
29th Annual GP2A Medicinal Chemistry Conference

 ,
,  ,
,  , , , ,
, , , ,  , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Aim and Scope of the Meeting
2. Keynote Lectures
2.1. From Activity-Based Protein Profiling to the Optimization of Covalent Inhibitors Targeting Dipeptidyl Peptidases (Kl01)
2.2. Programming Molecules for Therapeutic Applications (KL02)
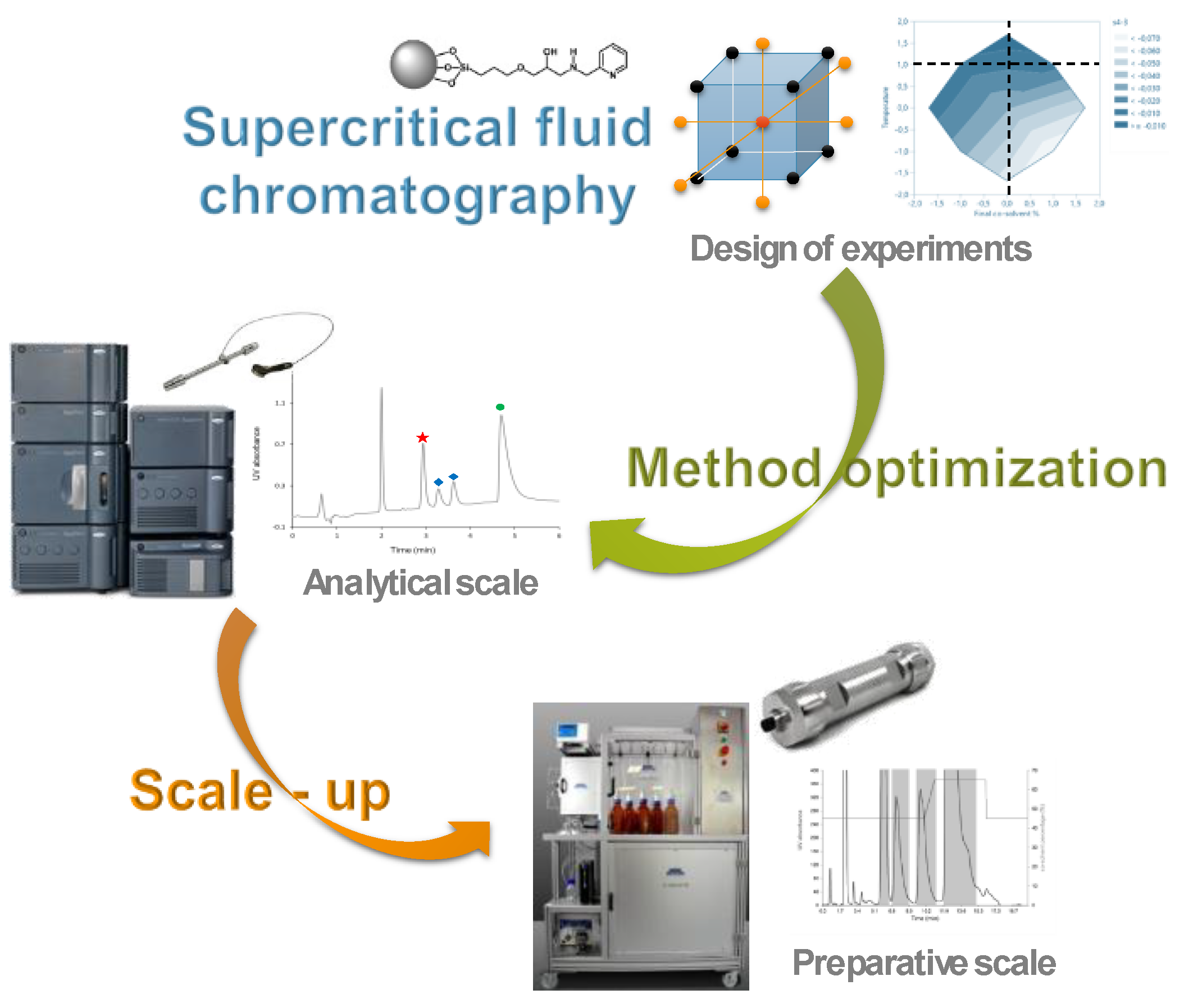
2.3. From Impurity Profiling to Purified Metabolites: Drug Discovery Supported by Analytical and Preparative Supercritical Fluid Chromatography (KL03)
2.4. Drug-Loaded Polymeric Systems as a Promising Tool for Cancer Management (KL04)
2.5. Discovery of Small Molecules to Manipulate Cell Fate In Vivo: Towards New Therapies for Degenerative Diseases (KL05)
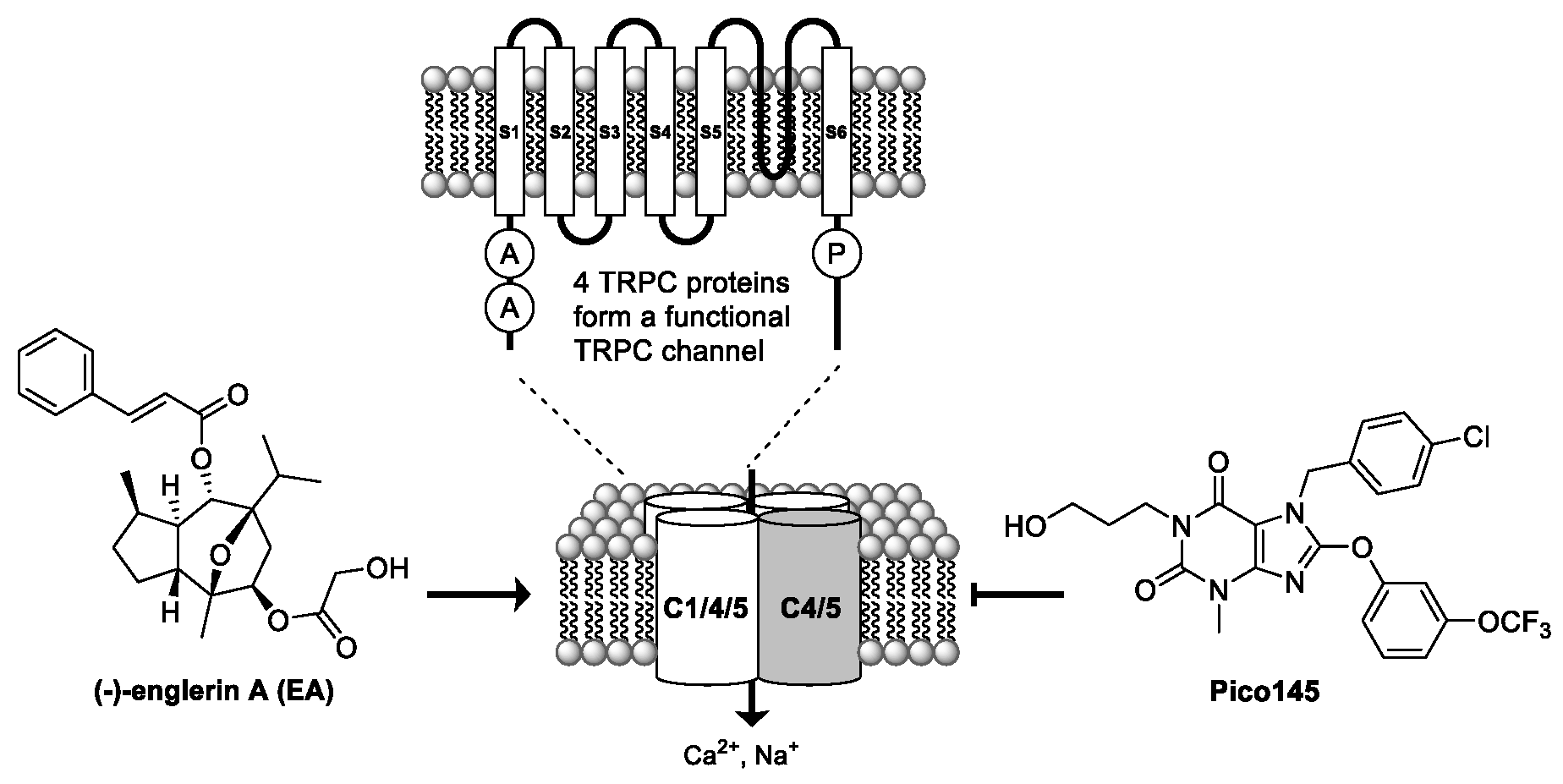
2.6. Understanding Potent Small-Molecule Modulation of TRPC1/4/5 Channels (KL06)
2.7. Fragment-Based Approaches to Inhibit Mycobacterium Tuberculosis Targets (KL07)
2.8. Chemical Approaches for the Study and Inhibition of Bacterial L,D-Transpeptidases (KL08)
2.9. Metal Complexes in Medicinal Chemistry (KL09)
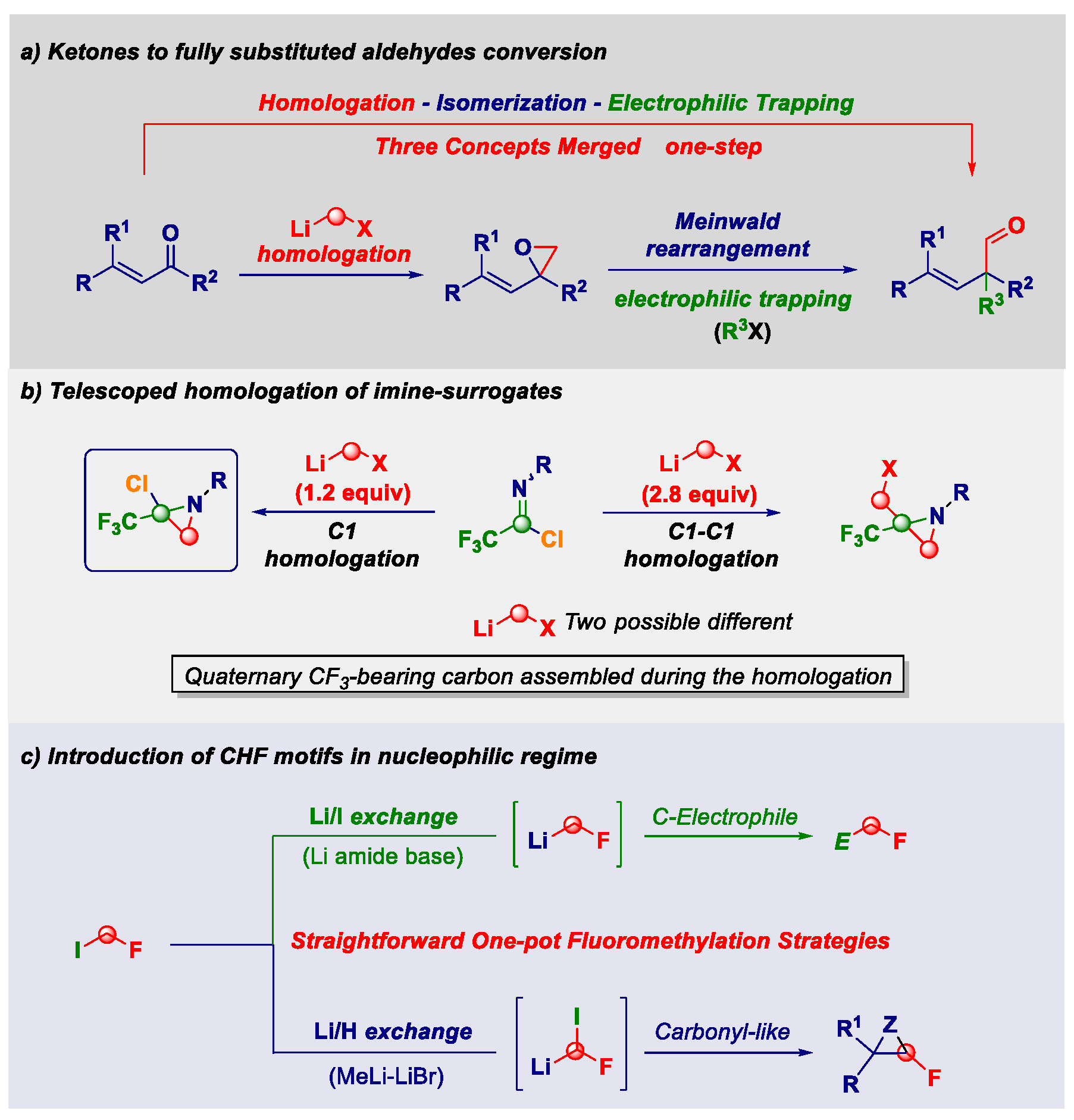
2.10. Designing New Synthetic Concepts for Imparting Molecular Complexity with C-1 Sources (KL10)
3. Young Researcher Communications
3.1. All for One and One for All: Design and Synthesis of Molecules with Neuronal Differentiation Properties (YRC01)
3.2. Synthesis and Biological Evaluation of Novel, Functionalized Bisindolylmaleimides (YRC02)
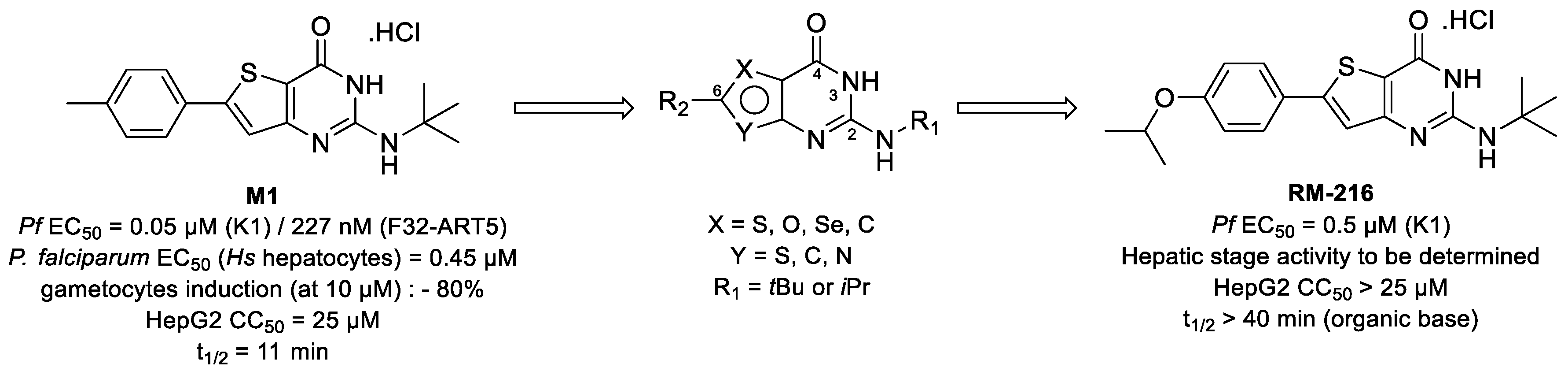
3.3. C6-Modulation and Scaffold Hopping of Thienopyrimidinone Antiplasmodial Hit with Multi-Stage Activity (YRC03)
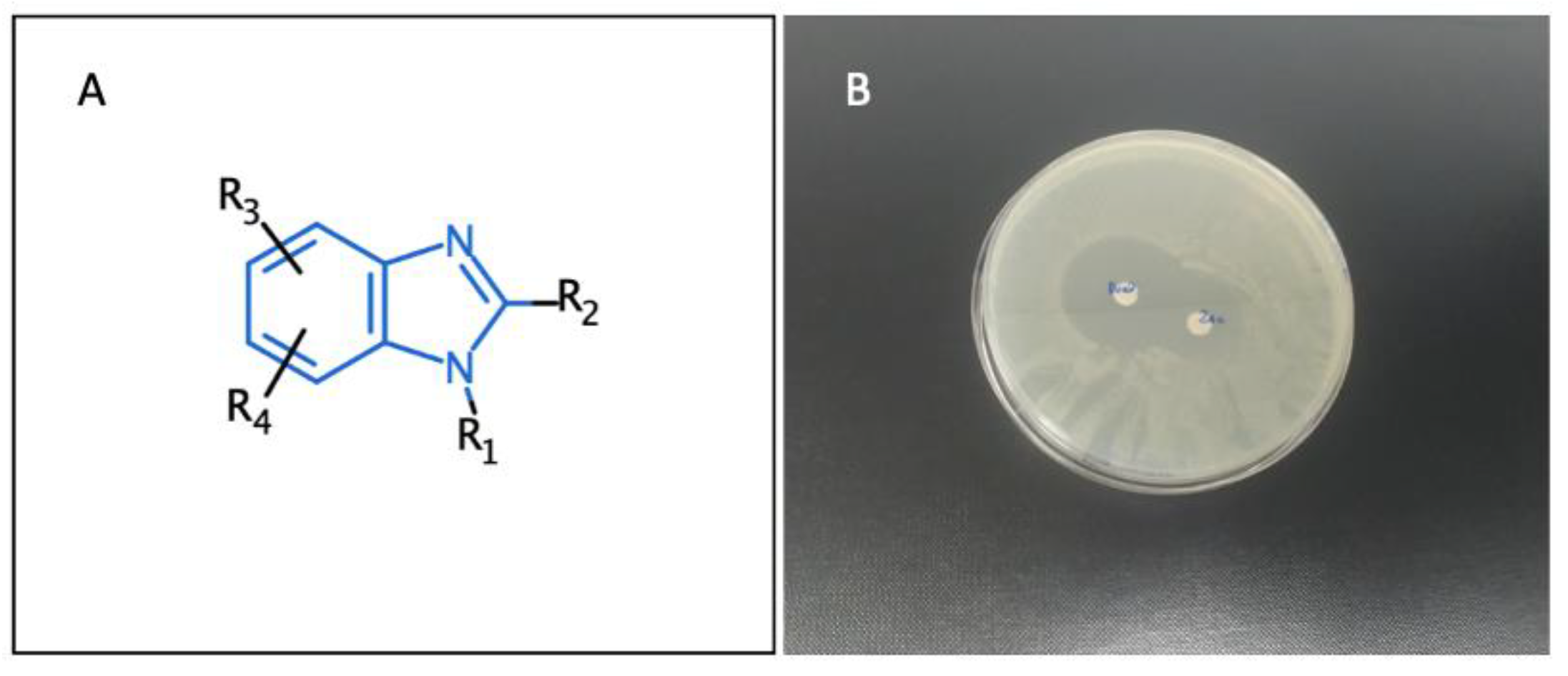
3.4. Benzimidazole Derivatives and Their Analogues as Myeloperoxidase Inhibitors (YRC04)
3.5. Synthesis and Chiral Resolution of Trans Configuration Anticancer β-Lactam Enantiomers (YRC05)
3.6. Peptide-Targeted Systems for Photodynamic Therapy (YRC06)
3.7. SAR of Methylsulfanylpyridine-Based Combretastatin Analogues as Promising Antimitotic Agents (YRC07)
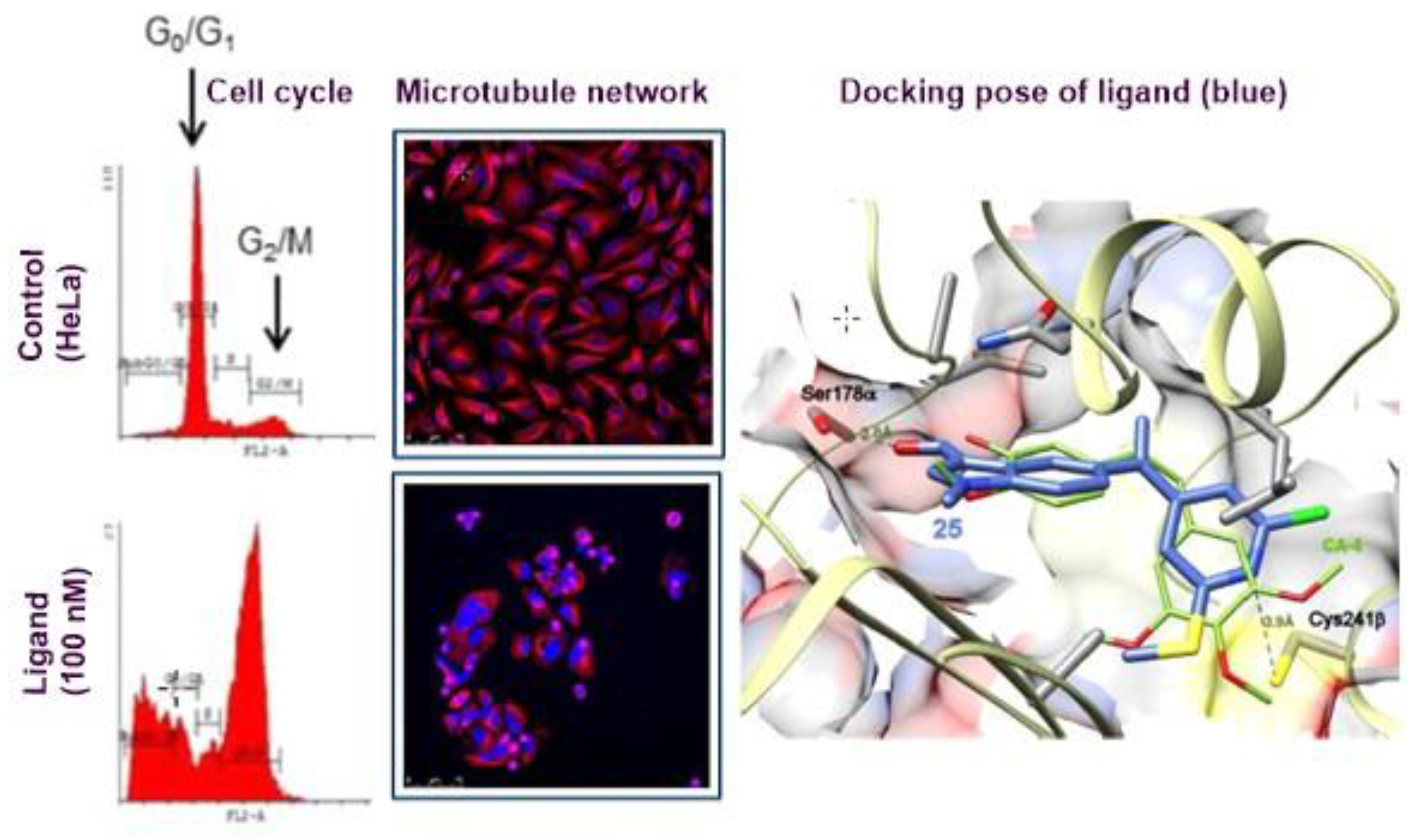
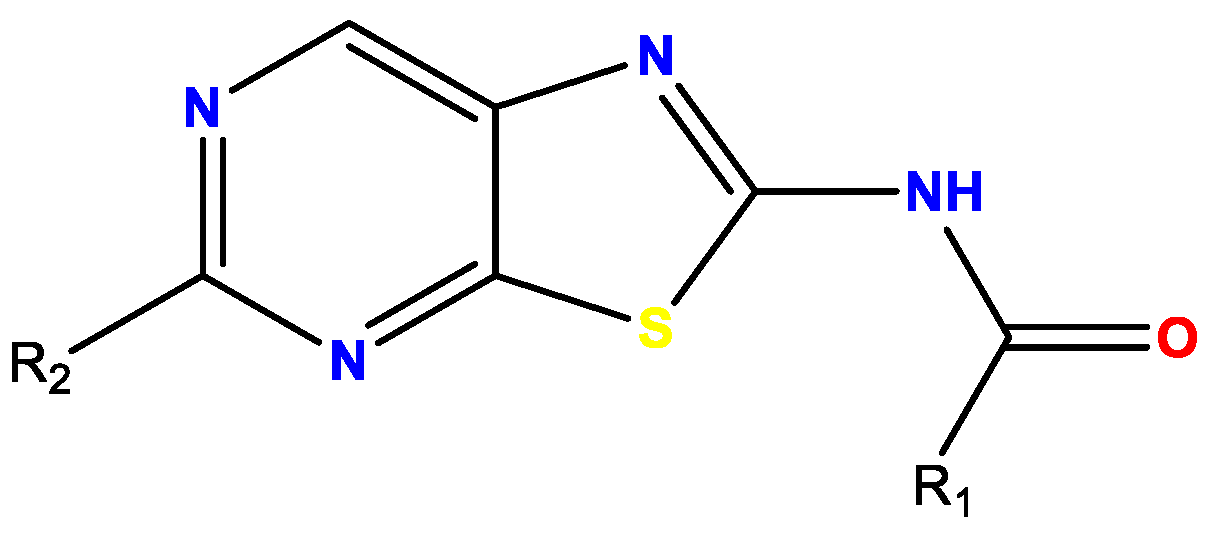
3.8. Discovery and Binding Study of Herap2 Modulators Identified via Hts (YRC08)
4. Posters
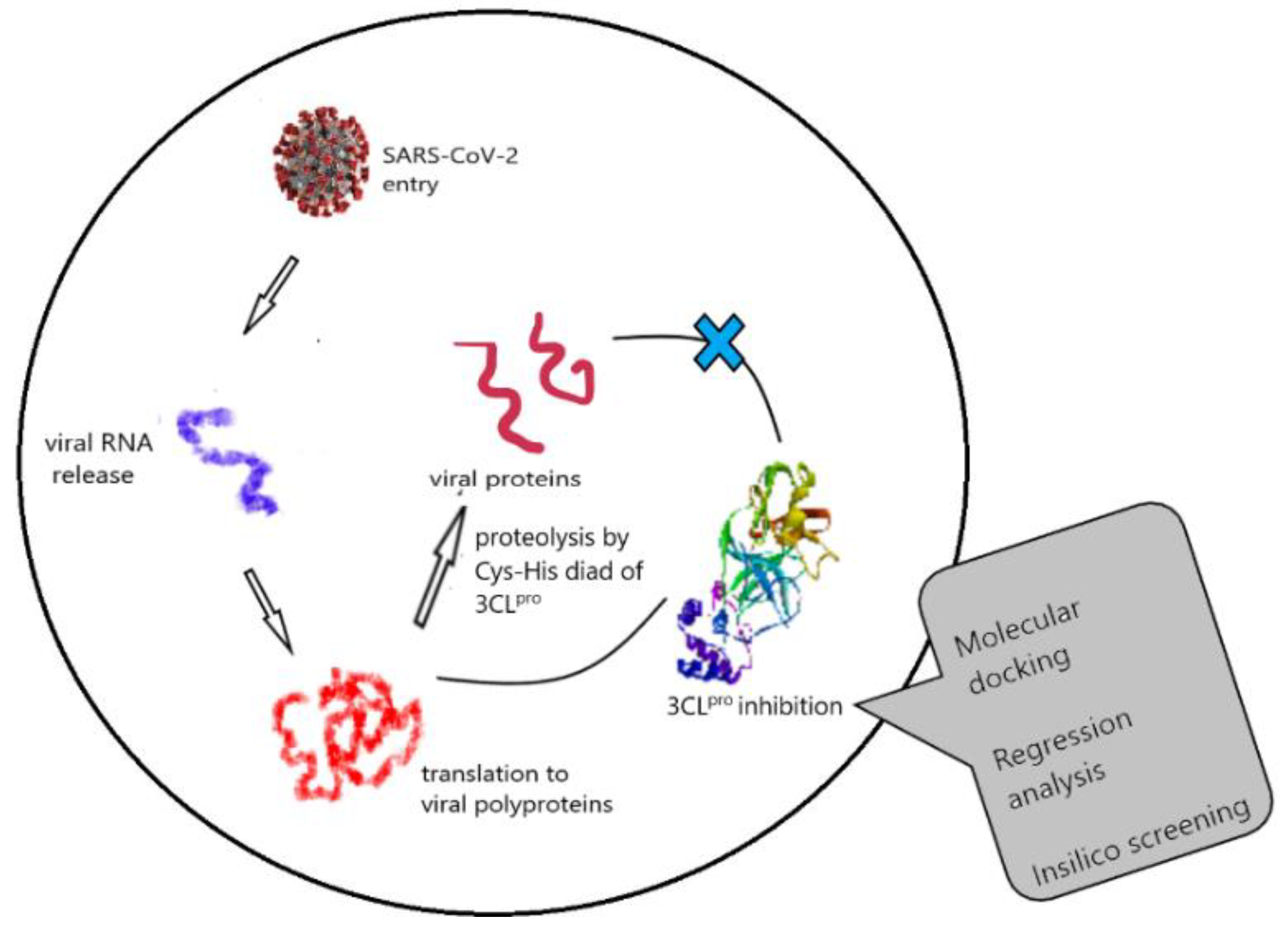
4.1. Theoretical Insights into the Anti-SARS-CoV-2 Activity of Chloroquine and Its Analogs and In Silico Screening of Main Protease Inhibitors (P01)
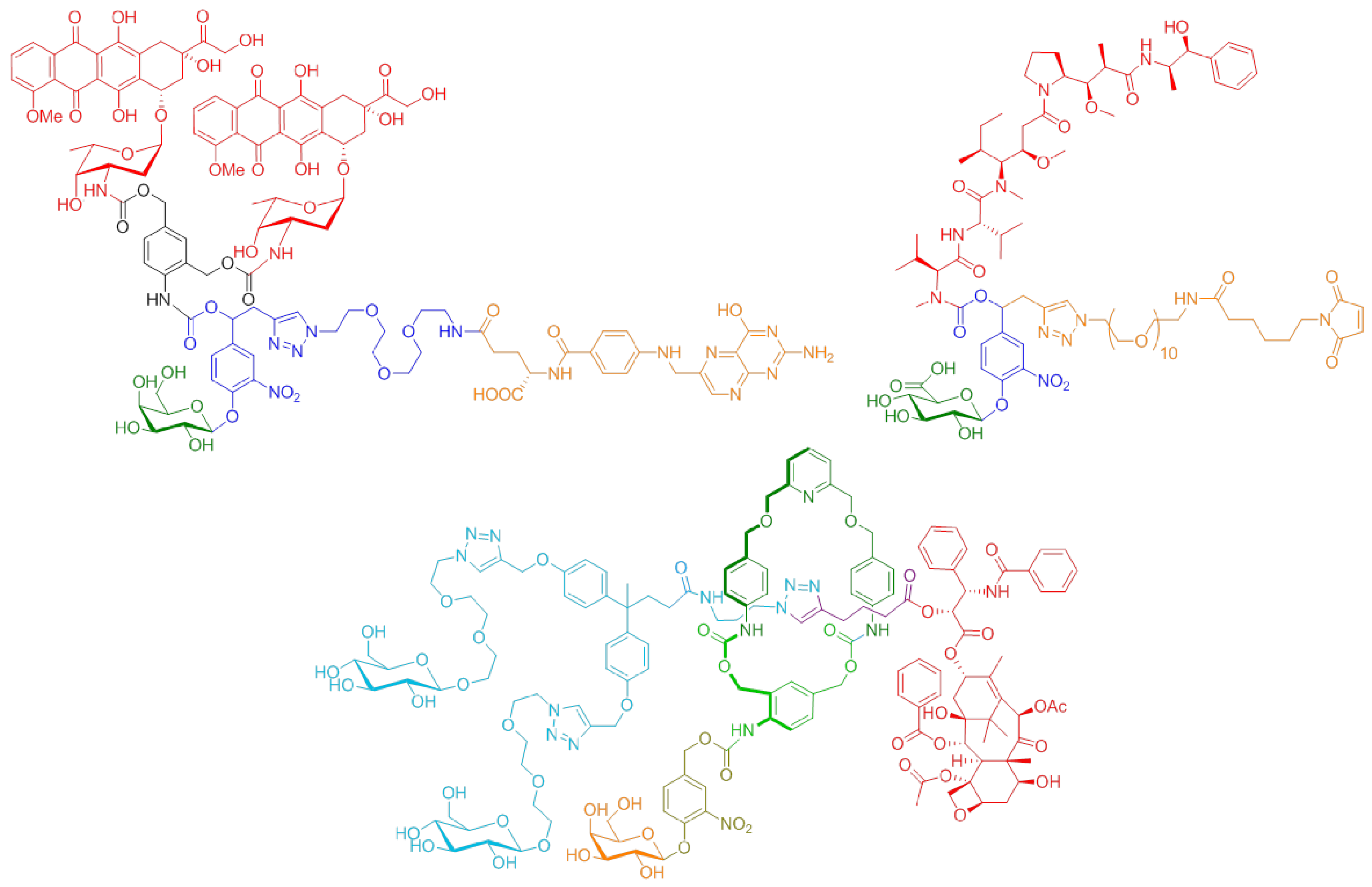
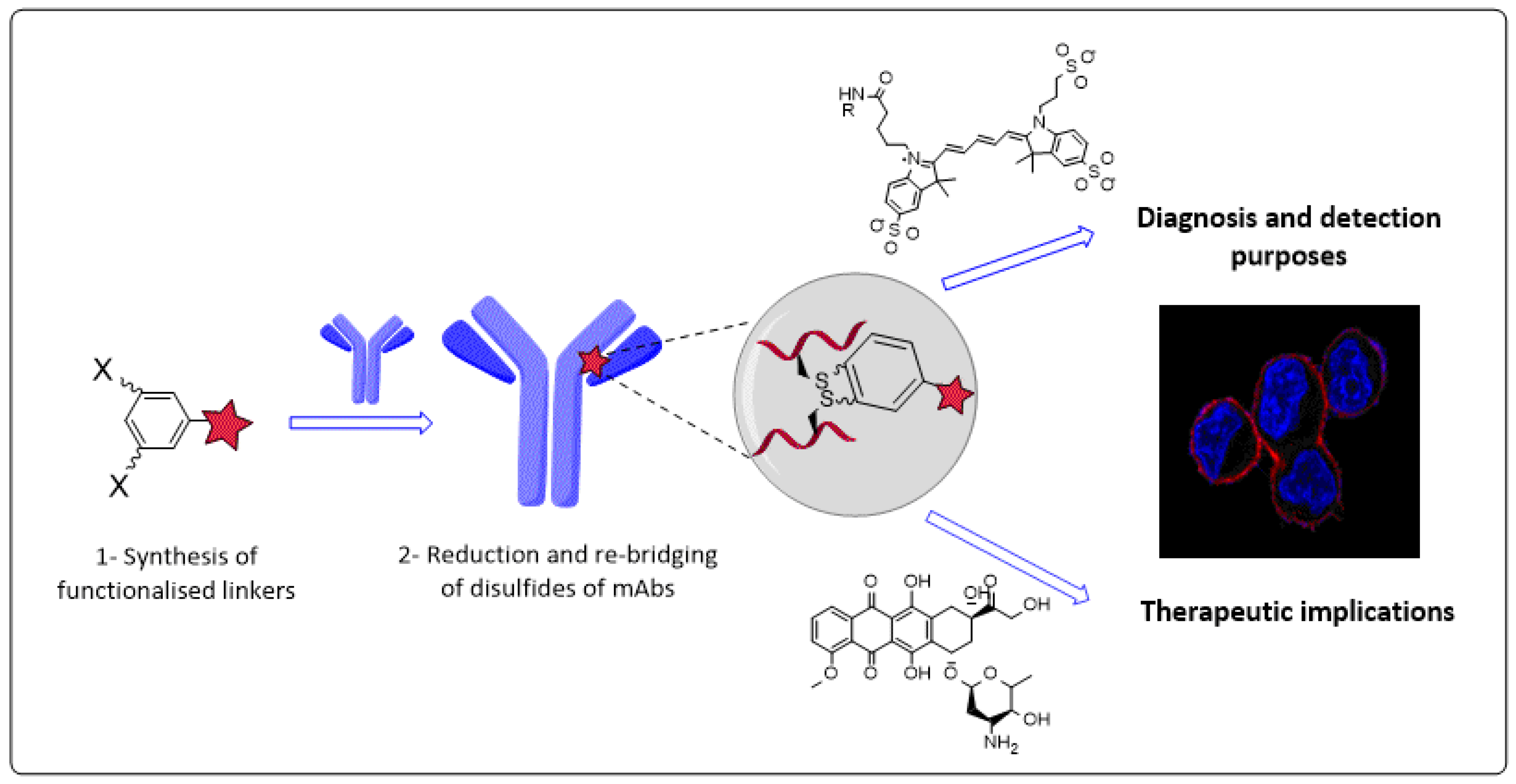
4.2. Novel Conjugation Approach to Attain Immunoconjugates of Monoclonal Antibodies (P02)
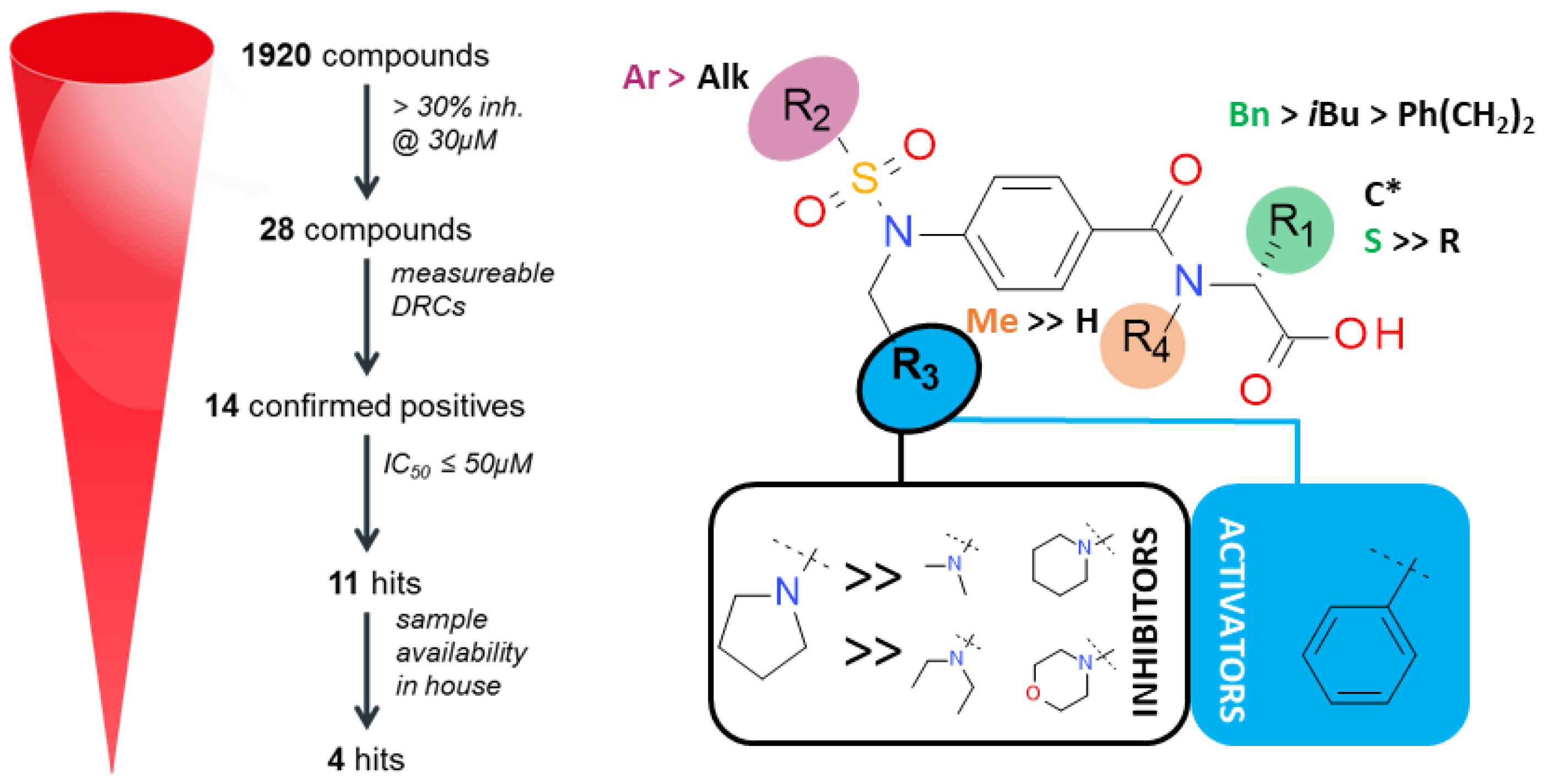
4.3. Inhibitors and Activators of Pyruvate Kinase Muscle Isozyme Splice Variant 2 (Pkm2): An Overview (P03)
4.4. Self-Assemblies of Azacitidine Prodrug: An Innovative Therapy against Myelodysplastic Syndromes (P04)
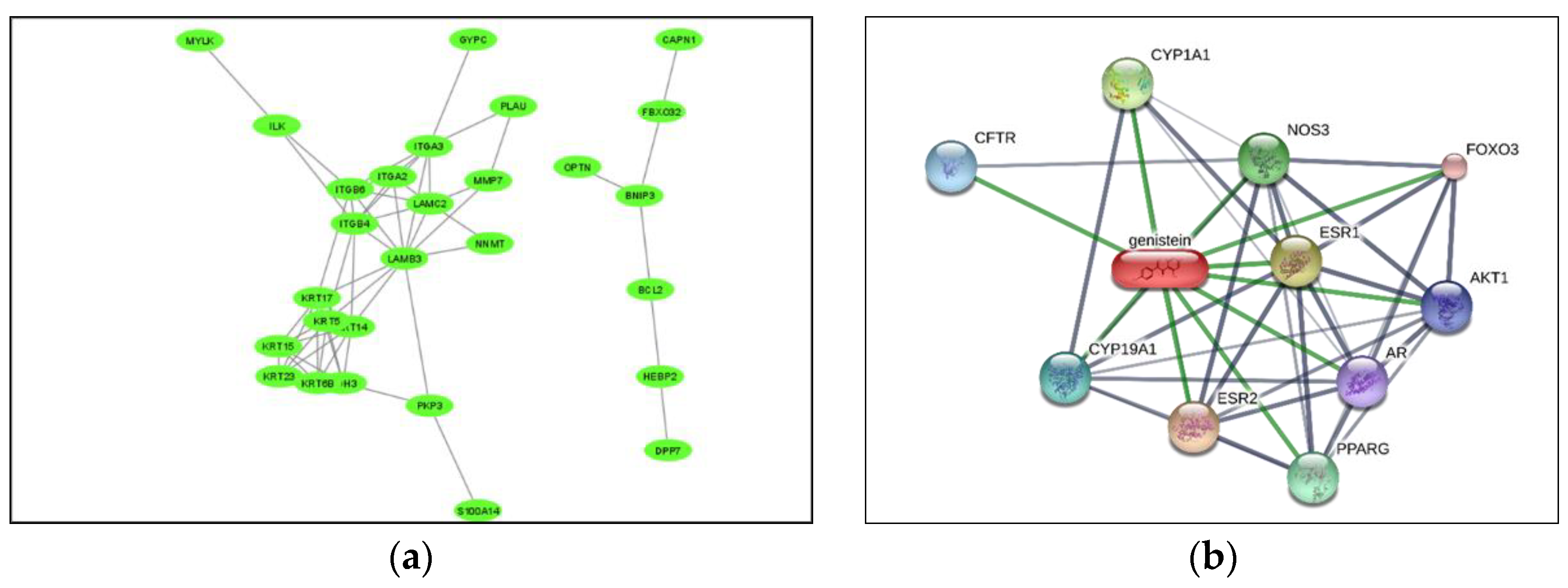
4.5. Genistein as Drug of Choice for Sars Cov2 Infection Using Drug-Gene Network (P05)
4.6. Design and Study of Potential FabZ Inhibitors as Antimicrobial Drugs (P06)
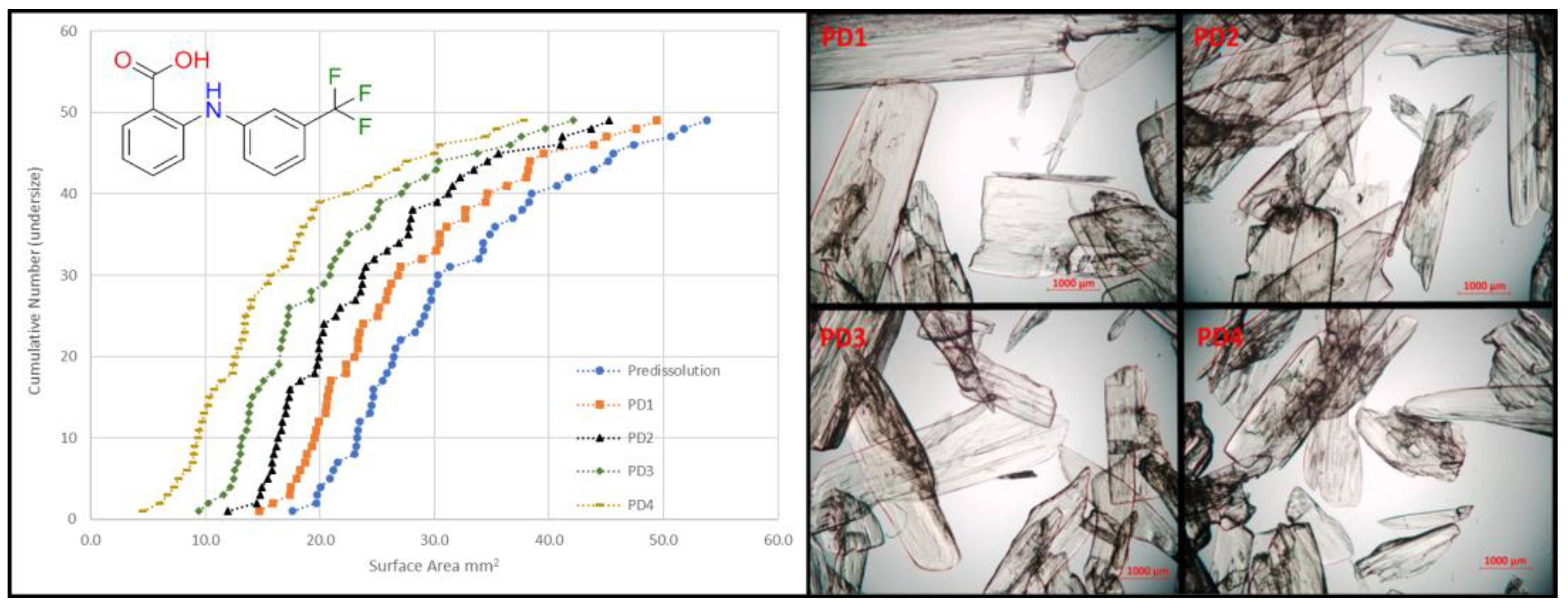
4.7. Determining Particle Impurity Distributions in Pharmaceutical Solids (P07)
4.8. The Synthesis of Enantiopure DSA Analogues for the Treatment of Cancer (P08)
4.9. Novel Synthesis of Pyrroloquinoxalines Using Sulfone Radicals (P09)
4.10. Synthesis of Potential Antitumor Agents Targeting DNA G-4 and Kinases (P10)
4.11. Novel Bioactive Benzimidazoles (P11)
4.12. Photocatalytic α-C–H Functionalization of Unprotected Primary Alkylamines (P12)
4.13. New Orthogonal Decoration of 4-Amino-pyrido[2,3-d]pyrimidin-7(8H)-ones (P13)
4.14. Tyrosine Selective Bioconjugation Using An Electro-Oxidative Methodology for Biomolecules Labeling (P14)
4.15. Modification of Carbon Surface to Enhance Removal of Cadmium from Aqueous Solution (P15)
4.16. From Innovation to the Market: Adding Value to the Compounds from Academic Research and Teaching (P16)
4.17. Antibiofilm Properties of Indolo[2,3-b]quinoline Derivatives (P17)
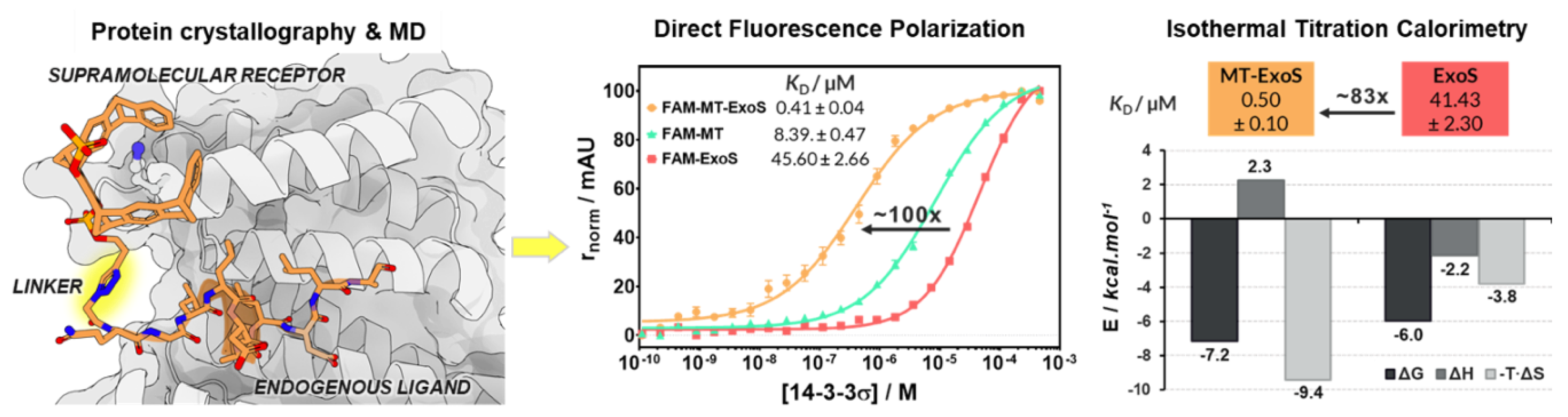
4.18. Supramolecular Enhancement of Natural 14–3-3 Protein Ligands (P18)
4.19. Taking Back Control of the Immune System (P19)
4.20. Chitosan-Based Electrospun for Wound-Healing Applications (P20)
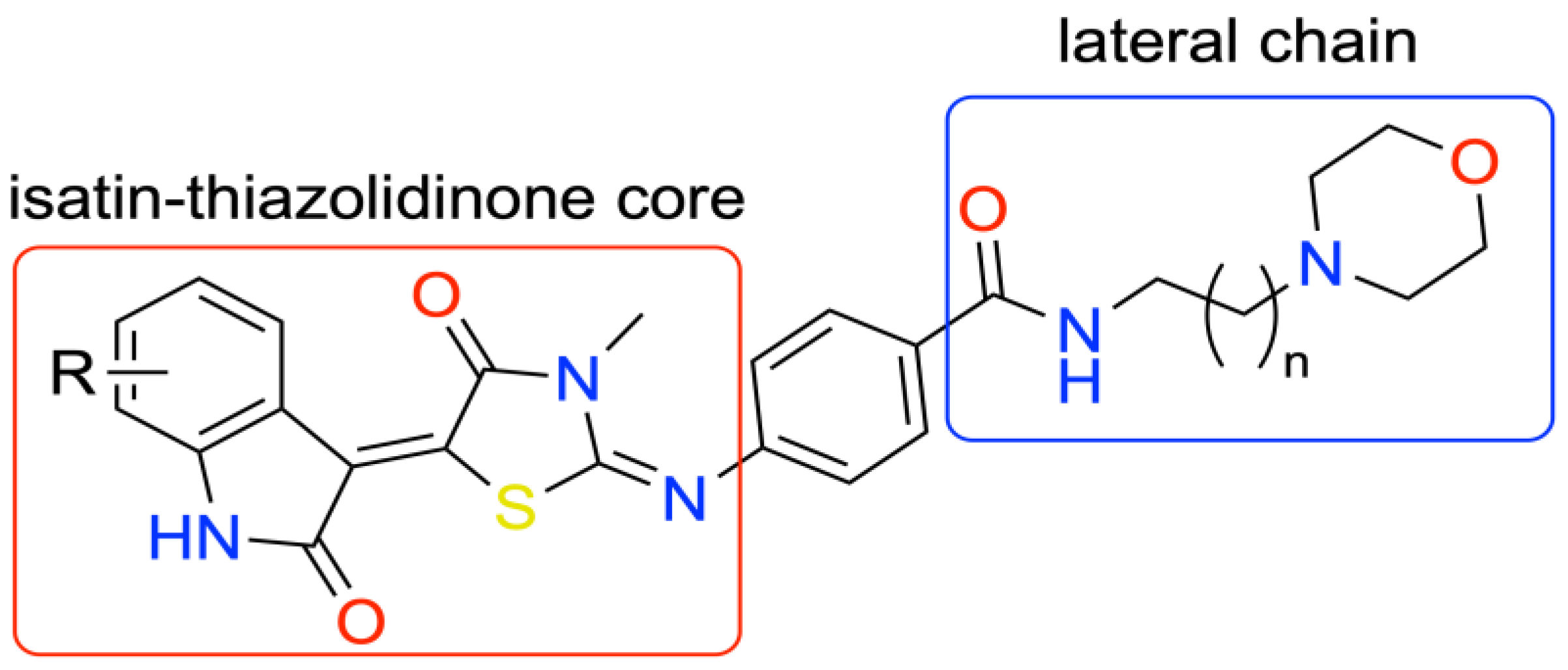
4.21. Design, Synthesis, Bioactivity, and Molecular Modelling Studies of Novel Heterocyclic Compounds with Antileishmanial Activity (P21)
4.22. Recombinant Synthesis of Human Trefoil Factor 2 Protein (P22)
4.23. The Royal Jelly Fatty Acid 10h2da Inhibits Migratory and Invasive Potential of Colorectal Cancer Cell Lines (P23)
4.24. Study of the Interactions of Caffeine-Derived Pt(II) and Pd(II) Complexes with Important Biomolecules (P34)
4.25. Synthesis of 1,4-Dihydropyrazolo[4,3-B]Indoles via Intramolecular C(Sp2)-N Bond Formation Involving Nitrene Insertion, Dft Study, and Their Anticancer Assessment (P25)
4.26. Synthesis of T20K Immunosuppressive Cyclotide (P26)
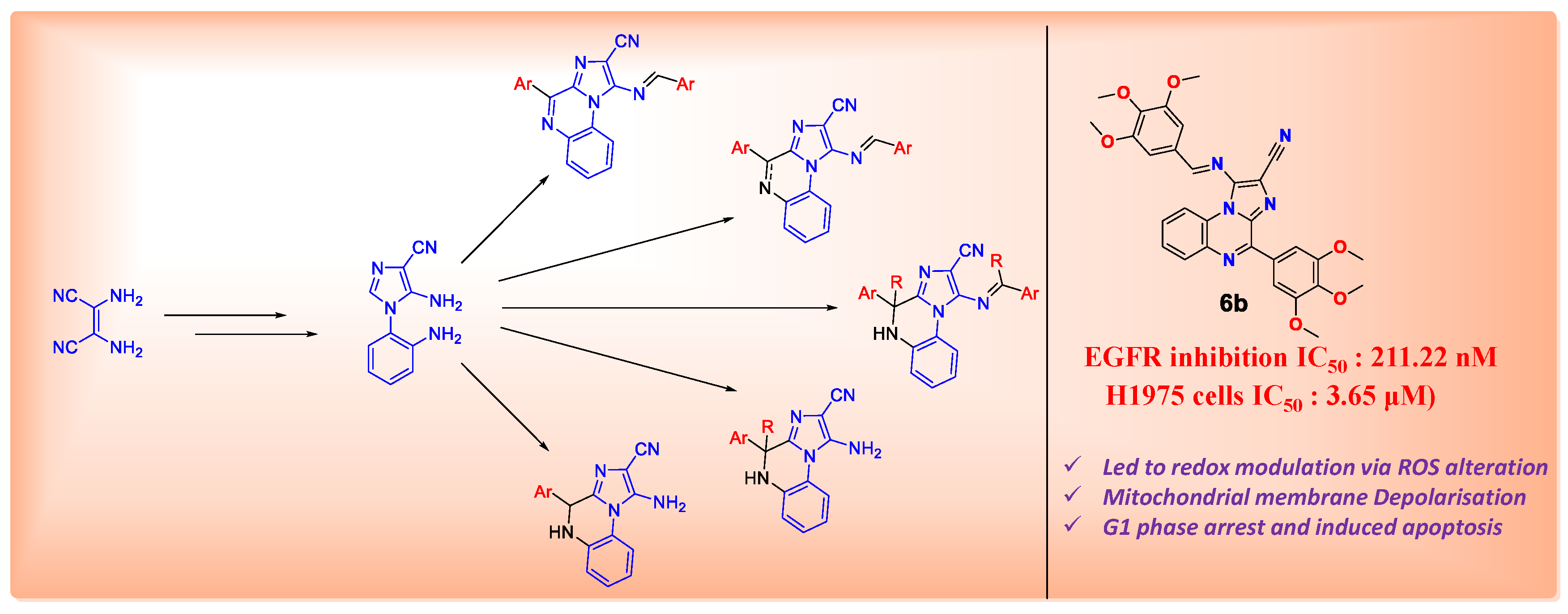
4.27. Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anticancer Assessment (P27)
4.28. Pyrazolones as Inhibitors of Immune Checkpoint Inhibitors Blocking Pd-1/Pd-L1 Interactions (P28)
4.29. Benzo[D]Thiazol-2(3h)-Ones as New Potent Selective Cb2 Agonists with Anti-Inflammatory Properties (P29)
4.30. Synthesis and Antipseudomonal Activities of New Iron Chelator–Ciprofloxacin Conjugates (P30)
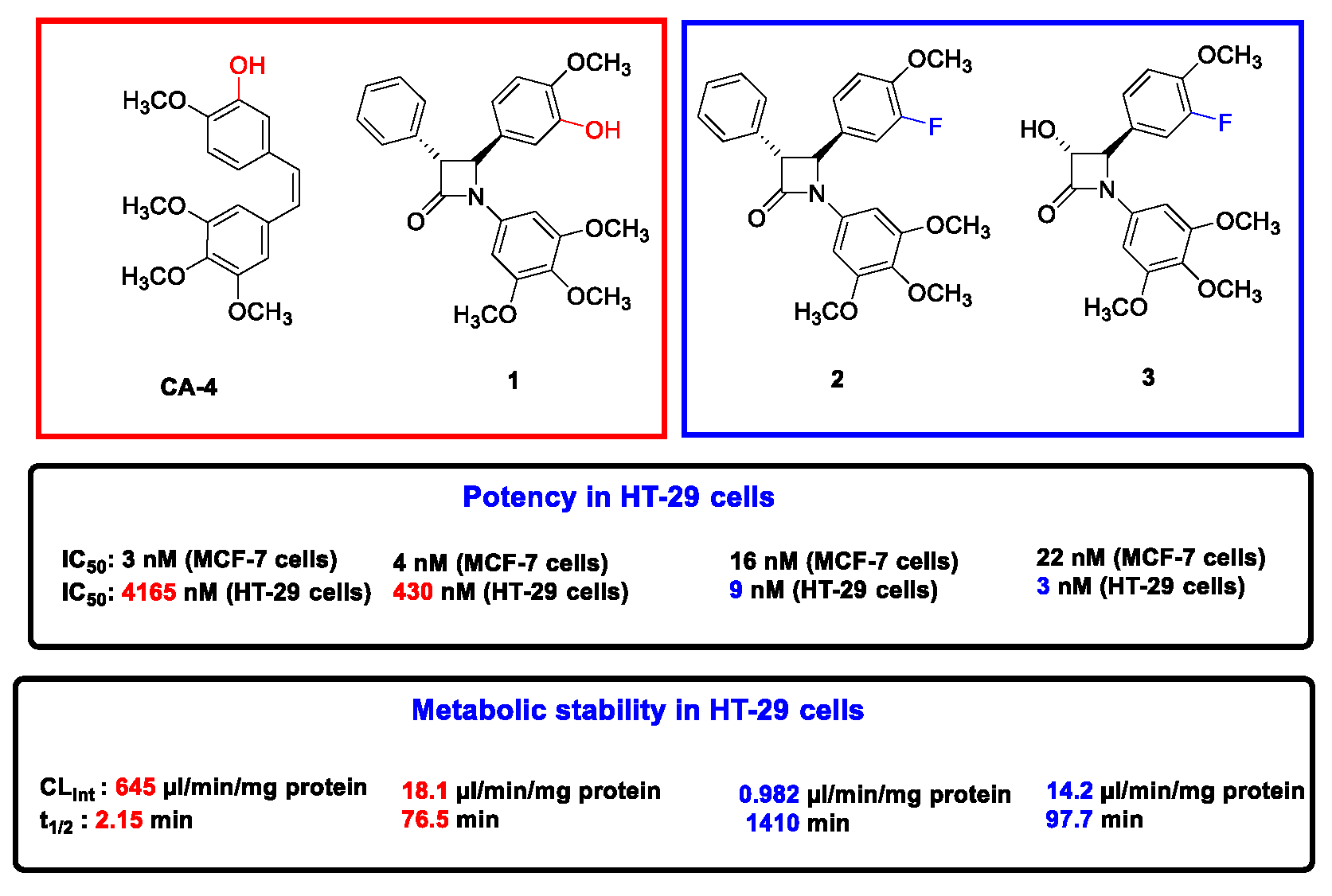
4.31. Investigation of CA-4 Metabolism and Related β-Lactam Analogues in Chemoresistant HT-29 Colon Cancer Cells (P31)
4.32. Using Metabolic Glycoengineering for Targeted Treatment of Cancer (P32)
4.33. Synthesis and Biological Evaluation of Covalent Inhibitors of Focal Adhesion Kinase against Human Malignant Glioblastoma (P33)
