
Peptide Inhibitors of Kv1.5: An Option for the Treatment of Atrial Fibrillation

 , ,
, ,  , and
, and {kind=link}
{kind=link}
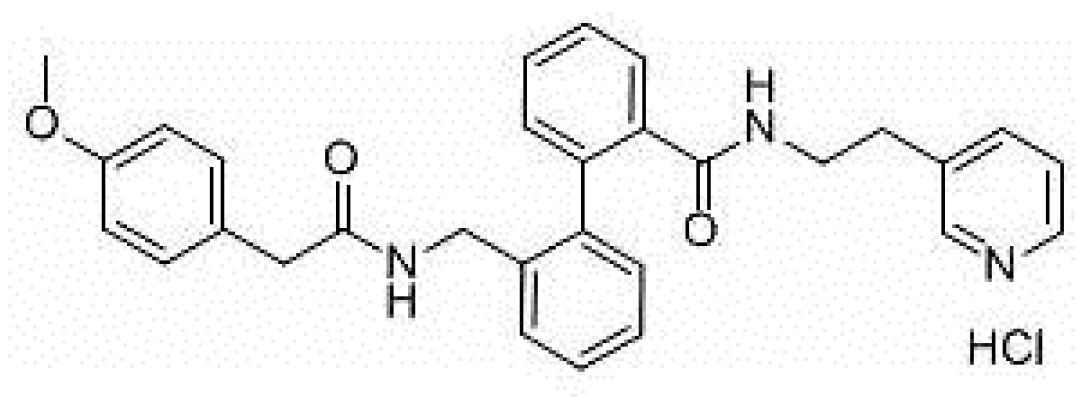
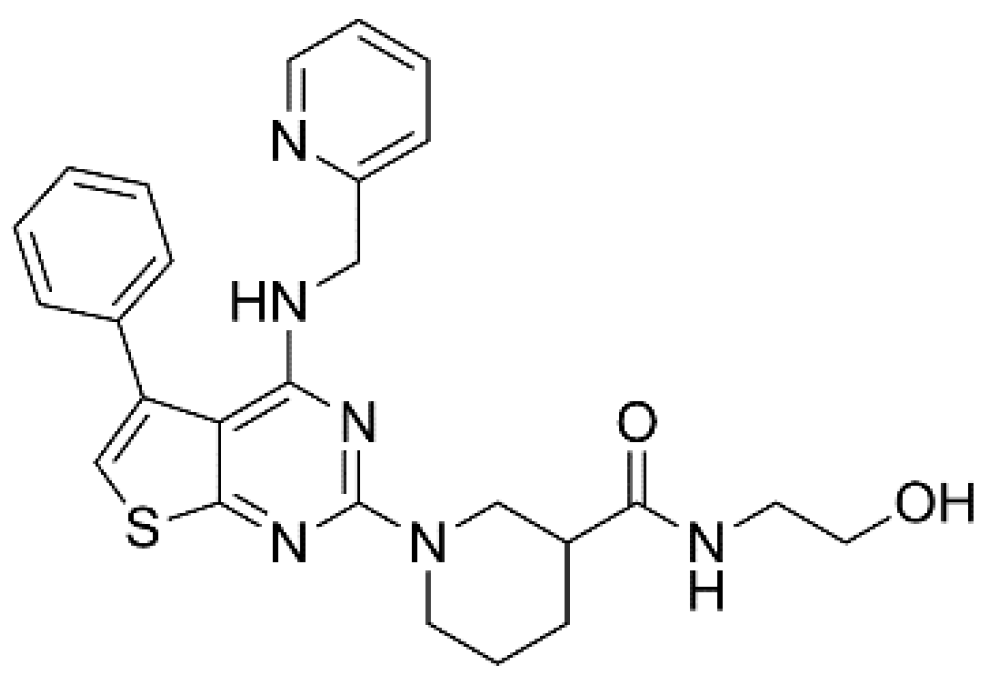{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
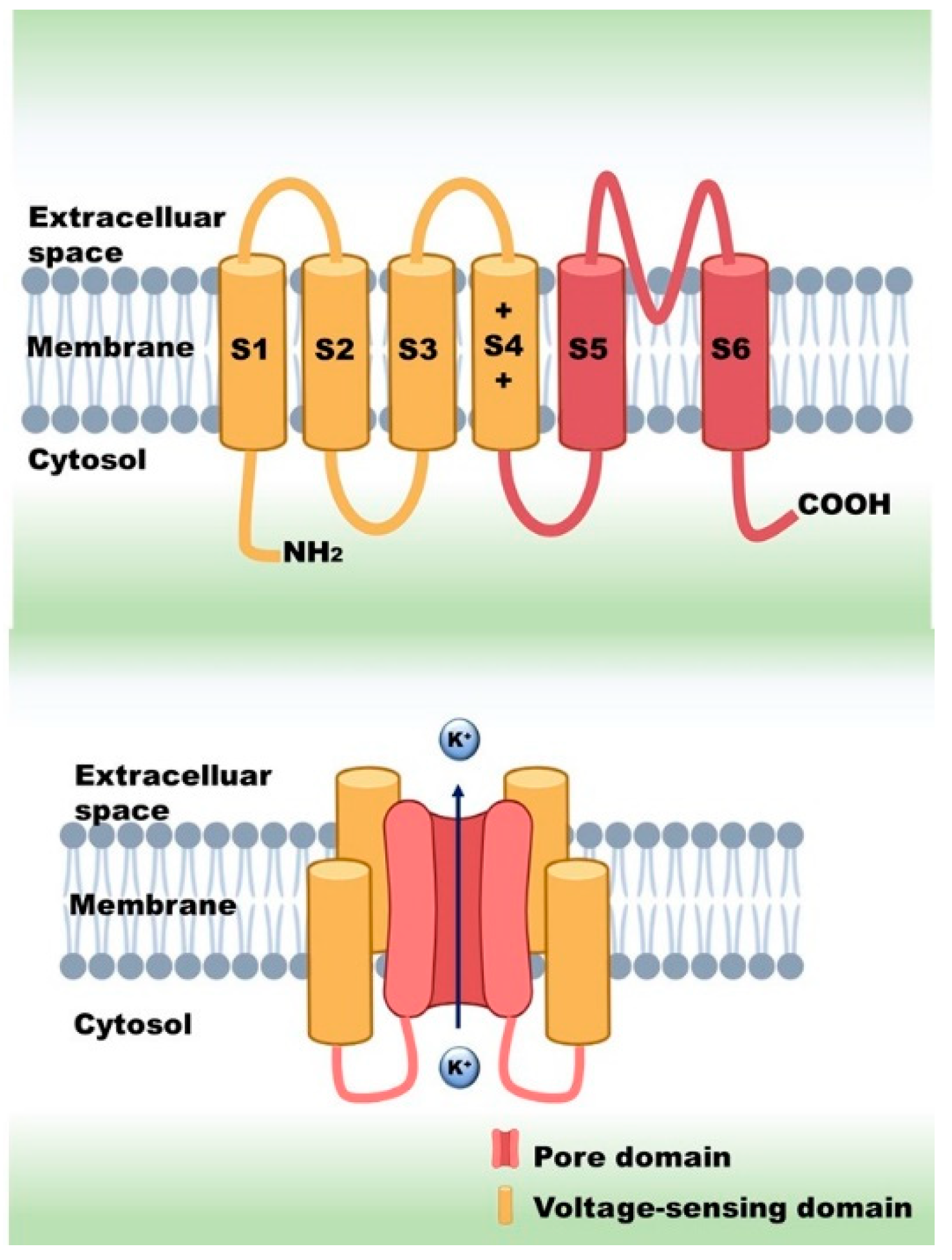
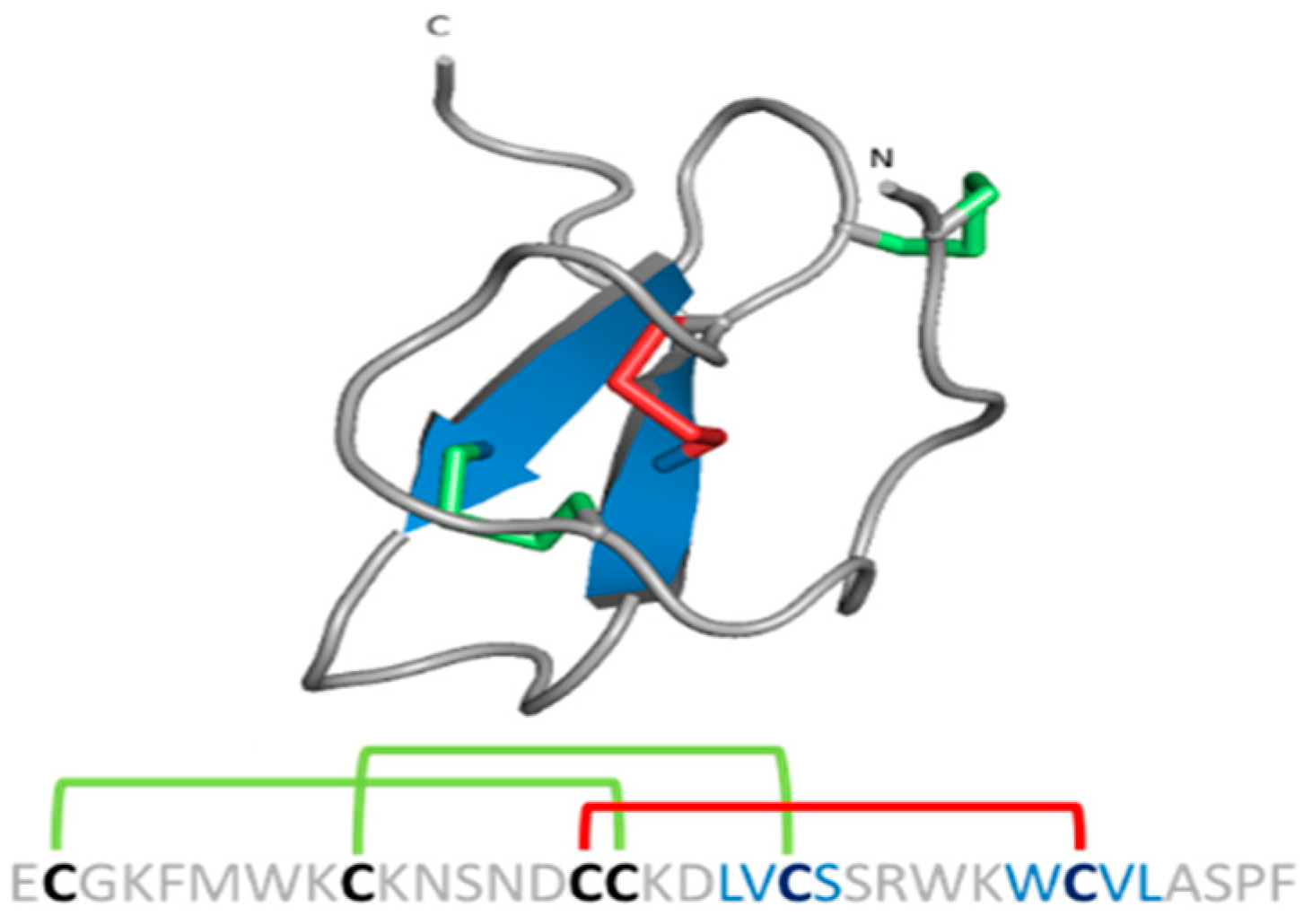{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diseases Related to Kv1.5
3. Atrial Fibrillation and Possible Pharmacological Treatments
- -
- Development and improvement of existing antiarrhythmic agents: Amiodarone derivates, Multi-channel blockers, etc.
- -
- Atrial selective therapeutic agents (ARDA): IKur blocker; IK,Ach blocker; INa, IKr blockers
- -
- Upstream therapy agents, drugs affecting structural remodeling; inflammation; hypertrophy; oxidative stress; etc.,
- -
- Gap junction modulators: Antiarrhythmic peptides affecting connexins Cx40 and Cx43
4. Peptide Modulators of the Kv Channels
4.1. Pore Blocker Peptides
Mechanism of Action of the Pore Blockers
4.2. Gating Modifiers Peptides
Mechanism of Action of the Gating Modifiers
5. Osu1 and Ts6: The Known Peptide Modulators of the Kv1.5
6. Selectivity of Kv1.5 Inhibitors
7. Improving Selectivity and Affinity of Peptide Toxins
7.1. Achieving the Native Peptide Scaffold
7.2. Uncovering Amino Acids Involved in Selectivity and Affinity
8. Testing Kv1.5 Modulation in AF Models
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | action potentials |
| APD | AP duration |
| ARDA | Atrial selective therapeutic agents |
| ChTx | Charybdotoxin |
| CMs | cardiomyocytes |
| EADs | probability of early afterdepolarizations |
| ERG | ether-á-go-go gene |
| ERP | effective refractory period |
| GOF | gain of function |
| hiAM | human immortalized atrial myocyte |
| iAM | immortalized atrial myocyte |
| IK,ACh | acetylcholine-sensitive inward rectifier potassium current |
| IKr | rapidly activating |
| IKs | slowly activating |
| IKur | ultra-rapidly activating |
| IL | Interleukin |
| Ito1 | the “transient outward” potassium current |
| Kv | voltage-gated potassium channels |
| KTx | potassium channel toxins |
| LOF | loss of function |
| MgTx | Margatoxin |
| PAH | pulmonary arterial hypertension |
| ProTx2 | Protoxin II |
| SPPS | solid-phase peptide synthesis |
| SR | sinus rhythm |
| TTX | tetrodotoxin |
| VGICs | Voltage-gated ion channels |
| Vmax | maximum speed of depolarization |
| VSD | voltage-sensing domain |
| VSP | voltage-sensing phosphatase |
References
- Alexander, S.P.; Catterall, W.A.; Kelly, E.; Marrion, N.; Peters, J.A.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. The Concise Guide to PHARMACOLOGY 2015/16: Voltage-gated ion channels. Br. J. Pharmacol. 2015, 172, 5904–5941. [Google Scholar] [CrossRef] [Green Version]
- Yellen, G. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998, 31, 239–295. [Google Scholar] [CrossRef]
- Swartz, K.J. Sensing voltage across lipid membranes. Nature 2008, 456, 891–897. [Google Scholar] [CrossRef] [Green Version]
- DeCoursey, T. The Voltage-Gated Proton Channel: A Riddle, Wrapped in a Mystery, inside an Enigma. Biochemistry 2015, 54, 3250–3268. [Google Scholar] [CrossRef]
- Murata, Y.; Iwasaki, H.; Sasaki, M.; Inaba, K.; Okamura, Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 2005, 435, 1239–1243. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Moran, M.M.; Chong, J.A.; Clapham, D.E. A voltage-gated proton-selective channel lacking the pore domain. Nature 2006, 440, 1213–1216. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Takagi, M.; Okamura, Y. A Voltage Sensor-Domain Protein Is a Voltage-Gated Proton Channel. Science 2006, 312, 589–592. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and Molecular Relationships of Voltage-Gated Potassium Channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef]
- Snyders, D.J. Structure and function of cardiac potassium channels. Cardiovasc. Res. 1999, 42, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Fedida, D.; Wible, B.; Wang, Z.; Fermini, B.; Faust, F.; Nattel, S.; Brown, A.M. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ. Res. 1993, 73, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Brunner, M.; Kodirov, S.; Mitchell, G.F.; Buckett, P.D.; Shibata, K.; Folco, E.J.; Baker, L.; Salama, G.; Chan, D.P.; Zhou, J.; et al. In vivo gene transfer of Kv1.5 normalizes action potential duration and shortens QT interval in mice with long QT phenotype. Am. J. Physiol. Circ. Physiol. 2003, 285, H194–H203. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, W.; Sun, H.; You, Q. Kv1.5 Inhibitors for Treatment of Atrial Fibrillation: A Tradeoff between Selectivity and Non-selectivity. Curr. Top. Med. Chem. 2016, 16, 1843–1854. [Google Scholar] [CrossRef]
