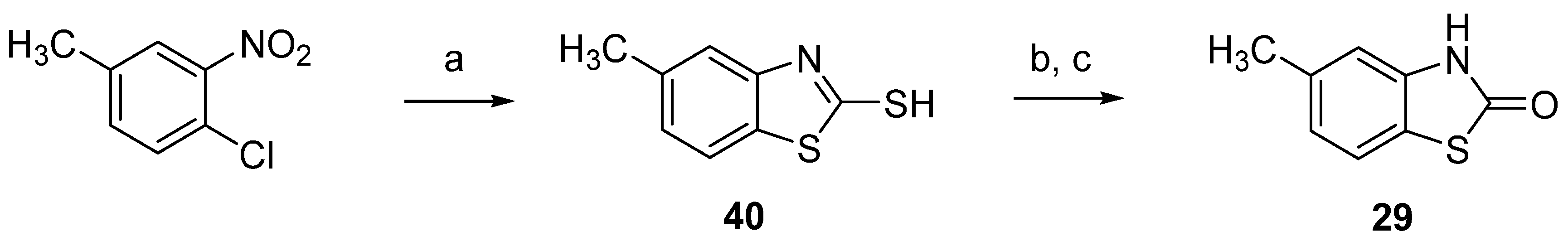
3.1. Chemistry
3.1.1. General Information
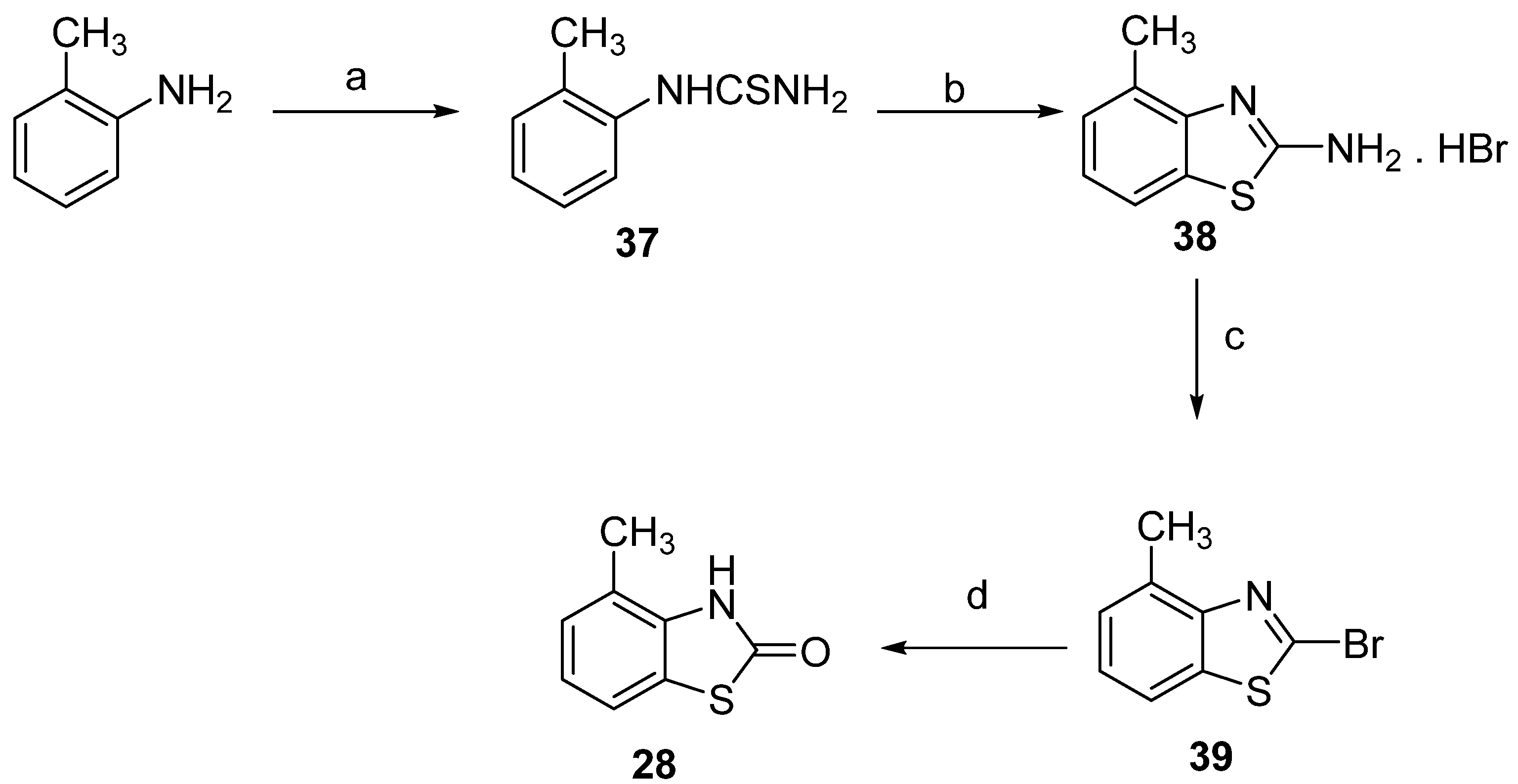
3.1.2. Synthesis of Dimethyl-2(3H)-Benzothiazolones 31–33, General Procedure
3,4-Dimethyl-2(3H)-Benzothiazolone (31)
3,5-Dimethyl-2(3H)-Benzothiazolone (32)
3,6-Dimethyl-2(3H)-Benzothiazolone (33)
3.1.3. Synthesis of Bromomethyl-3-Methyl-2(3H)-Benzothiazolones 34–36, General Procedure
4-Bromomethyl-3-Methyl-2(3H)-Benzothiazolone (34)
5-Bromomethyl-3-Methyl-2(3H)-Benzothiazolone (35)
6-Bromomethyl-3-Methyl-2(3H)-Benzothiazolone (36)
3.1.4. General Procedure for the Synthesis of Phosphonium Salts 11–13
[(3-Methyl-2(3H)-Benzothiazolone-4-yl)Methyl]Triphenylphosphonium Bromide (11)
[(3-Methyl-2(3H)-Benzothiazolone-5-yl)Methyl]Triphenylphosphonium Bromide (12)
[(3-Methyl-2(3H)-Benzothiazolone-6-yl)Methyl]Triphenylphosphonium Bromide (13)
3.1.5. 3-Methyl-2(3H)-Benzothiazolone-7-Carbaldehyde (10)
3.1.6. General Procedure for the Stilbene Syntheses 15–27
(E/Z)-4-(4-Methoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (15)
(E/Z)-4-(3,4-Dimethoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (16)
(E/Z)-4-(3,5-Dimethoxystyryl)-3-methyl-2(3H)-benzothiazolone (17)
(E/Z)-3-Methyl-4-(3,4,5-Trimethoxystyryl)-2(3H)-Benzothiazolone (18)
(E/Z)-5-(4-Methoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (19)
(E/Z)-5-(3,4-Dimethoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (20)
(E/Z)-5-(3,5-Dimethoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (21)
(E/Z)-3-Methyl-5-(3,4,5-Trimethoxystyryl)-2(3H)-Benzothiazolone (22)
(E/Z)-6-(4-Methoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (23)
(E/Z)-6-(3,4-Dimethoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (24)
(E/Z)-6-(3,5-Dimethoxystyryl)-3-Methyl-2(3H)-Benzothiazolone (25)
(E/Z)-3-Methyl-6-(3,4,5-Trimethoxystyryl)-2(3H)-Benzothiazolone (26)
(E/Z)-3-Methyl-7-(3,4,5-Trimethoxystyryl)-2(3H)-Benzothiazolone (27)
3.2. Crystallography and Docking
3.2.1. General
3.2.2. General Molecular Docking of the 22Z, 27Z, 26Z, and 25Z Derivatives in the Colchicine Binding Site of Tubulin
3.3. Biology
3.3.1. Cell Cultures
3.3.2. Cellular Treatment
3.3.3. Clonogenic Survival Assay
3.3.4. Western Blot Analysis
3.3.5. In Vitro Cell Migration Assays
3.3.6. Matrigel Tube Disruption Assay
3.3.7. Ex Vivo Tubulin Polymerization Assays
Ex Vivo Unlabeled Tubulin Polymerization
Colchicine-Binding Site Assay
Ex Vivo Rhodamin-Tubulin Polymerization
3.3.8. In Vitro Quantification of Polymeric vs. Soluble Tubulin Fractions
3.3.9. Cell Cycle Analysis
3.3.10. Immunocytochemistry
3.3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, G.; Gill, R.K.; Soni, R.; Bariwal, J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. [Google Scholar] [CrossRef]