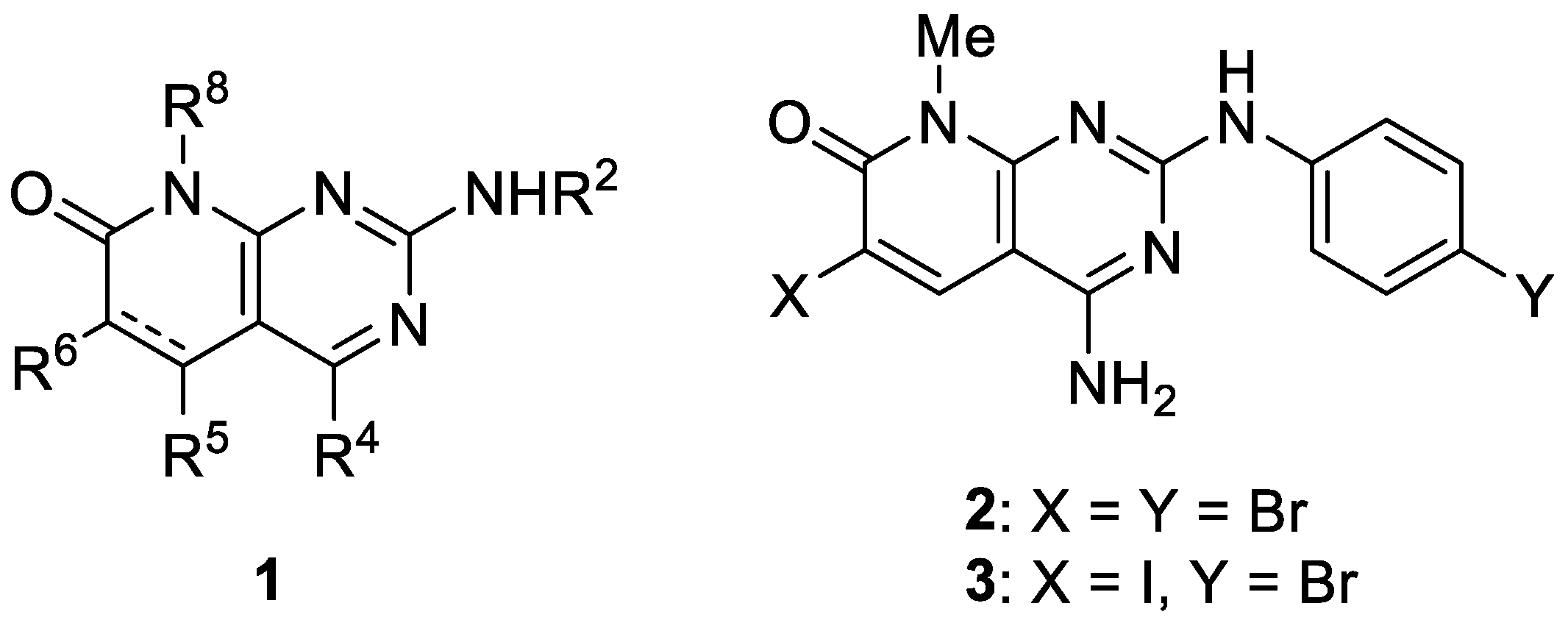
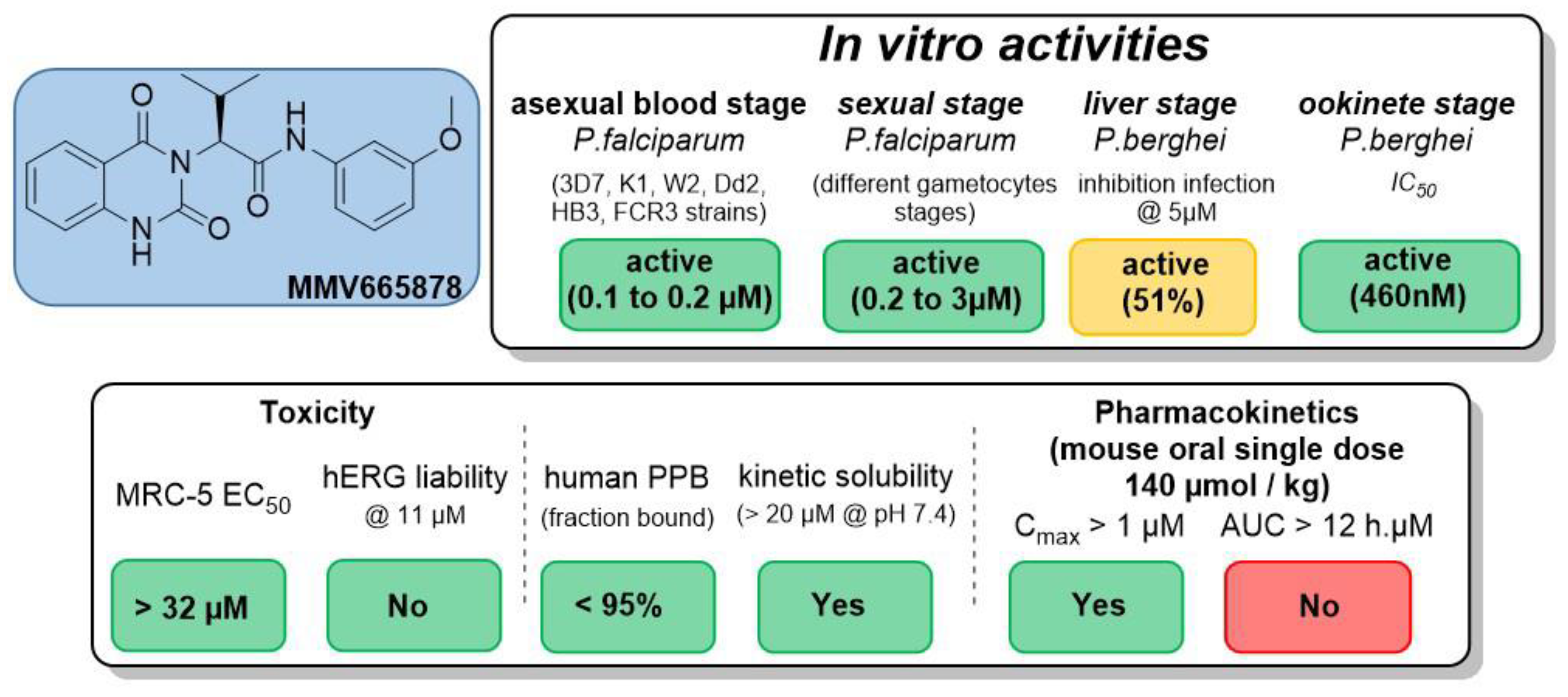
4.34. Optimization of a Novel Fast-Acting Transmission-Blocking Antimalarial Agent (P34)
4.35. Ruthenium (Ii) Octahedral Complexes Featuring Pyrimidine-Like Ligands: Cytotoxicity and DNA/Protein Interaction Studies (P35)
4.36. Samarium-Doped Anatase Tio2 Nanoparticles: Synthesis, Characterization, and Synergy between Sm Rare Earth Doping and Oxygen Vacancies (P36)
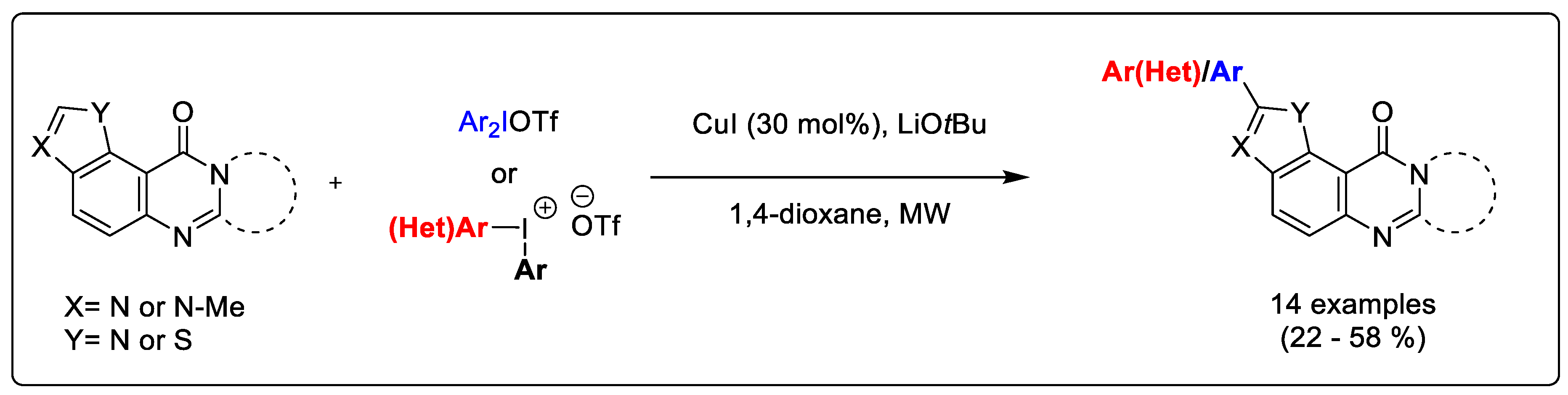
4.37. Copper-Catalyzed C-H Arylation of Fused Pyrimidinones Using Diaryliodoniums Salts under Microwave Irradiation (P37)
4.38. Sar Study of New Antikinetoplastid 3-Nitroimidazo[1,2-A]Pyridines (P38)
4.39. New Nitric-Oxide-Releasing Indomethacin Derivatives: Synthesis and Biological Evaluation (P39)
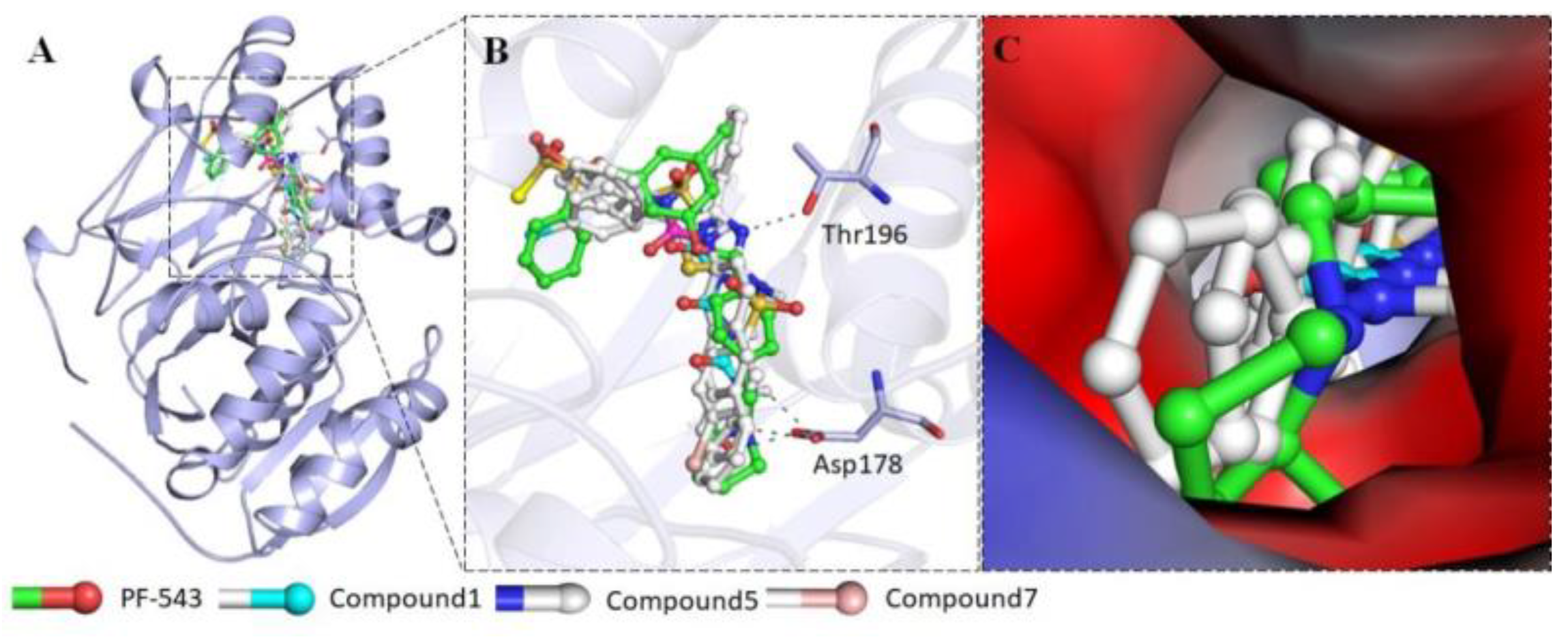
4.40. Fishing Potent Epac2 Inhibitors: An Interdisciplinary Approach (P40)
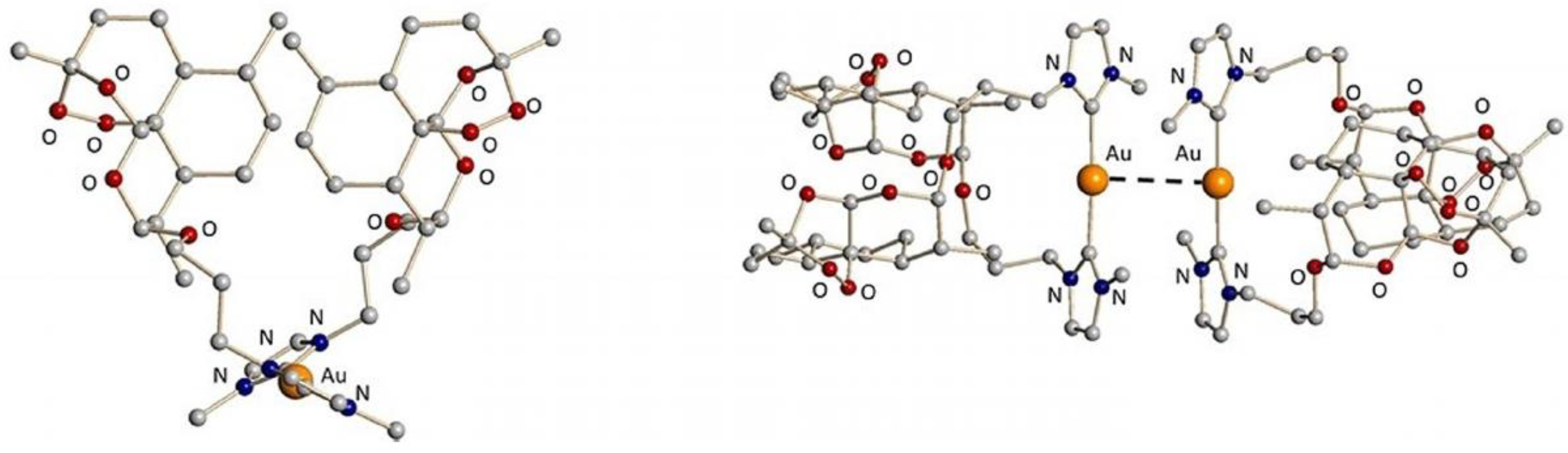
4.41. New Mononuclear Gold(Iii) Complexes—Study of the DNA/HSA/BSA-Binding Properties (P41)
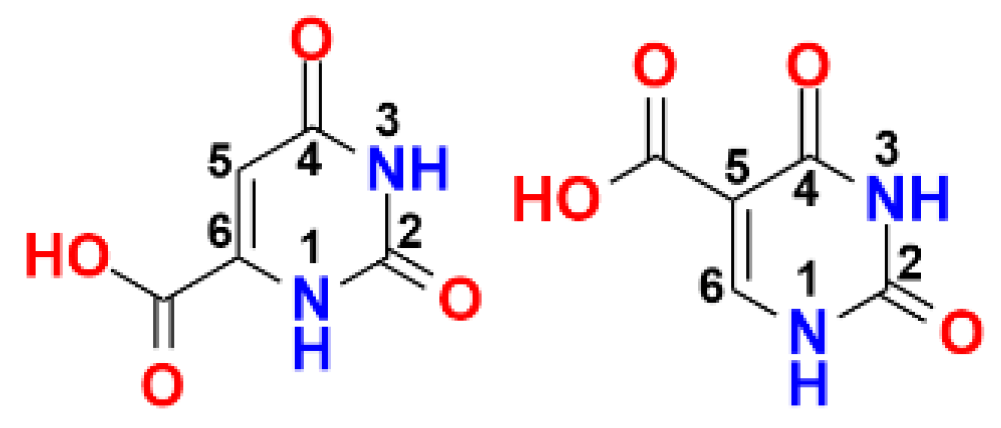
4.42. Aralkylpyrimidinetriones as Growth Inhibitors of Clostridioides Difficile (P42)
4.43. 1,3-. Dipolar Cycloaddition of Diazo Compounds to Activated Enynes (P43)
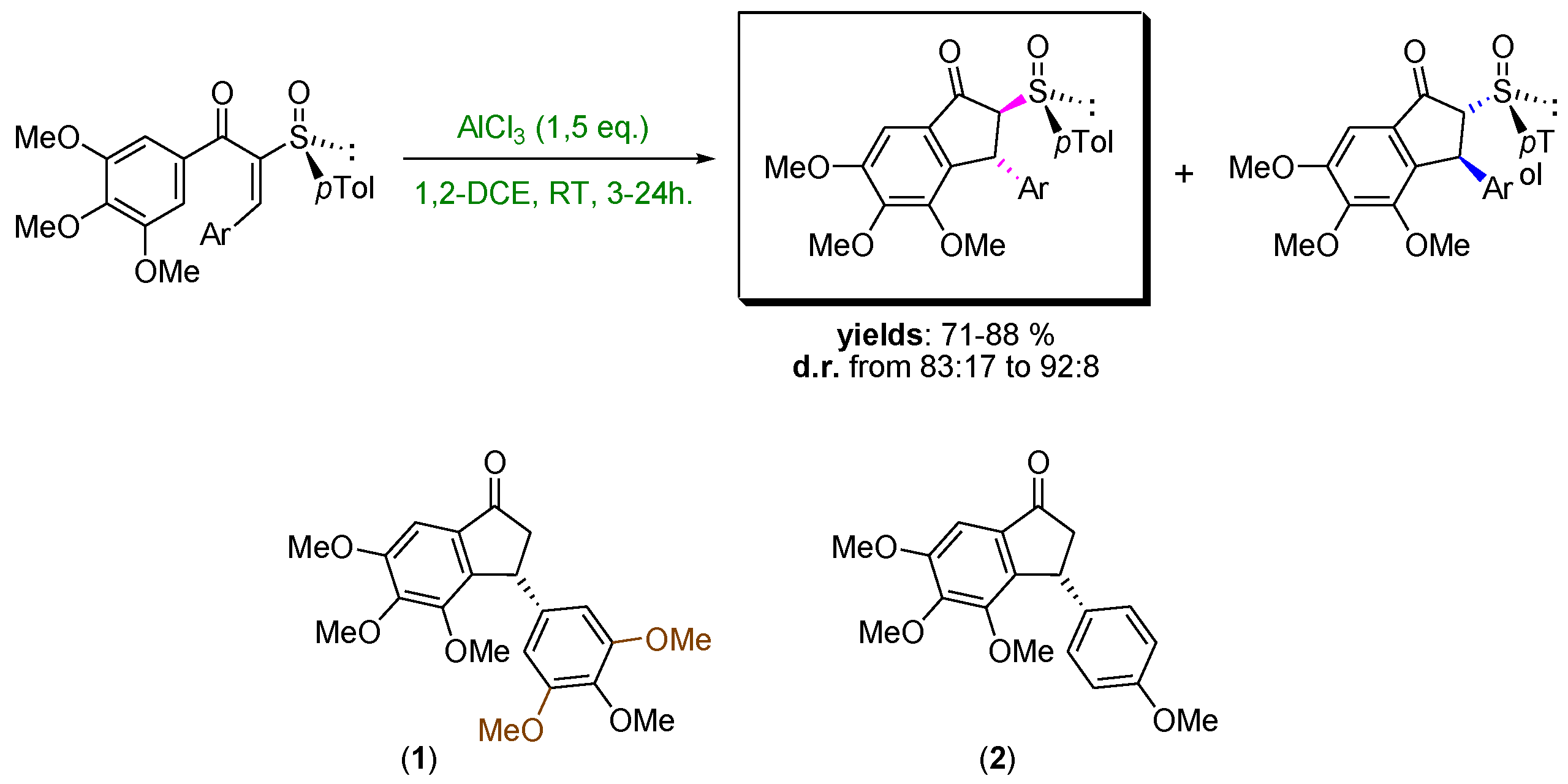
4.44. Torquoselective Nazarov Cyclization Mediated by a Chiral Sulfoxide (P44)
4.45. Design and Development of Novel Urea, Sulfonyltriurea, and Sulfonamide Derivatives as Potential Inhibitors of Sphingosine Kinase 1 (P45)
4.46. Development of Chromone Carboxamides as Quorum-Sensing Inhibitors for the Treatment of Cf-Related Multi-Species Biofilms (P46)
4.47. Removing Cancer’s Immortality: Targeting Telomerase (P47)
4.48. An Artemisinin-Derivative–(Nhc) Gold(I) Hybrid with Enhanced Cytotoxicity through Inhibition of Nrf2 Transcriptional Activity (P48)
4.49. New Approaches to the Synthesis of Pyoverdine D (P49)
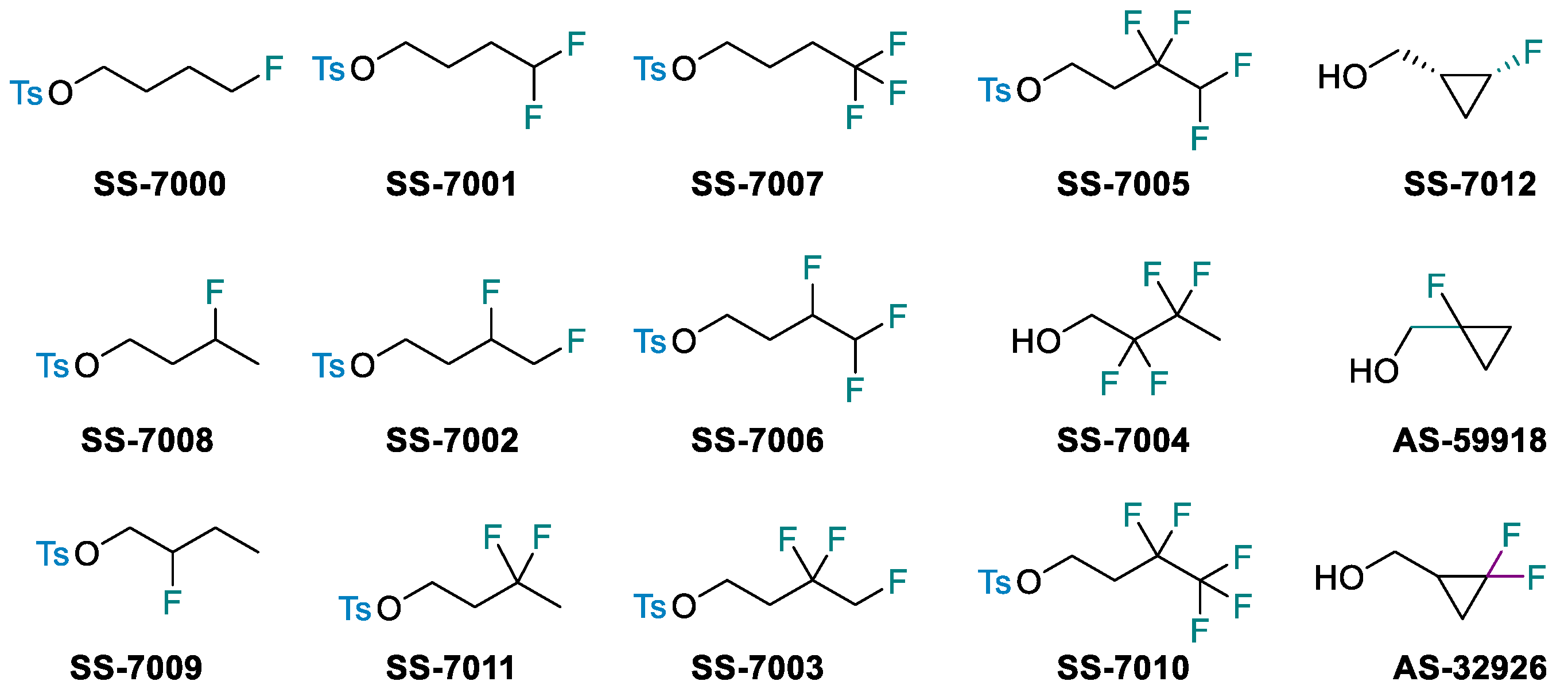
4.50. Commercialization of Small Fluorinated Hydrophobic Groups for Lipophilicity Tuning in Drug Development (P50)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lange, J.; Eddhif, B.; Tarighi, M.; Garandeau, T.; Péraudeau, E.; Clarhaut, J.; Renoux, B.; Papot, S.; Poinot, P. Volatile organic compound based probe for induced volatolomics of cancers. Angew. Chem. Int. Ed. 2019, 58, 17563–17566. [Google Scholar] [CrossRef]
- Porte, K.; Renoux, B.; Péraudeau, E.; Clarhaut, J.; Eddhif, B.; Poinot, P.; Gravel, E.; Doris, E.; Wijkhuisen, A.; Audisio, D.; et al. Controlled release of a micelle payload via sequential enzymatic and bioorthogonal reactions in living systems. Angew. Chem. Int. Ed. 2019, 58, 6366–6370. [Google Scholar] [CrossRef]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse polysarcosine-based highly-loaded antibody-drug conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef]
- Renoux, B.; Raes, F.; Legigan, T.; Péraudeau, E.; Eddhif, B.; Poinot, P.; Tranoy-Opalinski, I.; Alsarraf, J.; Koniev, O.; Kolodych, S.; et al. Targeting the tumour microenvironment with an enzyme-responsive drug delivery system for the efficient therapy of breast and pancreatic cancers. Chem. Sci. 2017, 8, 3427–3433. [Google Scholar] [CrossRef]
- Barat, R.; Legigan, T.; Tranoy-Opalinski, I.; Renoux, B.; Péraudeau, E.; Clarhaut, J.; Poinot, P.; Fernandes, A.E.; Aucagne, V.; Leigh, D.A.; et al. A mechanically interlocked molecular system programmed for the delivery of an anticancer drug. Chem. Sci. 2015, 6, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Alsarraf, J.; Péraudeau, E.; Poinot, P.; Tranoy-Opalinski, I.; Clarhaut, J.; Renoux, B.; Papot, S. A dendritic β-galactosidase-responsive folate–monomethylauristatin E conjugate. Chem. Commun. 2015, 51, 15792–15795. [Google Scholar] [CrossRef]
- Legigan, T.; Clarhaut, J.; Tranoy-Opalinski, I.; Monvoisin, A.; Renoux, B.; Thomas, M.; Le Pape, A.; Lerondel, S.; Papot, S. The first generation of β-galactosidase-responsive prodrugs designed for the selective treatment of solid tumors in prodrug monotherapy. Angew. Chem. Int. Ed. 2012, 51, 11606–11610. [Google Scholar] [CrossRef] [PubMed]
- Legigan, T.; Clarhaut, J.; Renoux, B.; Tranoy-Opalinski, I.; Monvoisin, A.; Berjeaud, J.M.; Guilhot, F.; Papot, S. Synthesis and antitumor efficacy of a β-glucuronidase-responsive albumin-binding prodrug of doxorubicin. J. Med. Chem. 2012, 55, 4516–4520. [Google Scholar] [CrossRef]
- Fernandes, A.; Viterisi, A.; Coutrot, F.; Potok, S.; Leigh, D.A.; Aucagne, V.; Papot, S. Rotaxane-based propeptides: Protection and enzymatic release of a bioactive pentapeptide. Angew. Chem. Int. Ed. 2009, 48, 6443–6447. [Google Scholar] [CrossRef]
- Lemasson, E.; Bertin, S.; Hennig, P.; Lesellier, E.; West, C. Comparison of ultra-high performance methods in liquid and supercritical fluid chromatography coupled to electrospray ionization-mass spectrometry for impurity profiling of drug candidates. J. Sep. Sci. 2016, 39, 212–233. [Google Scholar] [CrossRef]
- West, C. Current trends in supercritical fluid chromatography. Anal. Bioanal. Chem. 2018, 410, 6441–6457. [Google Scholar] [CrossRef]
- Noireau, A.; Lemasson, E.; Mauge, F.; Petit, A.M.; Bertin, S.; Hennig, P.; Lesellier, E.; West, C. Purification of drug degradation products supported by analytical and preparative supercritical fluid chromatography. J. Pharm. Biomed. Anal. 2019, 170, 40–47. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-loaded polymeric nanoparticles for cancer stem cell targeting. Front. Pharmacol. 2017, 8, 51. [Google Scholar] [CrossRef]