- Lip, G.Y.; Tse, H.F. Management of atrial fibrillation. Lancet 2007, 370, 604–618. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, J.R.; Biliczki, P.; Hohnloser, S.H.; Nattel, S. Atrial-Selective Approaches for the Treatment of Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Ford, J.W.; Milnes, J.T. New drugs targeting the cardiac ultra-rapid delayed-rectifier current (I Kur): Rationale, pharmacology and evidence for potential therapeutic value. J. Cardiovasc. Pharmacol. 2008, 52, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, J.R.; Nattel, S. Novel Approaches for Pharmacological Management of Atrial Fibrillation. Drugs 2009, 69, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Ravens, U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol. Ther. 2010, 128, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef]
- Christophersen, I.E.; Olesen, M.S.; Liang, B.; Andersen, M.N.; Larsen, A.P.; Nielsen, J.B.; Haunsø, S.; Olesen, S.-P.; Tveit, A.; Svendsen, J.H.; et al. Genetic variation in KCNA5: Impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 2013, 34, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations ofKCNA5in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol.-Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef]
- Fu, L.; Lv, Y.; Zhong, Y.; He, Q.; Liu, X.; Du, L. Tyrosine phosphorylation of Kv1.5 is upregulated in intrauterine growth retardation rats with exaggerated pulmonary hypertension. Braz. J. Med. Biol. Res. 2017, 50, e6237. [Google Scholar] [CrossRef]
- Macfarlane, S.N.; Sontheimer, H. Modulation of Kv1.5 Currents by Src Tyrosine Phosphorylation: Potential Role in the Differentiation of Astrocytes. J. Neurosci. 2000, 20, 5245–5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preussat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wölfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1.3 and Kv1.5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef]
- Bielanska, J.; Hernandez-Losa, J.; Perez-Verdaguer, M.; Moline, T.; Somoza, R.; Cajal, S.R.Y.; Condom, E.; Ferreres, J.C.; Felipe, A. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets 2009, 9, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Gracia, A.; Bielanska, J.; Hernández-Losa, J.; Castellví, J.; Ruiz-Marcellan, M.C.; Cajal, S.R.Y.; Condom, E.; Manils, J.; Soler, C.; Comes, N.; et al. Emerging role for the voltage-dependent K+ channel Kv1.5 in B-lymphocyte physiology: Expression associated with human lymphoma malignancy. J. Leukoc. Biol. 2013, 94, 779–789. [Google Scholar] [CrossRef]
- Chung, M.K.; Eckhardt, L.L.; Chen, L.Y.; Ahmed, H.M.; Gopinathannair, R.; Joglar, J.A.; Noseworthy, P.A.; Pack, Q.R.; Sanders, P.; Trulock, K.M. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement from the American Heart Association. Circulation 2020, 141, e750–e772. [Google Scholar] [CrossRef]
- Conen, D. Epidemiology of atrial fibrillation. Eur. Heart J. 2018, 39, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Wettwer, E.; Terlau, H. Pharmacology of voltage-gated potassium channel Kv1.5—Impact on cardiac excitability. Curr. Opin. Pharmacol. 2014, 15, 115–121. [Google Scholar] [CrossRef]
- Attali, B.; Chandy, K.G.; Giese, M.H.; Grissmer, S.; Gutman, G.A.; Jan, L.Y.; Lazdunski, M.; McKinnon, D.; Nerbonne, J.; Pardo, L.A.; et al. Voltage-gated potassium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, S.; Han, J. Venom-Derived Peptide Modulators of Cation-Selective Channels: Friend, Foe or Frenemy. Front. Pharmacol. 2019, 10, 58. [Google Scholar] [CrossRef]
- Wirth, K.J.; Paehler, T.; Rosenstein, B.; Knobloch, K.; Maier, T.; Frenzel, J.; Brendel, J.; Busch, A.E.; Bleich, M. Atrial effects of the novel K+-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc. Res. 2003, 60, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Christ, T.; Wettwer, E.; Voigt, N.; Hála, O.; Radicke, S.; Matschke, K.; Várro, A.; Dobrev, D.; Ravens, U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br. J. Pharmacol. 2008, 154, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Wettwer, E.; Hála, O.; Christ, T.; Heubach, J.F.; Dobrev, D.; Knaut, M.; Varró, A.; Ravens, U. Role of IKur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004, 110, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Wirth, K.J.; Steinmeyer, K.; Ruetten, H. Sensitization of Upper Airway Mechanoreceptors as a New Pharmacologic Principle to Treat Obstructive Sleep Apnea: Investigations with AVE0118 in Anesthetized Pigs. Sleep 2013, 36, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Shunmugam, S.R.; Sugihara, C.; Freemantle, N.; Round, P.; Furniss, S.; Sulke, N. A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers allowing continuous beat-to-beat monitoring of drug efficacy. J. Interv. Card. Electrophysiol. 2018, 51, 191–197. [Google Scholar] [CrossRef]