- La Regina, G.; Coluccia, A.; Naccarato, V.; Silvestri, R. Towards modern anticancer agents that interact with tubulin. Eur. J. Pharm. Sci. 2019, 131, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.-J.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. [Google Scholar] [CrossRef] [Green Version]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Liu, W.; Tian, L.; Mao, Y.; Song, C. Targeting Tubulin-colchicine Site for Cancer Therapy: Inhibitors, Antibody-Drug Conjugates and Degradation Agents. Curr. Top. Med. Chem. 2019, 19, 1289–1304. [Google Scholar] [CrossRef]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, S.; Zhao, X.; Feng, Y.; Bormans, G.; Swinnen, J.; Oyen, R.; Huang, G.; Ni, Y.; Li, Y. Predicting Clinical Efficacy of Vascular Disrupting Agents in Rodent Models of Primary and Secondary Liver Cancers: An Overview with Imaging-Histopathology Correlation. Diagnostics 2020, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Nagaiah, G.; Remick, S.C. Combretastatin A4 phosphate: A novel vascular disrupting agent. Future Oncol. 2010, 6, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Yin, T.; Liu, Y.; Peeters, R.; Feng, Y.; Yu, J.; Himmelreich, U.; Oyen, R.; Ni, Y. Vascular disrupting agent in pancreatic and hepatic tumour allografts: Observations of location-dependent efficacy by MRI, microangiography and histomorphology. Br. J. Cancer 2017, 117, 1529–1536. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Chaplin, D.J.; Walicke, P.A. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin. Investig. Drugs 2009, 18, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.; Ky, B.; Tewari, K.S.; Chaplin, D.J.; Walker, J. Clinical trial experience with CA4P anticancer therapy: Focus on efficacy, cardiovascular adverse events, and hypertension management. Gynecol. Oncol. Res. Pract. 2018, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaroch, K.; Karolak, M.; Górski, P.; Jaroch, A.; Krajewski, A.; Ilnicka, A.; Sloderbach, A.; Stefański, T.; Sobiak, S. Combretastatins: In vitro structure-activity relationship, mode of action and current clinical status. Pharmacol. Rep. 2016, 68, 1266–1275. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.E.; Herald, D.L.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Chapuis, J.C. Antineoplastic agents. 410. Asymmetric hydroxylation of trans-combretastatin A-4. J. Med. Chem. 1999, 42, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Malebari, A.M.; Wang, S.; Greene, T.F.; O’Boyle, N.M.; Fayne, D.; Khan, M.F.; Nathwani, S.M.; Twamley, B.; McCabe, T.; Zisterer, D.M.; et al. Synthesis and Antiproliferative Evaluation of 3-Chloroazetidin-2-ones with Antimitotic Activity: Heterocyclic Bridged Analogues of Combretastatin A-4. Pharmaceuticals 2021, 14, 1119. [Google Scholar] [CrossRef]