- Davies, S.G.; Kennewell, P.D.; Russell, A.J.; Seden, P.T.; Westwood, R.; Wynne, G.M. Stemistry: The control of stem cells in situ using chemistry. J. Med. Chem. 2015, 58, 2863–2894. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.V.; Perkins, K.J.; Dugdale, H.; Moir, L.; Vuorinen, A.; Chatzopoulou, M.; Squire, S.E.; Monecke, S.; Lomow, A.; Geese, M.; et al. Chemical proteomics and phenotypic profiling identifies the aryl hydrocarbon receptor as a molecular target of the utrophin modulator ezutromid. Angew. Chem. Int. Ed. 2020, 59, 2420–2428. [Google Scholar] [CrossRef]
- Babbs, A.; Berg, A.; Chatzopoulou, M.; Davies, K.E.; Davies, S.G.; Edwards, B.; Elsey, D.J.; Emer, E.; Guiraud, S.; Harriman, S.; et al. 2-Arylbenzo[d]oxazole phosphinate esters as second-generation modulators of utrophin for the treatment of Duchenne Muscular Dystrophy. J. Med. Chem. 2020, 63, 7880–7891. [Google Scholar] [CrossRef]
- Minard, A.; Bauer, C.C.; Wright, D.J.; Rubaiy, H.N.; Muraki, K.; Beech, D.J.; Bon, R.S. Remarkable progress with small-molecule modulation of TRPC1/4/5 channels: Implications for understanding the channels in health and disease. Cells 2018, 7, 52. [Google Scholar] [CrossRef]
- Minard, A.; Bauer, C.C.; Chuntharpursat-Bon, E.; Pickles, I.B.; Wright, D.J.; Ludlow, M.L.; Burnham, M.P.; Warriner, S.L.; Beech, D.J.; Muraki, K.; et al. Potent, selective, and subunit-dependent activation of TRPC5 channels by a xanthine derivative. Brit. J. Pharmacol. 2019, 176, 3924–3938. [Google Scholar] [CrossRef] [PubMed]
- Faïon, L.; Djaout, K.; Frita, R.; Pintiala, C.; Cantrelle, F.X.; Moune, M.; Vandeputte, A.; Bourbiaux, K.; Piveteau, C.; Herledan, A.; et al. Discovery of the first Mycobacterium tuberculosis MabA (FabG1) inhibitors through a fragment-based screening. Eur. J. Med. Chem. 2020, 200, 112440. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, B.; Machelart, A.; Tran, N.C.; Flipo, M.; Moune, M.; Leroux, F.; Piveteau, C.; Wohlkönig, A.; Wintjens, R.; Li, X.; et al. Fragment-based optimized EthR inhibitors with in vivo ethionamide boosting activity. ACS Infect. Dis. 2020, 6, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Dhar, N.; Dubée, V.; Ballell, L.; Cuinet, G.; Hugonnet, J.E.; Signorino-Gelo, F.; Barros, D.; Arthur, M.; McKinney, J.D. Rapid cytolysis of Mycobacterium tuberculosis by Faropenem, an orally bioavailable β-lactam antibiotic. Antimicrob. Agents Chemother. 2015, 59, 1308–1319. [Google Scholar] [CrossRef]
- Diacon, A.H.; van der Merwe, L.; Barnard, M.; von Groote-Bidlingmaier, F.; García-Basteiro, A.L.; Sevene, E.; Ballell, L.; Barros-Aguirre, D. β-lactams against tuberculosis--new trick for an old dog? N. Engl. J. Med. 2016, 375, 393–394. [Google Scholar] [CrossRef]
- Ngadjeua, F.; Braud, E.; Saidjalolov, S.; Iannazzo, L.; Schnappinger, D.; Ehrt, S.; Hugonnet, J.E.; Mengin-Lecreulx, D.; Patin, D.; Ethève-Quelquejeu, M.; et al. Critical impact of peptidoglycan precursor amidation on the activity of l,d-transpeptidases from Enterococcus faecium and Mycobacterium tuberculosis. Chem. Eur. J. 2018, 24, 5743–5747. [Google Scholar] [CrossRef]
- Heinemann, F.W.; Karges, J.; Gasser, G. Critical overview of the use of Ru(II) polypyridyl complexes as photosensitizers in one-photon and two-photon photodynamic therapy. Acc. Chem. Res. 2017, 50, 2727–2736. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Notaro, A.; Gasser, G. Monomeric and dimeric coordinatively saturated and substitutionally inert Ru(II) polypyridyl complexes as anticancer drug candidates. Chem. Soc. Rev. 2017, 46, 7317–7337. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Zarschler, K.; Pietzsch, H.J.; Stephan, H.; Gasser, G. New insights into the pretargeting approach to image and treat tumours. Chem. Soc. Rev. 2016, 45, 6415–6431. [Google Scholar] [CrossRef]
- Ong, Y.C.; Roy, S.; Andrews, P.C.; Gasser, G. Metal compounds against neglected tropical diseases. Chem. Rev. 2019, 119, 730–7396. [Google Scholar] [CrossRef]
- Brandt, M.; Cardinale, J.; Aulsebrook, M.L.; Gasser, G.; Mindt, T.L. An overview of PET radiochemistry, part 2: Radiometals. J. Nucl. Med. 2018, 59, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, L.; Monticelli, S.; Senatore, R.; Ielo, L.; Pace, V. Homologation chemistry with nucleophilic α-substituted organometallic reagents: Chemocontrol, new concepts and (solved) challenges. Chem. Comm. 2018, 54, 6692–6704. [Google Scholar] [CrossRef]
- Senatore, R.; Castoldi, L.; Ielo, L.; Holzer, W.; Pace, V. Expeditious and chemoselective synthesis of α-aryl and α-alkyl selenomethylketones via homologation chemistry. Org. Lett. 2018, 20, 2685–2688. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Castoldi, L.; Mazzeo, E.; Rui, M.; Langer, T.; Holzer, W. Efficient access to all-carbon quaternary and tertiary α-functionalized homoallyl-type aldehydes from ketones. Angew. Chem. Int. Ed. 2017, 56, 12677–12682. [Google Scholar] [CrossRef] [PubMed]
- Ielo, L.; Touqeer, S.; Roller, A.; Langer, T.; Holzer, W.; Pace, V. Telescoped, divergent, chemoselective C1 and C1-C1 homologation of imine surrogates: Access to quaternary chloro- and halomethyl-trifluoromethyl aziridines. Angew. Chem. Int. Ed. 2019, 58, 2479–2484. [Google Scholar] [CrossRef] [PubMed]
- Parisi, G.; Colella, M.; Monticelli, S.; Romanazzi, G.; Holzer, W.; Langer, T.; Degennaro, L.; Pace, V. Exploiting a “beast” in carbenoid chemistry: Development of a straightforward direct nucleophilic fluoromethylation strategy. J. Am. Chem. Soc. 2017, 139, 13648–13651. [Google Scholar] [CrossRef]
- Monticelli, S.; Colella, M.; Pillari, V.; Tota, A.; Langer, T.; Holzer, W.; Degennaro, L.; Renzo, L.; Pace, V. Modular and chemoselective strategy for the direct access to α-fluoroepoxides and aziridines via the addition of fluoroiodomethyllithium to carbonyl-like compounds. Org. Lett. 2019, 21, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Senatore, R.; Malik, M.; Spreitzer, W.; Holzer, W.; Pace, V. Direct and chemoselective electrophilic monofluoromethylation of heteroatoms (O-, S-, N-, P-, Se-) with fluoroiodomethane. Org. Lett. 2020, 22, 1345–1349. [Google Scholar] [CrossRef]