- Lagrutta, A.; Wang, J.; Fermini, B.; Salata, J.J. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedida, D.; Orth, P.M.; Chen, J.Y.; Lin, S.; Plouvier, B.; Jung, G.; Ezrin, A.M.; Beatch, G.N. The Mechanism of Atrial Antiarrhythmic Action of RSD1235. J. Cardiovasc. Electrophysiol. 2005, 16, 1227–1238. [Google Scholar] [CrossRef]
- Wettwer, E.; Christ, T.; Endig, S.; Rozmaritsa, N.; Matschke, K.; Lynch, J.J.; Pourrier, M.; Gibson, J.K.; Fedida, D.; Knaut, M.; et al. The new antiarrhythmic drug vernakalant: Ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc. Res. 2013, 98, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Dobrev, D.; Hamad, B.; Kirkpatrick, P. Vernakalant. Nat. Rev. Drug Discov. 2010, 9, 915–916. [Google Scholar] [CrossRef]
- Burashnikov, A.; Pourrier, M.; Gibson, J.K.; Lynch, J.J.; Antzelevitch, C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel: Comparison with ranolazine and dl-sotalol. Circ. Arrhythmia Electrophysiol. 2012, 5, 400–408. [Google Scholar] [CrossRef] [Green Version]
- van Middendorp, L.B.; Strik, M.; Houthuizen, P.; Kuiper, M.; Maessen, J.G.; Auricchio, A.; Prinzen, F.W. Electrophysiological and haemodynamic effects of vernakalant and flecainide in dyssynchronous canine hearts. Europace 2014, 16, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Camm, A.J.; Capucci, A.; Hohnloser, S.H.; Torp-Pedersen, C.; Van Gelder, I.C.; Mangal, B.; Beatch, G. A Randomized Active-Controlled Study Comparing the Efficacy and Safety of Vernakalant to Amiodarone in Recent-Onset Atrial Fibrillation. J. Am. Coll. Cardiol. 2011, 57, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cialdella, P.; Pedicino, D.; Santangeli, P. Novel Agents for the Acute Conversion of Atrial Fibrillation: Focus on Vernakalant. Recent Patents Cardiovasc. Drug Discov. 2011, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Niederdoeckl, J.; Janata, K.; Spiel, A.O.; Schuetz, N.; Schnaubelt, S.; Herkner, H.; Cacioppo, F.; Laggner, A.N.; Domanovits, H. Vernakalant and electrical cardioversion for AF–Safe and effective. IJC Heart Vasc. 2019, 24, 100398. [Google Scholar] [CrossRef]
- Hall, A.J.; Mitchell, A. Introducing Vernakalant into Clinical Practice. Arrhythmia Electrophysiol. Rev. 2019, 8, 70–74. [Google Scholar] [CrossRef]
- Bergeron, Z.L.; Bingham, J.P. Scorpion toxins specific for potassium (K+) channels: A historical overview of peptide bioengineering. Toxins 2012, 4, 1082–1119. [Google Scholar] [CrossRef] [Green Version]
- Rash, L.; Hodgson, W.C. Pharmacology and biochemistry of spider venoms. Toxicon 2002, 40, 225–254. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Jungo, F.; Bairoch, A. Tox-Prot, the toxin protein annotation program of the Swiss-Prot protein knowledgebase. Toxicon 2005, 45, 293–301. [Google Scholar] [CrossRef]
- Jungo, F.; Estreicher, A.; Bairoch, A.; Bougueleret, L.; Xenarios, I. Animal Toxins: How is Complexity Represented in Databases? Toxins 2010, 2, 262–282. [Google Scholar] [CrossRef]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero-Hernández, V.; Jiménez-Vargas, J.; Gurrola, G.; Valdivia, H.; Possani, L. Scorpion venom components that affect ion-channels function. Toxicon 2013, 76, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Kuzmenkov, A.I.; Krylov, N.; Chugunov, A.O.; Grishin, E.V.; Vassilevski, A.A. Kalium: A database of potassium channel toxins from scorpion venom. Database 2016, 2016, baw056. [Google Scholar] [CrossRef] [Green Version]
- Tytgat, J.; Chandy, K.G.; Garcia, M.L.; Gutman, G.A.; Martin-Eauclaire, M.F.; van der Walt, J.J.; Possani, L.D. A unified nomenclature for short-chain peptides isolated from scorpion venoms: Alpha-KTx molecular subfamilies. Trends Pharmacol. Sci. 1999, 20, 444–447. [Google Scholar] [CrossRef]
- de la Vega, R.C.R.; Possani, L.D. Current views on scorpion toxins specific for K+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B.; Aumelas, A.; Rodríguez, M.D.C.; Lanz-Mendoza, H.; Peigneur, S.; Diego-Garcia, E.; Martin-Eauclaire, M.-F.; Tytgat, J.; Possani, L.D. MeuTXKβ1, a scorpion venom-derived two-domain potassium channel toxin-like peptide with cytolytic activity. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 872–883. [Google Scholar] [CrossRef]
- Kuzmenkov, A.I.; Grishin, E.V.; Vassilevski, A.A. Diversity of Potassium Channel Ligands: Focus on Scorpion Toxins. Biochemistry 2015, 80, 1764–1799. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Panyi, G.; Varga, Z. Potassium Channel Blocking Peptide Toxins from Scorpion Venom. In Scorpion Venoms; Springer: Dordrecht, The Netherlands, 2015; pp. 493–527. [Google Scholar]