- Romagnoli, R.; Oliva, P.; Salvador, M.K.; Manfredini, S.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Ronca, R.; Maccarinelli, F.; et al. A facile synthesis of diaryl pyrroles led to the discovery of potent colchicine site antimitotic agents. Eur. J. Med. Chem. 2021, 214, 113229. [Google Scholar] [CrossRef]
- Gerova, M.S.; Stateva, S.R.; Radonova, E.M.; Kalenderska, R.B.; Rusew, R.I.; Nikolova, R.P.; Chanev, C.D.; Shivachev, B.L.; Apostolova, M.D.; Petrov, O.I. Combretastatin A-4 analogues with benzoxazolone scaffold: Synthesis, structure and biological activity. Eur. J. Med. Chem. 2016, 120, 121–133. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Hawwas, M.M.; Malebari, A.M.; Taher, E.S.; Omar, A.M.; O’Boyle, N.M.; McLoughlin, E.; Abdel-Samii, Z.K.; Elshaier, Y.A.M.M. Potent Quinoline-Containing Combretastatin A-4 Analogues: Design, Synthesis, Antiproliferative, and Anti-Tubulin Activity. Pharmaceuticals 2020, 13, 393. [Google Scholar] [CrossRef]
- Takahashi, S.; Nakano, K.; Yokota, T.; Shitara, K.; Muro, K.; Sunaga, Y.; Ecstein-Fraisse, E.; Ura, T. Phase 1 study of ombrabulin in combination with cisplatin (CDDP) in Japanese patients with advanced solid tumors. Jpn. J. Clin. Oncol. 2016, 46, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Zhang, D.; Jin, Q.; Jiang, C.; Wang, C.; Li, J.; Peng, F.; Huang, D.; Zhang, J.; Song, S. Combretastatin-A4 phosphate improves the distribution and antitumor efficacy of albumin-bound paclitaxel in W256 breast carcinoma model. Oncotarget 2016, 7, 58133–58141. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.D.; Banerjee, S.; Hall, M.; Clamp, A.R.; Zhou, C.; Hasan, J.; Orbegoso, C.; Taylor, S.; Tugwood, J.; Lyon, A.R.; et al. Pazopanib and Fosbretabulin in recurrent ovarian cancer (PAZOFOS): A multi-centre, phase 1b and open-label, randomised phase 2 trial. Gynecol. Oncol. 2020, 156, 545–551. [Google Scholar] [CrossRef]
- Ljubas, J.; Ovesen, T.; Rusan, M.A. Systematic Review of Phase II Targeted Therapy Clinical Trials in Anaplastic Thyroid Cancer. Cancers 2019, 11, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.J.; Sill, M.W.; Walker, J.L.; Darus, C.J.; Sutton, G.; Tewari, K.S.; Martin, L.P.; Schilder, J.M.; Coleman, R.L.; Balkissoon, J.; et al. Randomized Phase II Evaluation of Bevacizumab Versus Bevacizumab Plus Fosbretabulin in Recurrent Ovarian, Tubal, or Peritoneal Carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. J. Clin. Oncol. 2016, 34, 2279–2286. [Google Scholar] [CrossRef]