- Miele, M.; D’Orsi, R.; Sridharan, V.; Holzer, W.; Pace, V. Highly chemoselective difluoromethylative homologation of iso(thio)cyanates: Expeditious access to unprecedented α,α-difluoro(thio)amides. Chem. Comm. 2019, 55, 12960–12963. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and chemoselective synthesis of tertiary difluoroketones via weinreb amide homologation with a CHF2-carbene equivalent. Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Neurotrophin receptors in the pathogenesis, diagnosis and therapy of neurodegenerative diseases. Pharmacol. Res. 2017, 121, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.L.; Comaposada-Baró, R.; Vilar, M. Neurotrophins and neurotrophin receptors. In Hormonal Signaling in Biology and Medicine, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 83–106. [Google Scholar]
- Kozono, N.; Ohtani, A.; Shiga, T. Roles of the serotonin 5-HT4 receptor in dendrite formation of the rat hippocampal neurons in vitro. Brain Res. 2017, 1655, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef]
- Pascual-Brazo, J.; Castro, E.; Díaz, Á.; Valdizán, E.M.; Pilar-Cuéllar, F.; Vidal, R.; Treceño, B.; Pazos, Á. Modulation of neuroplasticity pathways and antidepressant-like behavioural responses following the short-term (3 and 7 days) administration of the 5-HT4 receptor agonist RS67333. Int. J. Neuropsychopharmacol. 2012, 15, 631–643. [Google Scholar] [CrossRef]
- Liao, J.J.L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef]
- Bansal, D.; Badhan, Y.; Gudala, K.; Schifano, F. Ruboxistaurin for the treatment of diabetic peripheral neuropathy: A systematic review of randomized clinical trials. Diabetes Metab. J. 2013, 37, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Winfield, H.J.; Cahill, M.M.; O’Shea, K.D.; Pierce, L.T.; Robert, T.; Ruchaud, S.; Bach, S.; Marchand, P.; McCarthy, F.O. Synthesis and anticancer activity of novel bisindolylhydroxymaleimide derivatives with potent GSK-3 kinase inhibition. Bioorg. Med. Chem. 2018, 26, 4209–4224. [Google Scholar] [CrossRef] [PubMed]
- WHO, (World Health Organization). World Malaria Report; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Ouji, M.; Augereau, J.M.; Paloque, L.; Benoit-Vical, F. Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24. [Google Scholar] [CrossRef]
- Cohen, A.; Suzanne, P.; Lancelot, J.C.; Verhaeghe, P.; Lesnard, A.; Basmaciyan, L.; Hutter, S.; Laget, M.; Dumètre, A.; Paloque, L.; et al. Discovery of new thienopyrimidinone derivatives displaying antimalarial properties toward both erythrocytic and hepatic stages of Plasmodium. Eur. J. Med. Chem. 2015, 95, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive oxygen species and neutrophil function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Soubhye, J.; Aldib, I.; Delporte, C.; Prévost, M.; Dufrasne, F.; Van Antwerpen, P. Myeloperoxidase as a target for the treatment of inflammatory syndromes: Mechanisms and structure activity relationships of inhibitors. Curr. Med. Chem. 2016, 23, 3975–4008. [Google Scholar] [CrossRef]
- Li, Y.; Ganesh, T.; Diebold, B.A.; Zhu, Y.; McCoy, J.W.; Smith, S.M.E.; Sun, A.; Lambeth, J.D. Thioxo-dihydroquinazolin-one compounds as novel inhibitors of myeloperoxidase. ACS Med. Chem. Lett. 2015, 6, 1047–1052. [Google Scholar] [CrossRef]
- Ward, J.; Spath, S.N.; Pabst, B.; Carpino, P.A.; Ruggeri, R.B.; Xing, G.; Speers, A.E.; Cravatt, B.F.; Ahn, K. Mechanistic characterization of a 2-thioxanthine myeloperoxidase inhibitor and selectivity assessment utilizing click chemistry–activity-based protein profiling. Biochemistry. 2013, 52, 9187–9201. [Google Scholar] [CrossRef]
- Soubhye, J.; Prévost, M.; Van Antwerpen, P.; Boudjeltia, K.Z.; Rousseau, A.; Furtmüller, P.G.; Obinger, C.; Vanhaeverbeek, M.; Ducobu, J.; Néve, J.; et al. Structure-based design, synthesis, and pharmacological evaluation of 3-(aminoalkyl)-5-fluoroindoles as myeloperoxidase inhibitors. J. Med. Chem. 2010, 53, 8747–8759. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Ott, S.; Farber, K.M.; Palazzo, T.A.; Conrad, W.E.; Haddadin, M.J.; Tantillo, D.J.; Cross, C.E.; Eiserich, J.P.; Kurth, M.J. Inhibition of myeloperoxidase: Evaluation of 2H-indazoles and 1H-indazolones. Bioorg. Med. Chem. 2014, 22, 6422–6429. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Singh, S.B.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef]
- Malebari, A.M.; Fayne, D.; Nathwani, S.M.; O’Connell, F.; Noorani, S.; Twamley, B.; O’Boyle, N.M.; O’Sullivan, J.; Zisterer, D.M.; Meegan, M.J. β-lactams with antiproliferative and antiapoptotic activity in breast and chemoresistant colon cancer cells. Eur. J. Med. Chem. 2020, 189, 112050. [Google Scholar] [CrossRef] [PubMed]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. β-Lactam analogues of combretastatin A-4 prevent metabolic inactivation by glucuronidation in chemoresistant HT-29 colon cancer cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef]
- Tewari, K.M.; Eggleston, I.M. Chemical approaches for the enhancement of 5-aminolevulinic acid-based photodynamic therapy and photodiagnosis. Photochem. Photobiol. Sci. 2018, 17, 1553–1572. [Google Scholar] [CrossRef]