- Diego-García, E.; Schwartz, E.F.; D’Suze, G.; González, S.A.R.; Batista, C.V.; García, B.I.; de la Vega, R.C.R.; Possani, L.D. Wide phylogenetic distribution of Scorpine and long-chain beta-KTx-like peptides in scorpion venoms: Identification of “orphan” components. Peptides 2007, 28, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vargas, J.; Restano-Cassulini, R.; Possani, L. Toxin modulators and blockers of hERG K+ channels. Toxicon 2012, 60, 492–501. [Google Scholar] [CrossRef]
- Corona, M.; Gurrola, G.B.; Merino, E.; Cassulini, R.R.; Valdez-Cruz, N.A.; García, B.; Ramírez-Domínguez, M.E.; Coronas, F.I.; Zamudio, F.Z.; Wanke, E.; et al. A large number of novel Ergtoxin-like genes and ERG K+-channels blocking peptides from scorpions of the genus Centruroides. FEBS Lett. 2002, 532, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Chagot, B.; Pimentel, C.; Dai, L.; Pil, J.; Tytgat, J.; Nakajima, T.; Corzo, G.; Darbon, H.; Ferrat, G. An unusual fold for potassium channel blockers: NMR structure of three toxins from the scorpion Opisthacanthus madagascariensis. Biochem. J. 2005, 388, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Dai, H.; Qiu, S.; Li, T.; He, Y.; Ma, Y.; Chen, Z.; Wu, Y.; Li, W.; Cao, Z. SdPI, The First Functionally Characterized Kunitz-Type Trypsin Inhibitor from Scorpion Venom. PLoS ONE 2011, 6, e27548. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Hu, Y.-T.; Yang, W.-S.; He, Y.-W.; Feng, J.; Wang, B.; Zhao, R.-M.; Ding, J.-P.; Cao, Z.-J.; Li, W.-X.; et al. Hg1, Novel Peptide Inhibitor Specific for Kv1.3 Channels from First Scorpion Kunitz-type Potassium Channel Toxin Family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremonez, C.M.; Maiti, M.; Peigneur, S.; Cassoli, J.S.; Dutra, A.A.A.; Waelkens, E.; Lescrinier, E.; Herdewijn, P.; De Lima, M.E.; Pimenta, A.M.C.; et al. Structural and Functional Elucidation of Peptide Ts11 Shows Evidence of a Novel Subfamily of Scorpion Venom Toxins. Toxins 2016, 8, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Vargas, J.M.; Possani, L.D.; Luna-Ramírez, K. Arthropod toxins acting on neuronal potassium channels. Neuropharmacology 2017, 127, 139–160. [Google Scholar] [CrossRef]
- Gao, B.; Harvey, P.J.; Craik, D.J.; Ronjat, M.; De Waard, M.; Zhu, S. Functional evolution of scorpion venom peptides with an inhibitor cystine knot fold. Biosci. Rep. 2013, 33, 513–527. [Google Scholar] [CrossRef]
- Banerjee, A.; Lee, A.; Campbell, E.B.; MacKinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2013, 2, e00594. [Google Scholar] [CrossRef]
- Mouhat, S.; Mosbah, A.; Visan, V.; Wulff, H.; Delepierre, M.; Darbon, H.; Grissmer, S.; De Waard, M.; Sabatier, J.-M. The functional dyad of scorpion toxin Pi1 is not itself a prerequisite for toxin binding to the voltage-gated Kv1.2 potassium channels. Biochem. J. 2004, 377, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Shen, J.; Briggs, J.M.; Fu, W.; Wu, J.; Zhang, Y.; Luo, X.; Chi, Z.; Ji, R.; Jiang, H.; et al. Brownian dynamics simulations of the recognition of the scorpion toxin P05 with the small-conductance calcium-activated potassium channels. J. Mol. Biol. 2002, 318, 417–428. [Google Scholar] [CrossRef]
- Pardo-Lopez, L.; Zhang, M.; Liu, J.; Jiang, M.; Possani, L.D.; Tseng, G.N. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ErgTx) to the channel’s outer vestibule. J. Mol. Biol. 2002, 277, 16403–16411. [Google Scholar]
- Li-Smerin, Y.; Swartz, K.J. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl. Acad. Sci. USA 1998, 95, 8585–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498. [Google Scholar] [PubMed]
- Redaelli, E.; Cassulini, R.R.; Silva, D.F.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagón, A.; et al. Target Promiscuity and Heterogeneous Effects of Tarantula Venom Peptides Affecting Na+ and K+ Ion Channels. J. Biol. Chem. 2010, 285, 4130–4142. [Google Scholar] [CrossRef] [Green Version]
- Diochot, S.; Drici, M.-D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Carlin, K.P.; Wu, G.; Ilyin, V.I.; Musza, L.L.; Blake, P.R.; Kyle, D.J. Studies Examining the Relationship between the Chemical Structure of Protoxin II and Its Activity on Voltage Gated Sodium Channels. J. Med. Chem. 2014, 57, 6623–6631. [Google Scholar] [CrossRef]
- Escoubas, P.; Diochot, S.; Célérier, M.L.; Nakajima, T.; Lazdunski, M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol. Pharmacol. 2002, 62, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Montandon, G.G.; Cassoli, J.S.; Peigneur, S.; Verano-Braga, T.; dos Santos, D.M.; Paiva, A.L.B.; de Moraes, É.R.; Kushmerick, C.; Borges, M.H.; Richardson, M.; et al. GiTx1(β/κ-theraphotoxin-Gi1a), a novel toxin from the venom of Brazilian tarantula Grammostola iheringi (Mygalomorphae, Theraphosidae): Isolation, structural assessments and activity on voltage-gated ion channels. Biochimie 2020, 176, 138–149. [Google Scholar] [CrossRef]