- Irfan, A.; Batool, F.; Zahra Naqvi, S.A.; Islam, A.; Osman, S.M.; Nocentini, A.; Alissa, S.A.; Supuran, C.T. Benzothiazole derivatives as anticancer agents. J. Enzyme Inhib. Med. Chem. 2020, 35, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef]
- Chander Sharma, P.; Sharma, D.; Sharma, A.; Bansal, K.K.; Rajak, H.; Sharma, S.; Thakur, V.K. New horizons in benzothiazole scaffold for cancer therapy: Advances in bioactivity, functionality, and chemistry. Appl. Mater Today 2020, 20, 100783. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Schmidt, J.M.; Hogan, F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1a). J. Med. Chem. 1995, 38, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zou, Y.; Sun, H.-Y.; Liu, X.-K.; Xiao, C.-F.; Sun, J.; He, S.-J.; Li, J. Practical and green synthesis of combretastatin A-4 and its prodrug CA4P using renewable biomass-based starting material. Synthesis 2011, 2, 217–222. [Google Scholar] [CrossRef]
- Gaukroger, K.; Hadfield, J.A.; Hepworth, L.A.; Lawrence, N.J.; McGown, A.T. Novel syntheses of cis and trans isomers of combretastatin A-4. J. Org. Chem. 2001, 66, 8135–8138. [Google Scholar] [CrossRef]
- Camacho-Dávila, A.A. Kumada–Corriu Cross Coupling Route to the Anti-Cancer Agent Combretastatin A-4. Synth. Commun. 2008, 38, 3823–3833. [Google Scholar] [CrossRef]
- Petrov, O.I.; Gerova, M.S.; Chanev, C.D.; Petrova, K.V. New efficient synthesis of combretastatin A-4 via colvin rearrangement. Synthesis 2011, 22, 3711–3715. [Google Scholar] [CrossRef]
- Boden, R. A mild method for preparing trans-alkenes: Crown ether catalysis of the Wittig reaction. Synthesis 1975, 784. [Google Scholar] [CrossRef]
- Petrov, O.I.; Kalcheva, V.B.; Antonova, A.T. C-Formylation of some 2(3H)-benzazolones and 2H-1,4-benzoxazin-3(4H)-one. Coll. Czechoslov. Chem. Commun. 1997, 62, 494–497. [Google Scholar] [CrossRef]
- Zhou, B.; Hong, H.; Wang, H.; Zhang, T.; Han, L.; Zhu, N. Efficient synthesis of benzothiazolone derivatives by a domino reaction of disulfide and COS under mild conditions. Eur. J. Org. Chem. 2018, 6, 983–6990. [Google Scholar] [CrossRef]
- Gerova, M.S.; Svetoslavov, F.E.; Shivachev, B.L.; Nikolova, R.P.; Petrov, O.I. Synthesis of 4-acetyl-2(3H)-benzothiazolone: Sulfur bioisostere of benzoxazolone allelochemicals. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 905–910. [Google Scholar] [CrossRef]
- Barnikow, G.; Bödeker, J. Über die S-N-Bindung, I. Bis-guanyl-disulfid-Salze als Zwischenstufe der Bildung von 1.2. 4-Thiadiazolidinen und 2-Amino-benzthiazolen. Chem. Ber. 1967, 100, 1394–1401. [Google Scholar] [CrossRef]