- Giuntini, F.; Bourré, L.; MacRobert, A.J.; Wilson, M.; Eggleston, I.M. Improved peptide prodrugs of 5-ALA for PDT: Rationalization of cellular accumulation and protoporphyrin IX production by direct determination of cellular prodrug uptake and prodrug metabolization. J. Med. Chem. 2009, 52, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.M.; Dondi, R.; Yaghini, E.; Pourzand, C.; MacRobert, A.J.; Eggleston, I.M. Peptide-targeted dendrimeric prodrugs of 5-aminolevulinic acid: A novel approach towards enhanced accumulation of protoporphyrin IX for photodynamic therapy. Bioorg. Chem. 2021, 109, 104667. [Google Scholar] [CrossRef]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef]
- Alvarez, R.; Gajate, C.; Puebla, P.; Mollinedo, F.; Medarde, M.; Peláez, R. Substitution at the indole 3 position yields highly potent indolecombretastatins against human tumor cells. Eur. J. Med. Chem. 2018, 158, 167–183. [Google Scholar] [CrossRef]
- Haroon, N.; Inman, R.D. Endoplasmic reticulum aminopeptidases: Biology and pathogenic potential. Nat. Rev. Rheumatol. 2010, 6, 461–467. Nat. Rev. Rheumatol. 2010, 6, 461–467. [Google Scholar] [CrossRef]
- Stratikos, E.; Stamogiannos, A.; Zervoudi, E.; Fruci, D. A role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 2014, 4, 363. [Google Scholar] [CrossRef] [PubMed]
- Medve, L.; Gealageas, R.; Vy, L.B.; Valentin, G.; Castillo-Aguilera, O.; Piveteau, C.; Rosell, M.; Fleau, C.; Warenghem, S.; Charton, J.; et al. Modulators of hERAP2 discovered by hight-throughput screening. Eur. J. Med. Chem. 2021, 5, 113053. [Google Scholar] [CrossRef] [PubMed]
- Achutha, A.S.; Pushpa, V.L.; Suchitra, S. Theoretical insights into the anti-SARS-CoV-2 activity of chloroquine and its analogs and in silico screening of main protease inhibitors. J. Proteome Res. 2020, 19, 4706–4717. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Mishra, S.P.; Pandey, U. Pyruvate kinase M2 and cancer: The role of PKM2 in promoting tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Rathod, B.; Chak, S.; Patel, S.; Shard, A. Tumor pyruvate kinase M2 modulators: A comprehensive account of activators and inhibitors as anticancer agents. RSC Med. Chem. 2021, 12, 1121–1141. [Google Scholar] [CrossRef]
- Issa, J.P.J. The myelodysplastic syndrome as a prototypical epigenetic disease. J. Blood 2013, 121, 3811–3817. [Google Scholar] [CrossRef] [PubMed]
- Peramo, A.; Mura, S.; Yesylevskyy, S.O.; Cardey, B.; Sobot, D.; Denis, S.; Ramseyer, C.; Desmaele, D.; Couvreur, P. Squalene versus cholesterol: Which is the best nanocarrier for the delivery to cells of the anticancer drug gemcitabine? C. R. Chim. 2018, 21, 974–986. [Google Scholar] [CrossRef]
- Dheer, D.; Nicolas, J.; Shankar, R. Cathepsin-sensitive nanoscale drug delivery systems for cancer therapy and other diseases. Adv. Drug Deliv. Rev. 2019, 151–152, 130–151. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Garber, H.R.; Zhang, M.; Sukhumalchandra, P.; Qiu, Y.; Jakher, H.; Perakis, A.A.; Becker, L.; Yoo, S.Y.; Dwyer, K.C.; et al. Cathepsin G is broadly expressed in acute myeloid leukemia and is an effective immunotherapeutic target. Leukemia 2016, 31, 234–237. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37 (Suppl. 2), W305–W311. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance, 2016, Final Report. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 12 November 2021).
- Maity, K.; Venkata, B.S.; Kapoor, N.; Surolia, N.; Surolia, A. Structural basis for the functional and inhibitory mechanisms ofb-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Plasmodium falciparum. J. Struct. Biol. 2011, 176, 238–249. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Zhang, Y.; Zhang, H.; Du, J.; Ding, J.; Guo, Y.; Jian, H.; Shen, X. Emodin targets the β-hydroxyacylacyl carrier protein dehydratase from Helicobacter pylori: Enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Micro. 2009, 9, 91–102. [Google Scholar] [CrossRef]
- Moynihan, H.; Horgan, D. Impurity occurrence and removal in crystalline products from process reactions. Org. Process Res. Dev. 2017, 21, 689–704. [Google Scholar] [CrossRef]
- Pysz, I.; Jackson, P.J.M.; Thurston, D.E. Chapter 1: Introduction to Antibody–Drug Conjugates (ADCs). In Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: Cambridge, UK, 2019; pp. 1–30. [Google Scholar] [CrossRef]
- Synthon Biopharmaceuticals BV. SYD985 vs. Physician’s Choice in Participants with HER2-Positive Locally Advanced or Metastatic Breast Cancer. Available online: www.clinicaltrials.gov/show/nct03262935 (accessed on 12 November 2021).
- Beekman, A.M.; Cominetti, M.M.D.; Searcey, M. Chapter 9: Duocarmycins as antibody–drug conjugate (ADC) payloads. In Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: Cambridge, UK, 2019; pp. 187–208. [Google Scholar] [CrossRef]
- Pillow, T.H.; Tercel, M. Chapter 11: Duocarmycin–PBD dimers as antibody–drug conjugate (ADC) Payloads. In Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: Cambridge, UK, 2019; pp. 241–258. [Google Scholar] [CrossRef]
- Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Acetylcholinesterase-inhibiting activity of pyrrole derivatives from a novel marine gliding bacterium, Rapidithrix thailandica. Mar. Drugs 2008, 6, 578–586. [Google Scholar] [CrossRef]
- Aguiar, A.C.C.; Panciera, M.; Simão dos Santos, E.F.; Singh, M.K.; Lopes Garcia, M.; Eduardo de Souza, G.; Nakabashi, M.; Costa, J.L.; Garcia, C.R.S.; Oliva, G.; et al. Discovery of marinoquinolines as potent and fast-acting Plasmodium falciparum inhibitors with in vivo activity. J. Med. Chem. 2018, 61, 5547–5568. [Google Scholar] [CrossRef]
- Huo, H.; Tang, X.Y.; Gong, Y.F. Metal-free synthesis of pyrrolo[1,2-a]quinoxalines mediated by TEMPO oxoammonium salts. Synthesis 2018, 50, 2727–2740. [Google Scholar] [CrossRef]
- Ma, X.; Vo, Y.; Banwell, M.G.; Willis, A.C. Total synthesis of marinoquinoline a using a palladium(0)-catalyzed Ullmann cross-coupling reaction. Asian J. Org. Chem. 2012, 1, 160–165. [Google Scholar] [CrossRef]