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar] [CrossRef]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S. SGTx1, a Kv Channel Gating-Modifier Toxin, Binds to the Interfacial Region of Lipid Bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Luo, Z.; Lei, S.; Li, S.; Li, X.; Yuan, C. Effects and mechanism of gating modifier spider toxins on the hERG channel. Toxicon 2021, 189, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-Venom Peptides as Therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [Green Version]
- Colgrave, M.L.; Craik, D.J. Thermal, Chemical, and Enzymatic Stability of the Cyclotide Kalata B1: The Importance of the Cyclic Cystine Knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, S.-I.; Kimura, T.; Nozaki, T.; Harada, H.; Shimada, I.; Osawa, M. Structural basis for the inhibition of voltage-dependent K+ channel by gating modifier toxin. Sci. Rep. 2015, 5, 14226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li-Smerin, Y.; Swartz, K.J. Localization and Molecular Determinants of the Hanatoxin Receptors on the Voltage-Sensing Domains of a K+ Channel. J. Gen. Physiol. 2000, 115, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Chen, X.; Deng, M.; Xiao, Y.; Wu, Y.; Liu, Z.; Zhou, S.; He, Y.; Liang, S. Interaction site for the inhibition of tarantula Jingzhaotoxin-XI on voltage-gated potassium channel Kv2.1. Toxicon 2016, 124, 8–14. [Google Scholar] [CrossRef]
- DeSimone, C.V.; Lu, Y.; Bondarenko, V.E.; Morales, M.J. S3b Amino Acid Substitutions and Ancillary Subunits Alter the Affinity of Heteropoda venatoria Toxin 2 for Kv4.3. Mol. Pharmacol. 2009, 76, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbinghaus, J.; Legros, C.; Nolting, A.; Guette, C.; Celerier, M.-L.; Pongs, O.; Bähring, R. Modulation of Kv4.2 channels by a peptide isolated from the venom of the giant bird-eating tarantula Theraphosa leblondi. Toxicon 2004, 43, 923–932. [Google Scholar] [CrossRef]
- Alvarado, D.; Cardoso-Arenas, S.; Corrales-García, L.-L.; Clement, H.; Arenas, I.; Montero-Dominguez, P.A.; Olamendi-Portugal, T.; Zamudio, F.; Csoti, A.; Borrego, J.; et al. A Novel Insecticidal Spider Peptide that Affects the Mammalian Voltage-Gated Ion Channel hKv1.5. Front. Pharmacol. 2021, 11, 1937. [Google Scholar] [CrossRef]
- Cerni, F.A.; Pucca, M.B.; Peigneur, S.; Cremonez, C.M.; Bordon, K.C.F.; Tytgat, J.; Arantes, E.C. Electrophysiological Characterization of Ts6 and Ts7, K+ Channel Toxins Isolated through an Improved Tityus serrulatus Venom Purification Procedure. Toxins 2014, 6, 892–913. [Google Scholar] [CrossRef] [Green Version]
- Martin-Eauclaire, M.F.; Pimenta, A.M.; Bougis, P.E.; De Lima, M.E. Potassium channel blockers from the venom of the Brazilian scorpion Tityus serrulatus (Lutz and Mello, 1922). Toxicon 2016, 119, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Zoccal, K.F.; da Silva Bitencourt, C.; Sorgi, C.A.; Bordon, K.D.C.F.; Sampaio, S.V.; Arantes, E.C.; Faccioli, L.H. Ts6 and Ts2 from Tityus serrulatus venom induce inflammation by mechanisms dependent on lipid mediators and cytokine production. Toxicon 2013, 61, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zoccal, K.F.; Bitencourt, C.D.S.; Secatto, A.; Sorgi, C.A.; Bordon, K.D.C.F.; Sampaio, S.V.; Arantes, E.C.; Faccioli, L.H. Tityus serrulatus venom and toxins Ts1, Ts2 and Ts6 induce macrophage activation and production of immune mediators. Toxicon 2011, 57, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Laghi-Pasini, F.; Acampa, M.; Srivastava, U.; Bertolozzi, I.; Giabbani, B.; Finizola, F.; Vanni, F.; Dokollari, A.; Natale, M.; et al. Systemic Inflammation Rapidly Induces Reversible Atrial Electrical Remodeling: The Role of Interleukin-6–Mediated Changes in Connexin Expression. J. Am. Heart Assoc. 2019, 8, e011006. [Google Scholar] [CrossRef] [PubMed]
- Loose, S.; MUller, J.; Wettwer, E.; El-Haou, S.; Jackson, C.; Tang, R.; Milnes, J.; Ravens, U.; Ford, J. Abstract 18323: Positive Frequency-Dependent Effects of Highly Selective Kv1.5 Blockers (XEN-D0103 & MK-0448) in Right Atrial Trabeculae from Patients in Sinus Rhythm. Circulation 2013, 128, A18323. [Google Scholar]
- Varga, Z.; Gurrola-Briones, G.; Papp, F.; de la Vega, R.C.R.; Pedraza-Alva, G.; Tajhya, R.B.; Gaspar, R.; Cardenas, L.; Rosenstein, Y.; Beeton, C.; et al. Vm24, a Natural Immunosuppressive Peptide, Potently and Selectively Blocks Kv1.3 Potassium Channels of Human T Cells. Mol. Pharmacol. 2012, 82, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Furman, B. Tetraethylammonium. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Schmitz, A.; Sankaranarayanan, A.; Azam, P.; Schmidt-Lassen, K.; Homerick, D.; Hänsel, W.; Wulff, H. Design of PAP-1, a Selective Small Molecule Kv1.3 Blocker, for the Suppression of Effector Memory T Cells in Autoimmune Diseases. Mol. Pharmacol. 2005, 68, 1254–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2008, 2, 407–412. [Google Scholar] [CrossRef]