- Dunbrook, R.; Zimmermann, M. A Method for the Preparation of 2-Mercaptobenzothiazole1. J. Am. Chem. Soc. 1934, 56, 2734–2736. [Google Scholar] [CrossRef]
- Teppema, J.; Sebrell, B. Researches on mercaptothiazoles. I. J. Am. Chem. Soc. 1927, 49, 1748–1758. [Google Scholar] [CrossRef]
- Efros, L.S.; Dawidenkow, L.R. Obtaining 2-benzothiazole sulfonic acid. Zh. Obsh Khim. 1951, 21, 2046–2048. [Google Scholar]
- Dekhane, D.V.; Pawar, S.S.; Gupta, S.V.; Shingare, M.S.; Thore, S.N. Synthesis of benzimidazolones, benzooxazolones, 2-amino-benzothiazoles from ethyl cyanoformate and o-phenylene diamines, o-aminophenols, oaminothiophenols promoted by lithium bromide. Lett. Org. Chem. 2011, 8, 406–411. [Google Scholar] [CrossRef]
- Furniss, B.S. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Pearson Education: India, Chennai, 2003. [Google Scholar]
- Chaudhuri, N.C. Convenient strategies for the preparation of modified 2 (3 H)-benzothiazolethiones. Synth. Commun. 1996, 26, 3783–3790. [Google Scholar] [CrossRef]
- Franchini, C.; Muraglia, M.; Corbo, F.; Florio, M.A.; Di Mola, A.; Rosato, A.; Matucci, R.; Nesi, M.; van Bambeke, F.; Vitali, C. Synthesis and biological evaluation of 2-mercapto-1,3-benzothiazole derivatives with potential antimicrobial activity. Arch. Pharm. 2009, 342, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Hrobárik, P.; Hrobáriková, V.; Sigmundová, I.; Zahradník, P.; Fakis, M.; Polyzos, I.; Persephonis, P. Benzothiazoles with tunable electron-withdrawing strength and reverse polarity: A route to triphenylamine-based chromophores with enhanced two-photon absorption. J. Org. Chem. 2011, 76, 8726–8736. [Google Scholar] [CrossRef]
- Matevosyan, R.O.; Abramova, N.I.; Donskikh, O.B. Chemistry of free radicals of the hydrazine series. LIV. Synthesis and study of the properties of α-(6-methylbenzothyazolyl-2)-α-phenyl-β-picrylhydrazyl and α-(2-methylbenzothyazolyl-6)-α-phenyl-β-picryhydrazyl. J. Org. Chem. USSR Engl. Transl. 1968, 4, 1405–1408. [Google Scholar]
- Hunter, F.R.; Parken, R.E. The unsaturation and tautomeric mobility of heterocyclic compounds. Part VI. The mobility of 5-substituted 1-hydroxybenzthiazoles, and the ultra-violet absorption of mobile and static derivatives of 1-hydroxybenzthiazole. J. Chem. Soc. 1935, 1755–1761. [Google Scholar] [CrossRef]
- Stanetty, P.; Krumpak, B. Novel synthesis of benzothiazole derivatives via directed lithiation and aryne-mediated cyclization followed by quenching with electrophiles. J. Org. Chem. 1996, 61, 5130–5133. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Aydın, A.; Akkurt, M.; Önkol, T.; Büyükgüngör, O. 3-[6-(2-Chlorobenzoyl)-2-oxo-2,3-dihydro-1,3-benzothiazol-3-yl] propanenitrile. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, o5933–o5934. [Google Scholar] [CrossRef]