- Patel, B.; Saviolaki, G.; Ayats, C.; Garcia, M.A.E.; Kapadia, T.; Hilton, S.T. Tuneable radical cyclisations: A tin-free approach towards tricyclic and spirocyclic heterocycles via a common precursor. RSC Adv. 2014, 4, 18930–18932. [Google Scholar] [CrossRef]
- Sun, K.; Chen, X.L.; Zhang, Y.L.; Li, K.; Huang, X.Q.; Peng, Y.Y.; Qua, L.B.; Yu, B. Metal-free sulfonyl radical-initiated cascade cyclization to access sulfonated indolo[1,2-a]quinolines. Chem. Commun. 2019, 55, 12615–12618. [Google Scholar] [CrossRef]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Boiani, M.; Gonzalez, M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini-Rev. Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef]
- Walia, R.; Hedaitullah, M.; Farha Naaz, S.; Iqbal, K.; Lamba, H.S. Benzimidazole derivatives—an overview. Int. J. Res. Pharm. Chem. 2011, 1, 565–574. [Google Scholar]
- Song, D.; Ma, S. Recent development of benzimidazole-containing antibacterial agents. ChemMedChem 2016, 11, 646–659. [Google Scholar] [CrossRef]
- Ryder, A.S.H.; Cunningham, W.B.; Ballantyne, G.; Mules, T.; Kinsella, A.G.; Turner-Dore, J.; Alder, C.M.; Edwards, L.J.; McKay, B.S.J.; Grayson, M.N.; et al. Photocatalytic α-tertiary amine synthesis via C-H alkylation of unmasked primary amines. Angew. Chem. Int. Ed. 2020, 59, 14986–14991. [Google Scholar] [CrossRef]
- Grayson, J.D.; Cresswell, A.J. γ-Amino phosphonates via the photocatalytic α-C–H alkylation of primary amines. Tetrahedron 2021, 81, 131896. [Google Scholar] [CrossRef]
- Galve, I.; Ondoño, R.; de Rocafiguera, C.; Puig de la Bellacasa, R.; Batllori, X.; Puigjaner, C.; Font-Bardia, M.; Vallcorba, O.; Teixidó, J.; Borrell, J.I. A captured room temperature stable Wheland intermediate as a key structure for the orthogonal decoration of 4-amino-pyrido[2,3-d]pyrimidin-7(8H)-ones. Org. Biomol. Chem. 2020, 48, 9810–9815. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dorta, D.; Thobie-Gautier, D.C.; Croyal, M.; Bouzelha, M.; Mevel, M.; Deniaud, D.; Boujtita, M.; Gouin, S.G. Electrochemically promoted tyrosine-click-chemistry for protein labeling. J. Am. Chem. Soc. 2018, 140, 17120–17126. [Google Scholar] [CrossRef]
- Pudlo, M.; Allart-Simon, I.; Tinant, B.; Gérard, S.; Sapi, J. First domino radical cyclisation/Smiles rearrangement combination. Chem. Comm. 2012, 48, 2442–2444. [Google Scholar] [CrossRef] [PubMed]
- Allart-Simon, I.; Gérard, S.; Sapi, J. Radical Smiles rearrangement: An update. Molecules 2016, 21, 878–889. [Google Scholar] [CrossRef]
- Lamret, F.; Colin, M.; Mongaret, C.; Gangloff, S.C.; Reffuveille, F. Antibiotic tolerance of Staphylococcus aureus biofilm in periprosthetic joint infections and antibiofilm strategies. Antibiotics 2020, 9, 547. [Google Scholar] [CrossRef]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Van Dun, S.; Ottmann, C.; Milroy, L.G.; Brunsveld, L. Supra-molecular Chemistry Targeting Proteins. J. Am. Chem. Soc. 2017, 139, 13960–13968. [Google Scholar] [CrossRef]
- Guillory, X.; Hadrovic, I.; de Vink, P.J.; Brunsveld, L.; Schrader, T.; Ottmann, C. Supramolecular enhancement of natural 14-3-3 protein ligands. J. Am. Chem. Soc. 2021, 143, 13495–13500. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer. BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, L.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive macrocyclic inhibitors of the PD-1/PD-L1 immune checkpoint. Angew. Chem. Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Beekman, A.M.; O’Connell, M.A.; Howell, L.A. Peptide-directed binding for the discover of modulators of α-helix-mediated protein-protein interactions: Proof-of-concept studies with the apoptosis regulator Mcl-1. Angew. Chem. Int. Ed. 2017, 56, 10446–10450. [Google Scholar] [CrossRef]
- Barnes, C.; Sell, S.; Boland, E.; Simpson, D.; Bowling, G. Nanofibers technology: Designing the next generation of tissue engineering scaffolds. Adv. Drug Deliv. Rev. 2007, 59, 1413–1433. [Google Scholar] [CrossRef]
- Dwivedi, C.; Pandey, H.; Pandey, A.C.; Ramteke, P.W. Nanofibre based smart pharmaceutical scaffolds for wound repair and regenerations. Curr. Pharm. Des. 2016, 11, 1460–1471. [Google Scholar] [CrossRef]
- Kamble, P.; Sadarani, B.; Majumdar, A.; Bhullar, S. Nanofiber based drug delivery systems for skin: A promising therapeutic approach. J. Deliv. Sci. Technol. 2017, 41, 124–133. [Google Scholar] [CrossRef]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 1997, 114, S101–S110. [Google Scholar] [CrossRef]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef]
- Ozbilgin, A.; Durmuskahya, C.; Kayalar, H.; Ertabaklar, H.; Gunduz, C.; Ostan-Ural, I.; Zeyrek, F.; Kurt, O.; Cavus, I.; Balcıoglu, C.; et al. Antileishmanial activity of selected turkish medicinal plants. Trop. J. Pharm. Res. 2014, 13, 2047–2055. [Google Scholar] [CrossRef]
- Kjellev, S. The trefoil factor family—small peptides with multiple functionalities. Cell. Mol. Life Sci. 2009, 66, 1350–1369. [Google Scholar] [CrossRef]
- Braga Emidio, N.; Hoffmann, W.; Brierley, S.M.; Muttenthaler, M. Trefoil factor family: Unresolved questions and clinical perspectives. Trends Biochem Sci. 2019, 44, 387–390. [Google Scholar] [CrossRef]
- Šeklić, D.S.; Stanković, M.S.; Milutinović, M.G.; Topuzović, M.D.; Štajn, A.Š.; Marković, S.D. Cytotoxic, antimigratory and pro/antioxidative activities of extracts from medicinal mushrooms on colon cancer cell lines. Arch. Biol. Sci. 2016, 68, 93–105. [Google Scholar] [CrossRef]