- Ahuja, S.; Mukund, S.; Deng, L.; Khakh, K.; Chang, E.; Ho, H.; Shriver, S.; Young, C.; Lin, S.; Johnson, J.P.; et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350, aac5464. [Google Scholar] [CrossRef] [Green Version]
- Gilquin, B.; Braud, S.; Eriksson, M.A.L.; Roux, B.; Bailey, T.D.; Priest, B.T.; Garcia, M.L.; Ménez, A.; Gasparini, S. A Variable Residue in the Pore of Kv1 Channels Is Critical for the High Affinity of Blockers from Sea Anemones and Scorpions. J. Biol. Chem. 2005, 280, 27093–27102. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Gomez, B.; Watanabe, I.; Thornhill, W.B. Amino acids in the pore region of Kv1 potassium channels dictate cell-surface protein levels: A possible trafficking code in the Kv1 subfamily. Biochem. J. 2005, 388, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sunchen, S.; He, D.; Qin, C.; Zuo, Z.; Shen, B.; Cao, Z.; Hong, W.; Miao, L. ImKTx96, a peptide blocker of the Kv1.2 ion channel from the venom of the scorpion Isometrus maculates. Peptides 2019, 123, 170172. [Google Scholar] [CrossRef]
- Lin, S.; Wang, X.; Hu, X.; Zhao, Y.; Zhao, M.; Zhang, J.; Cui, Y. Recombinant Expression, Functional Characterization of Two Scorpion Venom Toxins with Three Disulfide Bridges from the Chinese Scorpion Buthus martensii Karsch. Protein Pept. Lett. 2017, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.U.; Tajti, G.; Gaspar, A.; Szanto, T.G.; Borrego, J.; Panyi, G. Optimization of Pichia pastoris Expression System for High-Level Production of Margatoxin. Front. Pharmacol. 2021, 12, 733610. [Google Scholar] [CrossRef] [PubMed]
- Geron, M. Production and Purification of Recombinant Toxins. In Snake and Spider Toxins; Humana: New York, NY, USA, 2020; pp. 73–84. [Google Scholar]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Factories 2012, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessette, P.H.; Åslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klint, J.; Senff, S.; Saez, N.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.; Rash, L.; Mobli, M.; King, G.F. Production of Recombinant Disulfide-Rich Venom Peptides for Structural and Functional Analysis via Expression in the Periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [Green Version]
- De Marco, A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Factories 2009, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Demain, A.L.; Vaishnav, P. Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 2009, 27, 297–306. [Google Scholar] [CrossRef]
- De-Bona, E.; Chaves-Moreira, D.; Batista, T.B.D.; da Justa, H.C.; Rossi, G.R.; Antunes, B.C.; Matsubara, F.H.; Minozzo, J.C.; Wille, A.C.M.; Veiga, S.S.; et al. Production of a novel recombinant brown spider hyaluronidase in baculovirus-infected insect cells. Enzym. Microb. Technol. 2021, 146, 109759. [Google Scholar] [CrossRef]
- Szolajska, E.; Poznanski, J.; Ferber, M.L.; Michalik, J.; Gout, E.; Fender, P.; Bailly, I.; Dublet, B.; Chroboczek, J. Poneratoxin, a neurotoxin from ant venom. Structure and expression in insect cells and construction of a bio-insecticide. Eur. J. Biochem. 2004, 271, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Borgia, J.A.; Fields, G.B. Chemical synthesis of proteins. Trends Biotechnol. 2000, 18, 243–251. [Google Scholar] [CrossRef]
- Jensen, J.E.; Durek, T.; Alewood, P.F.; Adams, D.J.; King, G.F.; Rash, L.D. Chemical synthesis and folding of APETx2, a potent and selective inhibitor of acid sensing ion channel 3. Toxicon 2009, 54, 56–61. [Google Scholar] [CrossRef]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Na v 1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Xu, H.; Li, T.; Rohou, A.; Arthur, C.P.; Tzakoniati, F.; Wong, E.; Estevez, A.; Kugel, C.; Franke, Y.; Chen, J.; et al. Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 2019, 176, 702–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego, J.; Clement, H.; Corrales-García, L.-L.; Arenas, I.; Corzo, G. Key amino acid residues involved in mammalian and insecticidal activities of Magi4 and Hv1b, cysteine-rich spider peptides from the δ-atracotoxin family. Amino Acids 2020, 52, 465–475. [Google Scholar] [CrossRef]
- Netirojjanakul, C.; Miranda, L.P. Progress and challenges in the optimization of toxin peptides for development as pain therapeutics. Curr. Opin. Chem. Biol. 2017, 38, 70–79. [Google Scholar] [CrossRef]