- Dölling, W.; Kischkies, K.; Heinemann, F.; Hartung, H. Synthesen von α, β-ungesättigten 2-(2-Oxo-benzazolin-3-yl)-3-hydroxy-dithio-carbonsäure-alkylestern und entsprechend substituierten Keten-S, S-acetalen sowie deren Kristall-und Molekülstruktur. Monatsh. Chem. 1993, 124, 707–719. [Google Scholar] [CrossRef]
- Chantrapromma, S.; Suwunwong, T.; Boonnak, N.; Fun, H.-K. (2E)-1-(Pyridin-2-yl)-3-(2,4,5-trimethoxyphenyl) prop-2-en-1-one. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, o1076–o1077. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Nicholls, R.A.; Emsley, P.; Grazulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A stereochemical description generator for ligands. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017, 73, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Mahal, K.; Biersack, B.; Schruefer, S.; Resch, M.; Ficner, R.; Schobert, R.; Mueller, T. Combretastatin A-4 derived 5-(1-methyl-4-phenyl-imidazol-5-yl)indoles with superior cytotoxic and anti-vascular effects on chemoresistant cancer cells and tumors. Eur. J. Med. Chem. 2016, 118, 9–20. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural Basis of cis- and trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Kwak, Y.S.; Joo, S.H.; Gansukh, E.; Mistry, B.M.; Keum, Y.S. Synthesis and anticancer activities of polymethylenedioxy analogues of combretastatin A-2. Appl. Biol. Chem. 2019, 62, 25. [Google Scholar] [CrossRef]
- Conesa-Milián, L.; Falomir, E.; Murga, J.; Carda, M.; Meyen, E.; Liekens, S.; Alberto Marco, J. Synthesis and biological evaluation of carbamates derived from aminocombretastatin A-4 as vascular disrupting agents. Eur. J. Med. Chem. 2018, 147, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hura, N.; Sawant, A.V.; Kumari, A.; Guchhait, S.K.; Panda, D. Combretastatin-Inspired Heterocycles as Antitubulin Anticancer Agents. ACS Omega 2018, 3, 9754–9769. [Google Scholar] [CrossRef]
- Han, J.; Lim, W.; You, D.; Jeong, Y.; Kim, S.; Lee, J.E.; Shin, T.H.; Lee, G.; Park, S. Chemoresistance in the Human Triple-Negative Breast Cancer Cell Line MDA-MB-231 Induced by Doxorubicin Gradient Is Associated with Epigenetic Alterations in Histone Deacetylase. J. Oncol. 2019, 2019, 1345026. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315. [Google Scholar] [CrossRef]