- He, T.; Zhou, H.; Li, C.; Chen, Y.; Chen, X.; Li, C.; Mao, J.; Lyu, J.; Meng, Q.H. Methylglyoxal suppresses human colon cancer cell lines and tumor growth in a mouse model by impairing glycolytic metabolism of cancer cells associated with down-regulation of c-Myc expression. Cancer Biol. Ther. 2016, 7, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Bugarčić, Ž.D.; Bogojeski, J.; Petrović, B.; Hochreuther, S.; Van Eldik, R. Mechanistic studies on the reactions of platinum(II) complexes with nitrogen- and sulfur-donor biomolecules. Dalton Trans. 2012, 41, 12329–12345. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Mehta, V.; Wani, A.A.; Arora, S.; Bharatam, V.P.; Sharon, A.; Singh, S.; Kumar, R. Synthesis of 1,4-dihydropyrazolo[4,3-b]indoles via intramolecular C(sp2)-N bond formation involving nitrene insertion, DFT study and their anticancer assessment. Bioorg. Chem. 2021, 114, 105114. [Google Scholar] [CrossRef] [PubMed]
- Thell, K.; Hellinger, R.; Sahin, E.; Michenthaler, P.; Gold-Binder, M.; Haider, T.; Kuttke, M.; Liutkevičiūtė, Z.; Göransson, U.; Gründemann, C.; et al. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3960–3965. [Google Scholar] [CrossRef]
- Cheneval, O.; Schroeder, C.I.; Durek, T.; Walsh, P.; Huang, Y.-H.; Liras, S.; Price, D.A.; Craik, D.J. Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem. 2014, 79, 5538–5544. [Google Scholar] [CrossRef]
- Kumar, M.; Joshi, G.; Arora, S.; Singh, T.; Biswas, S.; Sharma, N.; Bhat, Z.R.; Tikoo, K.; Singh, S.; Kumar, R. Design and synthesis of non-covalent imidazo[1,2-a]quinoxaline-based inhibitors of EGFR and their anti-cancer assessment. Molecules 2021, 26, 1490–1512. [Google Scholar] [CrossRef] [PubMed]
- Goshi, G.; Chauhan, M.; Kumar, R.; Thakur, A.; Sharma, S.; Singh, R.; Wani, A.A.; Sharon, A.; Bharatam, P.V.; Kumar, R. Cyclocondensation reactions of an electron deactivated 2-aminophenyl tethered imidazole with mono/1,2-biselectrophiles: Synthesis and DFT studies on the rationalisation of imidazo[1,2-a]quinoxaline versus benzo[f]imidazo[1,5-a][1,3,5]triazepine selectivity switches. Org. Chem Front. 2018, 5, 3526–3533. [Google Scholar] [CrossRef]
- Zhuansun, Y.; Huang, F.; Du, Y.; Lin, L.; Chen, R.; Li, J. Anti-PD-1/PD-L1 antibody versus conventional chemotherapy for previously-treated, advanced non-small-cell lung cancer: A meta-analysis of randomized controlled trials. J. Thorac. Dis. 2017, 9, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulation. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Wright, K.; Rooney, N.; Feeney, M.; Tate, J.; Robertson, D.; Welham, M.; Ward, S. Differential expression of cannabinoid receptors in the human colon: Cannabinoids promote epithelial wound healing. Gastroenterology 2005, 129, 437–453. [Google Scholar] [CrossRef]
- Leleu-Chavain, N.; Body-Malapel, M.; Spencer, J.; Chavatte, P.; Desreumaux, P.; Millet, R. Recent advances in the development of selective CB2 agonists as promising anti-inflammatory agents. Curr. Med. Chem. 2012, 19, 3457–3474. [Google Scholar] [CrossRef] [PubMed]
- Leleu-Chavain, N.; Desreumaux, P.; Chavatte, P. Millet, R. Therapeutical potential of CB2 receptors in immune-related diseases. Curr. Mol. Pharm. 2013, 6, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Leleu-Chavain, N.; Baudelet, D.; Heloire, V.M.; Rocha, D.E.; Renault, N.; Barczyk, A.; Djouina, M.; Body-Malapel, M.; Carato, P.; Millet, R. Benzo[d]thiazol-2(3H)-ones as new potent selective CB2 agonists with anti-inflammatory properties. Eur. J. Med. Chem. 2019, 165, 347–362. [Google Scholar] [CrossRef]
- WHO Global Priority List of Antibiotic-Resistant Bacteria to Guide Research. 2017, pp. 1–7. Available online: https://www.aidsdatahub.org/sites/default/files/resource/who-global-priority-list-antibiotic-resistant-bacteria.pdf (accessed on 12 November 2021).
- Antimicrobial Resistance in the EU/EEA (EARS-Net)-Annual Epidemiological Report 2019. 2020, pp. 1–28. Available online: https://www.ecdc.europa.eu/en/publications-data/surveillance-antimicrobial-resistance-europe-2019 (accessed on 12 November 2021).
- Souli, M.; Galani, I.; Giamarellou, H. Emergence of extensively drug-resistant and pandrug-resistant Gram-negative bacilli in Europe. Eurosurveillance 2008, 13, 1–11. [Google Scholar] [CrossRef]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In vitro antibacterial properties of cefiderocol, a novel siderophore cephalosporin, against Gram-negative bacteria. Antimicrob. Agents Chemother. 2018, 62, e01454–e17. [Google Scholar] [CrossRef]
- Vickers, P.J.; Townsend, A.J.; Cowan, K.H. Mechanisms of resistance to antineoplastic drugs. In Developments in Cancer Chemotherapy; CRC Press: Boca Raton, FL, USA, 2019; pp. 117–152. [Google Scholar]
- Greene, L.M.; O’Boyle, N.M.; Nolan, D.P.; Meegan, M.J.; Zisterer, D.M. The vascular targeting agent Combretastatin-A4 directly induces autophagy in adenocarcinoma-derived colon cancer cells. Biochem. Pharmacol. 2012, 84, 612–624. [Google Scholar] [CrossRef]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation—Potential for therapeutics and diagnostics. Nat. Rev. Drug Disc. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.C.; Yu, C.; Kato, D.L.; Bertozzi, C.R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. USA 2003, 100, 14846–14851. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Yen-Pon, E.; Li, B.; Acebrón-Garcia-de-Eulate, M.; Tomkiewicz-Raulet, C.; Dawson, J.; Lietha, D.; Frame, M.C.; Coumoul, X.; Garbay, C.; Etheve-Quelquejeu, M.; et al. Structure-based design, synthesis, and characterization of the first irreversible inhibitor of focal adhesion kinase. ACS Chem. Biol. 2018, 8, 2067–2073. [Google Scholar] [CrossRef]
- Li, B.; Li, Y.; Tomkiewicz-Raulet, C.; Dao, P.; Lietha, D.; Yen-Pon, E.; Du, Z.; Coumoul, X.; Garbay, C.; Etheve-Quelquejeu, M.; et al. Design, synthesis, and biological evaluation of covalent inhibitors of focal adhesion kinase (FAK) against human malignant glioblastoma. J. Med. Chem. 2020, 63, 12707–12724. [Google Scholar] [CrossRef]