- Horovitz, A. Double-mutant cycles: A powerful tool for analyzing protein structure and function. Fold. Des. 1996, 1, R121–R126. [Google Scholar] [CrossRef] [Green Version]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikouee, A.; Khabiri, M.; Grissmer, S.; Ettrich, R. Charybdotoxin and Margatoxin Acting on the Human Voltage-Gated Potassium Channel hKv1.3 and Its H399N Mutant: An Experimental and Computational Comparison. J. Phys. Chem. B 2012, 116, 5132–5140. [Google Scholar] [CrossRef] [PubMed]
- Corzo, G.; Papp, F.; Varga, Z.; Barraza, O.; Espino-Solis, P.G.; de la Vega, R.C.R.; Gaspar, R.; Panyi, G.; Possani, L.D. A selective blocker of Kv1.2 and Kv1.3 potassium channels from the venom of the scorpion Centruroides suffusus suffusus. Biochem. Pharmacol. 2008, 76, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Gigolaev, A.M.; Kuzmenkov, A.I.; Peigneur, S.; Tabakmakher, V.; Pinheiro-Junior, E.; Chugunov, A.O.; Efremov, R.G.; Tytgat, J.; Vassilevski, A.A. Tuning Scorpion Toxin Selectivity: Switching From KV1.1 to KV1.3. Front. Pharmacol. 2020, 11, 1010. [Google Scholar] [CrossRef] [PubMed]
- Tabakmakher, V.M.; Kuzmenkov, A.I.; Gigolaev, A.M.; Pinheiro-Junior, E.L.; Peigneur, S.; Efremov, R.G.; Tytgat, J.; Vassilevski, A.A. Artificial Peptide Ligand of PotassiumChannel KV1.1 with High Selectivity. J. Evol. Biochem. Physiol. 2021, 57, 386–403. [Google Scholar] [CrossRef]
- Bartok, A.; Fehér, K.; Bodor, A.; Rákosi, K.; Tóth, G.K.; Kövér, K.E.; Panyi, G.; Varga, Z. An engineered scorpion toxin analogue with improved Kv1.3 selectivity displays reduced conformational flexibility. Sci. Rep. 2015, 5, 18397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmenkov, A.I.; Peigneur, S.; Chugunov, A.; Tabakmakher, V.; Efremov, R.G.; Tytgat, J.; Grishin, E.V.; Vassilevski, A.A. C-Terminal residues in small potassium channel blockers OdK1 and OSK3 from scorpion venom fine-tune the selectivity. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2017, 1865, 465–472. [Google Scholar] [CrossRef]
- Luna-Ramirez, K.; Csoti, A.; McArthur, J.R.; Chin, Y.K.; Anangi, R.; Najera, R.D.C.; Possani, L.D.; King, G.F.; Panyi, G.; Yu, H.; et al. Structural basis of the potency and selectivity of Urotoxin, a potent Kv1 blocker from scorpion venom. Biochem. Pharmacol. 2020, 174, 113782. [Google Scholar] [CrossRef]
- Delgado-Prudencio, G.; Possani, L.D.; Becerril, B.; Ortiz, E. The Dual α-Amidation System in Scorpion Venom Glands. Toxins 2019, 11, 425. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Kennedy, K.; De Blas, G.A.; Orta, G.; Pavarotti, M.A.; Arias, R.J.; de la Vega-Beltrán, J.L.; Li, Q.; Dai, H.; Perozo, E.; et al. Role of human Hv1 channels in sperm capacitation and white blood cell respiratory burst established by a designed peptide inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E11847–E11856. [Google Scholar] [CrossRef] [Green Version]
- Erlanson, D.A. Introduction to Fragment-Based Drug Discovery. Top. Curr. Chem. 2011, 317, 1–32. [Google Scholar]
- Price, A.J.; Howard, S.; Cons, B.D. Fragment-based drug discovery and its application to challenging drug targets. Essays Biochem. 2017, 61, 475–484. [Google Scholar] [PubMed]
- Varga, Z.; Tajti, G.; Panyi, G. The Kv1.3 K+ channel in the immune system and its “precision pharmacology” using peptide toxins. Biol. Futur. 2021, 72, 75–83. [Google Scholar] [CrossRef]
- Shen, B.; Cao, Z.; Yingliang, W.; Sabatier, J.-M.; Wu, Y. Treating autoimmune disorders with venom-derived peptides. Expert Opin. Biol. Ther. 2017, 17, 1065–1075. [Google Scholar] [CrossRef]
- Gubič, Š.; Hendrickx, L.A.; Toplak, Ž.; Sterle, M.; Peigneur, S.; Tomašič, T.; Pardo, L.A.; Tytgat, J.; Zega, A.; Mašič, L.P. Discovery of Kv1.3 ion channel inhibitors: Medicinal chemistry approaches and challenges. Med. Res. Rev. 2021, 41, 2423–2473. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Volkers, L.; Jangsangthong, W.; Bart, C.I.; Engels, M.C.; Zhou, G.; Schalij, M.J.; Ypey, D.L.; Pijnappels, D.A.; Vries, A.A.F.D. Generation and primary characterization of iAM-1, a versatile new line of conditionally immortalized atrial myocytes with preserved cardiomyogenic differentiation capacity. Cardiovasc. Res. 2018, 114, 1848–1859. [Google Scholar] [CrossRef]
- Harlaar, N.; Liu, J.; Volkers, L.; Ramkisoensing, A.A.; Schalij, M.J.; Klautz, R.J.M.; Van Brakel, T.J.; Pijnappels, D.A.; Vries, A.A.F.D. P1229Massive expansion of native human atrial cardiomyocytes through immortogenetics: Generation of the hiAM cell lines. Eur. Heart J. 2019, 40, ehz748-0187. [Google Scholar] [CrossRef]