- Tarade, D.; Ma, D.; Pignanelli, C.; Mansour, F.; Simard, D.; van den Berg, S.; Gauld, J.; McNulty, J.; Pandey, S. Structurally simplified biphenyl combretastatin A4 derivatives retain in vitro anti-cancer activity dependent on mitotic arrest. PLoS ONE 2017, 12, e0171806. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Shaik, T.B.; Malik, M.S.; Syed, R.; Mallipeddi, P.L.; Vardhan, M.V.P.S.V.; Kamal, A. Design and synthesis of cis-restricted benzimidazole and benzothiazole mimics of combretastatin A-4 as antimitotic agents with apoptosis inducing ability. Bioorg. Med. Chem. Lett. 2016, 26, 4527–4535. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Gao, Q.L.; Wu, B.W.; Zhu, T.; Cui, X.X.; Jin, C.J.; Wang, S.Y.; Wang, S.H.; Fu, D.J.; Liu, H.M.; et al. Discovery of tertiary amide derivatives incorporating benzothiazole moiety as anti-gastric cancer agents in vitro via inhibiting tubulin polymerization and activating the Hippo signaling pathway. Eur. J. Med. Chem. 2020, 203, 112618. [Google Scholar] [CrossRef]
- Fortin, S.; Lacroix, J.; Côté, M.-F.; Moreau, E.; Petitclerc, E.; C-Gaudreault, R. Quick and simple detection technique to assess the binding of antimicrotubule agents to the colchicine-binding site. Biol. Proced. Online 2010, 12, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merks, R.M.; Brodsky, S.V.; Goligorksy, M.S.; Newman, S.A.; Glazier, J.A. Cell elongation is key to in silico replication of in vitro vasculogenesis and subsequent remodeling. Dev. Biol. 2006, 289, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Guidolin, D.; Vacca, A.; Nussdorfer, G.G.; Ribatti, D. A new image analysis method based on topological and fractal parameters to evaluate the angiostatic activity of docetaxel by using the Matrigel assay in vitro. Microvasc. Res. 2004, 67, 17–124. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Martin, M.; Bouchet, B.P.; Guillen-Navarro, M.J.; Martínez-Diez, M.; Martinez-Leal, J.F.; Akhmanova, A.; Aviles, P. Plocabulin, a novel tubulin-binding agent, inhibits angiogenesis by modulation of microtubule dynamics in endothelial cells. BMC Cancer 2018, 18, 164. [Google Scholar] [CrossRef] [Green Version]
- Umemura, T.; Morino, H.; Watanabe, T.; Uematsu, T. Method for Preparing 4-Substituted-N-methylbenzothiazolone derivatives. US4293702A, 6 October 1981. Available online: https://patents.google.com/patent/US4293702A/en (accessed on 1 November 2021).
- Sexton, W.A. Reactions of benzthiazole derivatives. Part I. The reactivity of methylthiol group in quaternary salts of 1-methylthiobenzthiazole. J. Chem. Soc. 1939, 470–473. [Google Scholar] [CrossRef]
- Bruneau, A.R.P.; Crawley, G.C.; Oldham, K. Bicyclic Heterocyclic Derivatives as 5-lipoxygenase Inhibitors. US5179115A, 12 January 1993. Available online: https://patents.google.com/patent/US5179115A/en (accessed on 5 November 2021).