- Laksman, Z.; Wauchop, M.; Lin, E.; Protze, S.; Lee, J.; Yang, W.; Izaddoustdar, F.; Shafaattalab, S.; Gepstein, L.; Tibbits, G.F.; et al. Modeling Atrial Fibrillation using Human Embryonic Stem Cell-Derived Atrial Tissue. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Barré-Sinoussi, F.; Montagutelli, X. Animal models are essential to biological research: Issues and perspectives. Future Sci. OA 2015, 1, FSO63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saljic, A.; Jespersen, T.; Buhl, R. Anti-arrhythmic investigations in large animal models of atrial fibrillation. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Michael, G.; Dobrev, D.; Nattel, S. Animal models for atrial fibrillation: Clinical insights and scientific opportunities. Europace 2009, 12, 160–172. [Google Scholar] [CrossRef]
- Schüttler, D.; Bapat, A.; Kääb, S.; Lee, K.; Tomsits, P.; Clauss, S.; Hucker, W.J. Animal Models of Atrial Fibrillation. Circ. Res. 2020, 127, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Christ, T.; Magyar, J. New Strategies for the Treatment of Atrial Fibrillation. Pharmaceuticals 2021, 14, 926. [Google Scholar] [CrossRef] [PubMed]
- de Haan, S.; Greiser, M.; Harks, E.; Blaauw, Y.; van Hunnik, A.; Verheule, S.; Allessie, M.; Schotten, U. AVE0118, blocker of the transient outward current (Ito) and ultrarapid delayed rectifier current (IKur), fully restores atrial contractility after cardioversion of atrial fibrillation in the goat. Circulation 2006, 114, 1234–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oros, A.; Volders, P.G.; Beekman, J.D.; van der Nagel, T.; Vos, M.A. Atrial-specific drug AVE0118 is free of torsades de pointes in anesthetized dogs with chronic complete atrioventricular block. Heart Rhythm 2006, 3, 1339–1345. [Google Scholar] [CrossRef]
- Blaauw, Y.; Schotten, U.; Van Hunnik, A.; Neuberger, H.; Allessie, M. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc. Res. 2007, 75, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, K.J.; Brendel, J.; Steinmeyer, K.; Linz, D.K.; Rütten, H.; Gögelein, H. In Vitro and In Vivo Effects of the Atrial Selective Antiarrhythmic Compound AVE1231. J. Cardiovasc. Pharmacol. 2007, 49, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Rivard, L.; Shiroshita-Takeshita, A.; Maltais, C.; Ford, J.; Pinnock, R.; Madge, D.; Nattel, S. Electrophysiological and atrial antiarrhythmic effects of a novel IKur/Kv1.5 blocker in dogs. Heart Rhythm 2005, 2, S180. [Google Scholar] [CrossRef]
- Ford, J.; Milnes, J.; El Haou, S.; Wettwer, E.; Loose, S.; Matschke, K.; Tyl, B.; Round, P.; Ravens, U. The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm 2016, 13, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Stump, G.L.; Wallace, A.A.; Regan, C.P.; Lynch, J.J. In Vivo Antiarrhythmic and Cardiac Electrophysiologic Effects of a Novel Diphenylphosphine Oxide IKur Blocker (2-Isopropyl-5-methylcyclohexyl) Diphenylphosphine Oxide. J. Pharmacol. Exp. Ther. 2005, 315, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- van Hunnik, A.; Lau, D.H.; Zeemering, S.; Kuiper, M.; Verheule, S.; Schotten, U. Antiarrhythmic effect of vernakalant in electrically remodeled goat atria is caused by slowing of conduction and prolongation of postrepolarization refractoriness. Heart Rhythm 2016, 13, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Diness, J.G.; Skibsbye, L.; Simó-Vicens, R.; Santos, J.L.; Lundegaard, P.; Citerni, C.; Sauter, D.R.P.; Bomholtz, S.H.; Svendsen, J.H.; Olesen, S.-P.; et al. Termination of Vernakalant-Resistant Atrial Fibrillation by Inhibition of Small-Conductance Ca2+-Activated K+ Channels in Pigs. Circ. Arrhythmia Electrophysiol. 2017, 10, e005125. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; De Blasio, E.; Beatch, G.N.; Wang, W.Q. RSD1235: A novel antiarrhythmic agent with a unique electrophysiological profile that terminates AF in dogs. Eur. Heart J. 2001, 22, 448. [Google Scholar]
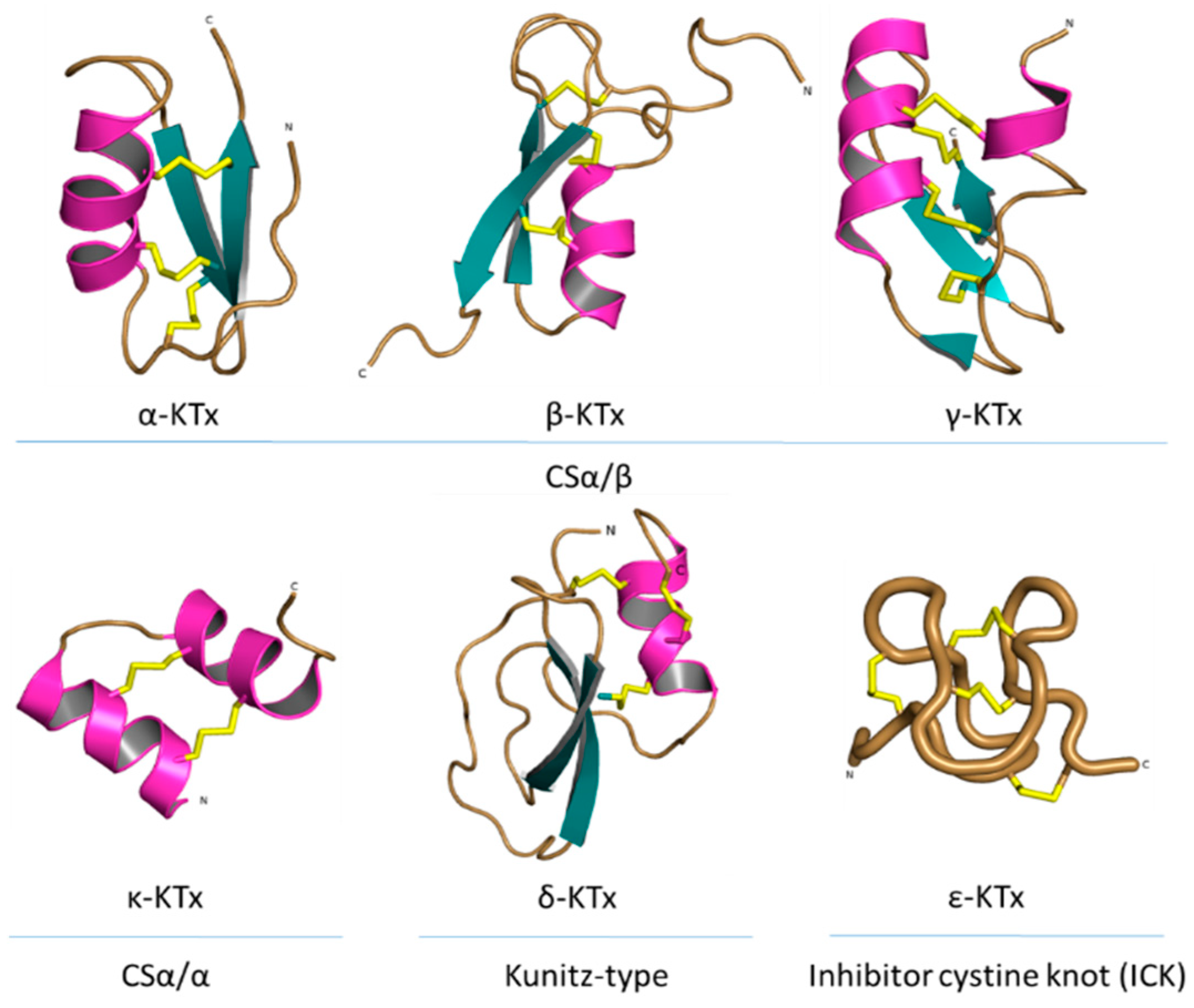
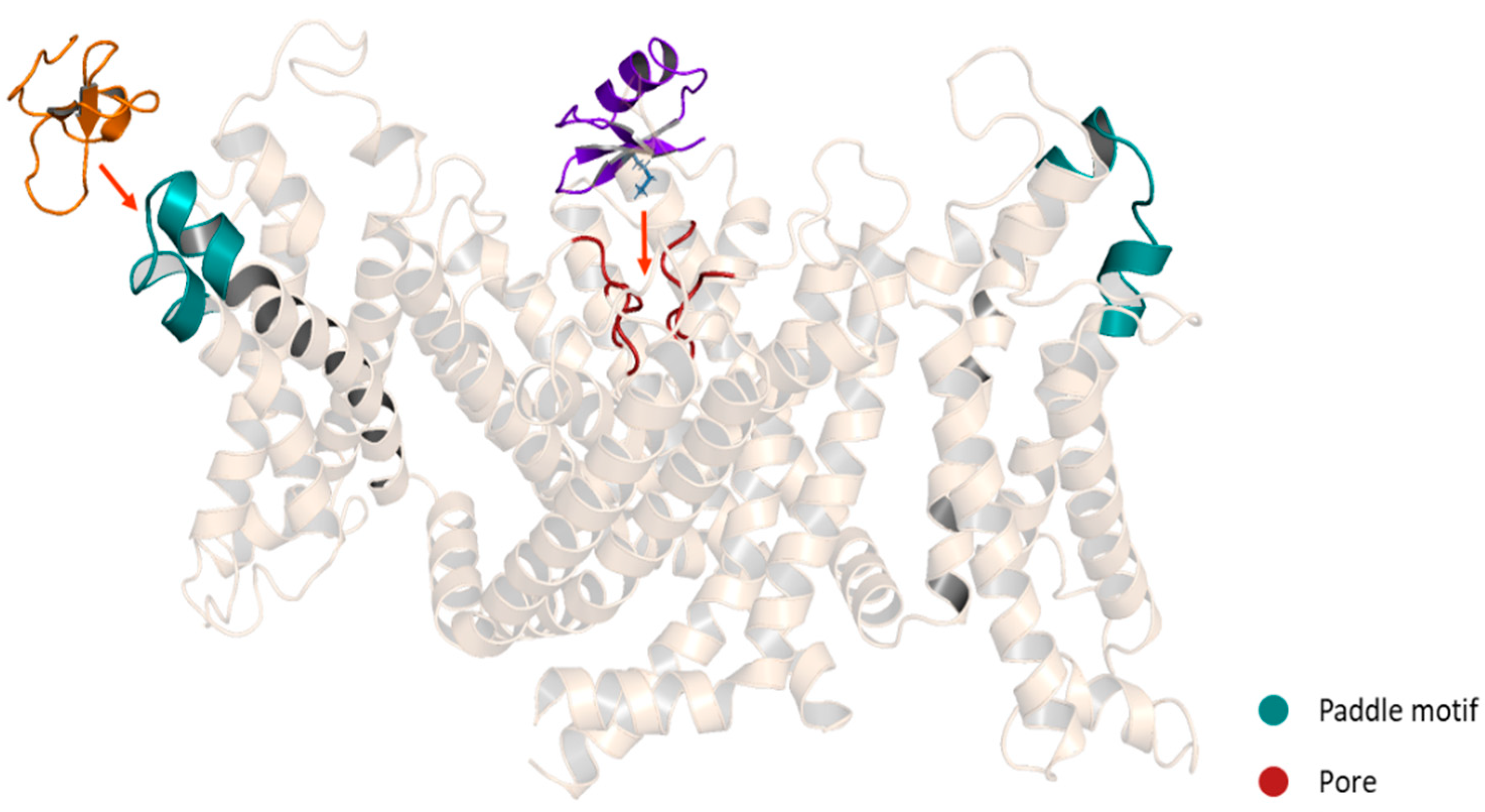

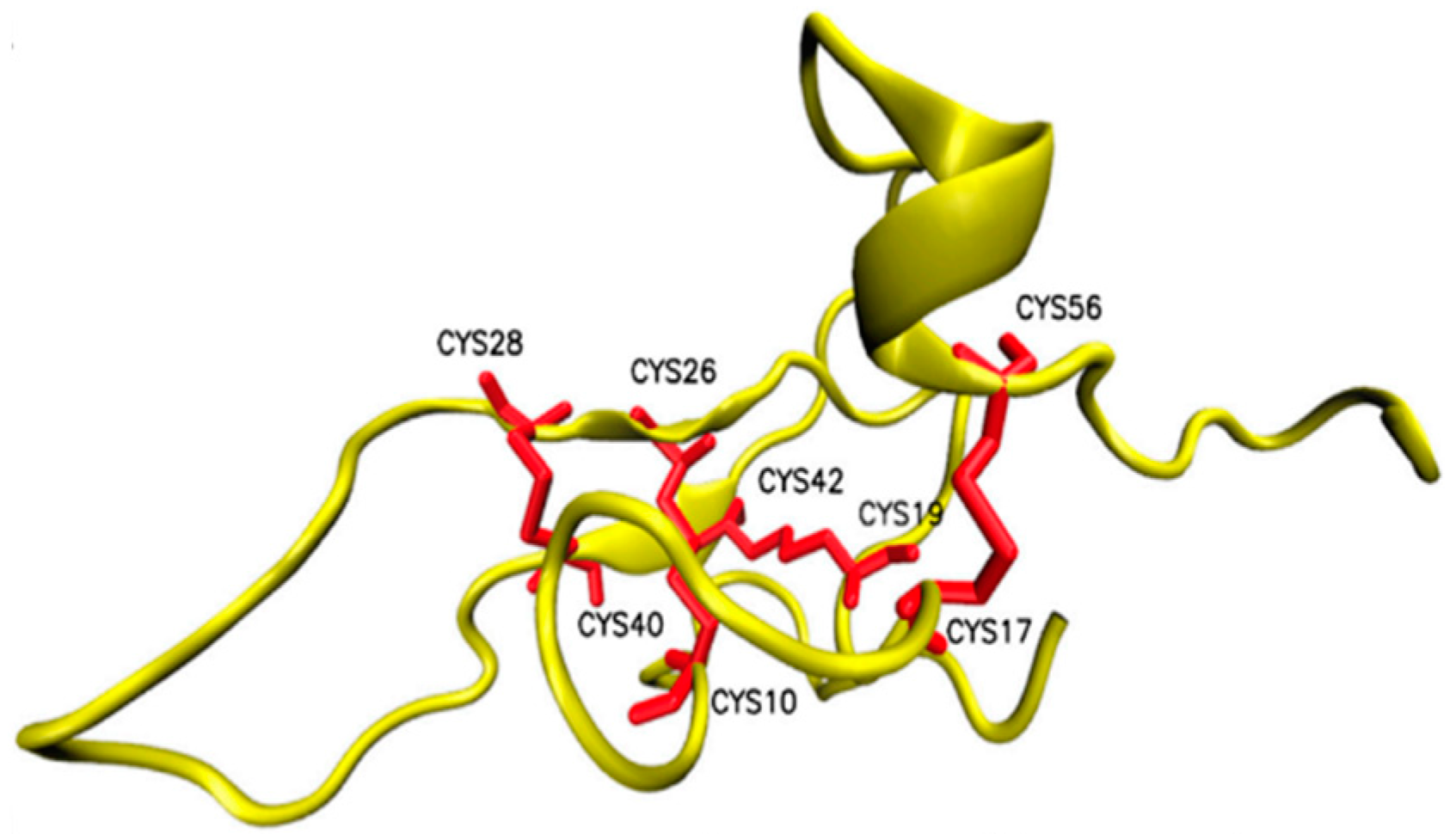
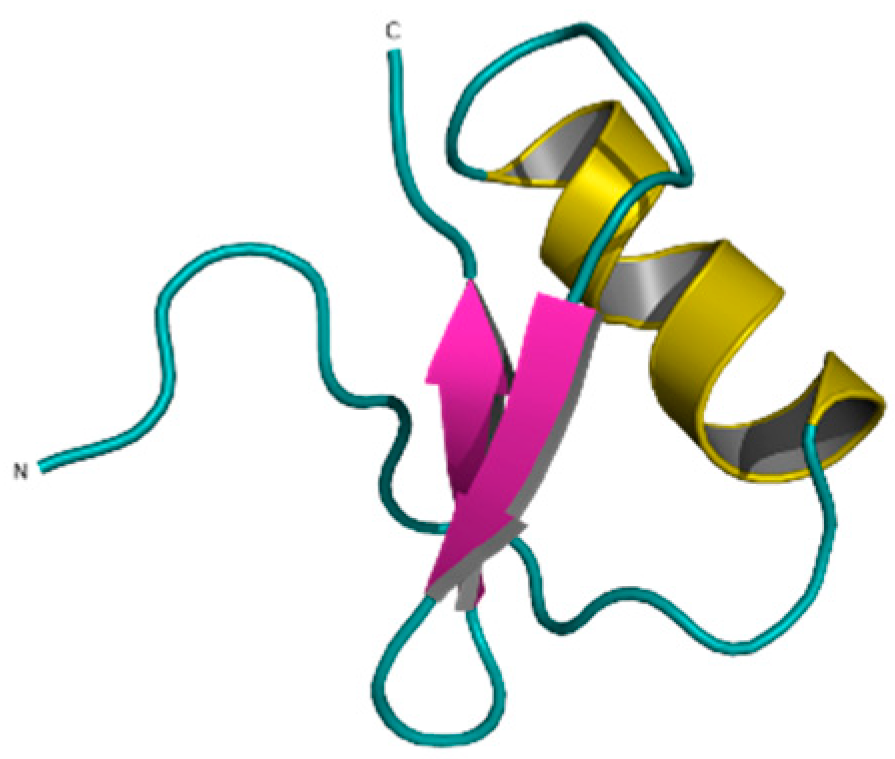
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego, J.; Feher, A.; Jost, N.; Panyi, G.; Varga, Z.; Papp, F. Peptide Inhibitors of Kv1.5: An Option for the Treatment of Atrial Fibrillation. Pharmaceuticals 2021, 14, 1303. https://doi.org/10.3390/ph14121303
Borrego J, Feher A, Jost N, Panyi G, Varga Z, Papp F. Peptide Inhibitors of Kv1.5: An Option for the Treatment of Atrial Fibrillation. Pharmaceuticals. 2021; 14(12):1303. https://doi.org/10.3390/ph14121303
Chicago/Turabian StyleBorrego, Jesús, Adam Feher, Norbert Jost, Gyorgy Panyi, Zoltan Varga, and Ferenc Papp. 2021. "Peptide Inhibitors of Kv1.5: An Option for the Treatment of Atrial Fibrillation" Pharmaceuticals 14, no. 12: 1303. https://doi.org/10.3390/ph14121303
APA StyleBorrego, J., Feher, A., Jost, N., Panyi, G., Varga, Z., & Papp, F. (2021). Peptide Inhibitors of Kv1.5: An Option for the Treatment of Atrial Fibrillation. Pharmaceuticals, 14(12), 1303. https://doi.org/10.3390/ph14121303