- Pettit, G.R.; Singh, S.B.; Niven, M.L.; Hamel, E.; Schmidt, J.M. Isolation, structure, and synthesis of combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly, derived from Combretum caffrum. J. Nat. Prod. 1987, 50, 119–131. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- D’Souza, S.J.; Pajak, A.; Balazsi, K.; Dagnino, L. Ca2+ and BMP-6 signaling regulate E2F during epidermal keratinocyte differentiation. J. Biol. Chem. 2001, 276, 23531–23538. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Padmaja, P.; Rao, G.K.; Indrasena, A.; Reddy, B.V.S.; Patel, N.; Shaik, A.B.; Reddy, N.; Dubey, P.K.; Bhadra, M.P. Synthesis and biological evaluation of novel pyrano [3, 2-c] carbazole derivatives as antitumor agents inducing apoptosis via tubulin polymerization inhibition. Org. Biomol. Chem. 2015, 13, 1404–1414. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Zhao, Z.; Zhang, C.; Yang, X.; Wang, Y. Connexin 43 enhances paclitaxel cytotoxicity in colorectal cancer cell lines. Exp. Ther. Med. 2017, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolova, M.D.; Cherian, M.G. Delay of M-phase onset by aphidicolin can retain the nuclear localization of zinc and metallothionein in 3T3-L1 fibroblasts. J. Cell. Physiol. 2000, 183, 247–253. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ring A | Position in Ring B | Yield (%) | ||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| 15Z 15E | H | OCH3 | H | 4 | 81 |
| 16Z 16E | OCH3 | OCH3 | H | 4 | 84 |
| 17Z 17E | OCH3 | H | OCH3 | 4 | 84 |
| 18Z 18E | OCH3 | OCH3 | OCH3 | 4 | 74 |
| 19Z 19E | H | OCH3 | H | 5 | 70 |
| 20Z 20E | OCH3 | OCH3 | H | 5 | 68 |
| 21Z 21E | OCH3 | H | OCH3 | 5 | 68 |
| 22Z 22E | OCH3 | OCH3 | OCH3 | 5 | 57 |
| 23Z 23E | H | OCH3 | H | 6 | 71 |
| 24Z 24E | OCH3 | OCH3 | H | 6 | 85 |
| 25Z 25E | OCH3 | H | OCH3 | 6 | 85 |
| 26Z 26E | OCH3 | OCH3 | OCH3 | 6 | 87/76 a |
| 27Z 27E | OCH3 | OCH3 | OCH3 | 7 | 81 |
| Compound | IC50 a ± SE (µM) | Compound | IC50 ± SE (µM) |
|---|---|---|---|
| 15Z | 22.03 ± 4.57 | 22Z | 9.43 ± 0.92 |
| 16Z | 25.78 ± 3.11 | 23Z | 19.76 ± 3.45 |
| 17Z | 19.50 ± 2.71 | 24Z | 9.28 ± 1.13 |
| 18Z | 15.64 ± 1.16 | 25Z | 1.88 ± 0.18 |
| 19Z | 11.96 ± 2.49 | 26Z | 0.13 ± 0.01 |
| 20Z | 14.58 ± 1.75 | 26E | 38.20 ± 5.35 |
| 21Z | 10.77 ± 0.62 | 27Z | 0.30 ± 0.05 |
| CA-4 | 0.0033 ± 0.0002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atanasov, G.; Rusew, R.I.; Gelev, V.M.; Chanev, C.D.; Nikolova, R.; Shivachev, B.L.; Petrov, O.I.; Apostolova, M.D. New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-benzothiazolones. Pharmaceuticals 2021, 14, 1331. https://doi.org/10.3390/ph14121331
Atanasov G, Rusew RI, Gelev VM, Chanev CD, Nikolova R, Shivachev BL, Petrov OI, Apostolova MD. New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-benzothiazolones. Pharmaceuticals. 2021; 14(12):1331. https://doi.org/10.3390/ph14121331
Chicago/Turabian StyleAtanasov, Gjorgji, Rusi I. Rusew, Vladimir M. Gelev, Christo D. Chanev, Rosica Nikolova, Boris L. Shivachev, Ognyan I. Petrov, and Margarita D. Apostolova. 2021. "New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-benzothiazolones" Pharmaceuticals 14, no. 12: 1331. https://doi.org/10.3390/ph14121331
APA StyleAtanasov, G., Rusew, R. I., Gelev, V. M., Chanev, C. D., Nikolova, R., Shivachev, B. L., Petrov, O. I., & Apostolova, M. D. (2021). New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-benzothiazolones. Pharmaceuticals, 14(12), 1331. https://doi.org/10.3390/ph14121331