- Van Voorhis, W.C.; Adams, J.H.; Adelfio, R.; Ahyong, V.; Akabas, M.H.; Alano, P.; Alday, A.; Alemán Resto, Y.; Alsibaee, A.; Alzualde, A.; et al. Open Source Drug Discovery with the Malaria Box Compound Collection for Neglected Diseases and Beyond. PLOS Pathog. 2016, 12, e1005763. [Google Scholar] [CrossRef]
- Maistralis, G.; Koutsodimou, A.; Katsaros, N. Transition metal orotic acid complexes. Transit. Met. Chem. 2000, 25, 166–173. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition metal complexes and photodynamic therapy from a tumor-centered approach: Challenges, opportunities, and highlights from the development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Ouled Amor, C.; Kais, E.; Virlan, C.; Pui, A.; Elaloui, E. Effect of dysprosium ion (Dy3+) doping on morphological, crystal growth and optical properties of TiO2 particles and thin films. Phys. B Condens. Matter 2019, 560, 67–74. [Google Scholar] [CrossRef]
- Khade, G.V.; Suwarnkar, M.B.; Gavade, N.L.; Garadkar, K.M. Sol–gel microwave assisted synthesis of Sm-doped TiO2 nanoparticles and their photocatalytic activity for the degradation of Methyl Orange under sunlight. J. Mater. Sci. Mater. Electron. 2016, 27, 6425–6432. [Google Scholar] [CrossRef]
- Harari, M.; Couly, F.; Fruit, C.; Besson, T. Pd-Catalyzed and copper assisted regioselective sequential C2 and C7 arylation of thiazolo[5,4-f]quinazolin-9(8H)-one with aryl halides. Org. Lett. 2016, 18, 3282–3285. [Google Scholar] [CrossRef]
- Couly, F.; Harari, M.; Dubouilh-Benard, C.; Bailly, L.; Petit, E.; Diharce, J.; Bonnet, P.; Meijer, L.; Fruit, C.; Besson, T. Development of kinase inhibitors via metal-catalyzed C-H arylation of 8-alkyl-thiazolo[5,4- f]-quinazolin-9-ones designed by fragment-growing studies. Molecules 2018, 23, 2181. [Google Scholar] [CrossRef]
- Fruit, C.; Couly, F.; Bhansali, R.; Rammohan, M.; Lindberg, M.F.; Crispino, J.D.; Meijer, L.; Besson, T. Biological characterization of 8-cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a promising inhibitor of DYRK1A. Pharmaceuticals 2019, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Pilania, M.; Arun, V.; Pooniya, S. C–H arylation of azaheterocycles: A direct ligand-free and Cu-catalyzed approach using diaryliodonium salts. Org. Biomol. Chem. 2014, 12, 6340–6344. [Google Scholar] [CrossRef]
- Pacheco-Benichou, A.; Besson, T.; Fruit, C. Diaryliodoniums salts as coupling partners for transition-metal catalyzed C- and N-arylation of heteroarenes. Catalysts 2020, 10, 483–516. [Google Scholar] [CrossRef]
- Fersing, C.; Basmaciyan, L.; Boudot, C.; Pedron, J.; Hutter, S.; Cohen, A.; Castera-Ducros, C.; Primas, N.; Laget, M.; Casanova, M.; et al. Nongenotoxic 3-nitroimidazo[1,2-a]pyridines are NTR1 substrates that display potent in vitro antileishmanial activity. ACS Med. Chem. Lett. 2019, 10, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Boudot, C.; Castera-Ducros, C.; Pinault, E.; Hutter, S.; Paoli-Lombardo, R.; Primas, N.; Pedron, J.; Line Seguy, L.; Bourgeade-Delmas, S.; et al. 8-Alkynyl-3-nitroimidazopyridines display potent antitrypanosomal activity against both T. b. brucei and cruzi. Eur. J. Med. Chem. 2020, 202, 112558. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Boudot, C.; Paoli-Lombardo, R.; Primas, N.; Pinault, E.; Hutter, S.; Castera-Ducros, C.; Kabri, Y.; Pedron, J.; Bourgeade-Delmas, S.; et al. Antikinetoplastid SAR study in 3-nitroimidazopyridine series: Identification of a novel non-genotoxic and potent anti-T. b. brucei hit-compound with improved pharmacokinetic properties. Eur. J. Med. Chem. 2020, 206, 112668. [Google Scholar] [CrossRef] [PubMed]
- Tatakihara, V.L.H.; Malvezi, A.D.; Panis, C.; Cecchini, R.; Zanluqui, N.G.; Yamauchi, L.M.; Martins, M.I.L.; da Silva, R.V.; Yamada-Ogatta, S.F.; Rizzo, L.V.; et al. Nitric oxide-releasing indomethacin enhances susceptibility to Trypanosoma cruzi infection acting in the cell invasion and oxidative stress associated with anemia. Chem. Biol. Interact. 2015, 227, 104–111. [Google Scholar] [CrossRef]
- Hughes, M.N. Chemistry of nitric oxide and related species. Methods Enzymol. 2008, 436, 3–19. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.K.; Bhardwaj, T.R. Therapeutic role of nitric oxide as emerging molecule. Biomed. Pharmacother. 2017, 85, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Lanas, A. Role of nitric oxide in the gastrointestinal tract. Arthritis Res. Ther. 2008, 10 (Suppl. 2), S4. [Google Scholar] [CrossRef]
- Santana, A.P.M.; Tavares, B.M.; Lucetti, L.T.; Gouveia, F.S.; Ribeiro, R.A.; Soares, P.M.G.; Sousa, E.H.S.; Lopes, L.G.F.; Medeiros, J.-V.R.; Souza, M.H.L.P. The nitric oxide donor cis-[Ru(bpy)2(SO3)NO](PF6) increases gastric mucosa protection in mice—Involvement of the soluble guanylate cyclase/KATP pathway. Nitric Oxide 2015, 45, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, U.; Khan, M.I.; Ashraf, S.; Hameed, A.; Hafizur, R.M.; Rafique, R.; Khan, K.M.; Ul-Haq, Z. Identification of novel Epac2 antagonists through in silico and in vitro analyses. Eur. J. Pharm. Sci. 2020, 153, 105492. [Google Scholar] [CrossRef]
- Hameed, A.; Hafizur, R.M.; Hussain, N.; Raza, S.A.; Rehman, M.; Ashraf, S.; Ul-Haq, Z.; Khan, F.; Abbas, G.; Choudhary, M.I. Eriodictyol stimulates insulin secretion through cAMP/PKA signaling pathway in mice islets. Eur. J. Pharmacol. 2018, 820, 245–255. [Google Scholar] [CrossRef]
- Radisavljević, S.; Petrović, B. Gold(III) complexes: An overview on their kinetics, interactions with DNA/BSA, cytotoxic activity, and computational calculations. Front. Chem. 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.Y.; Haque, R.A.; Razali, M.R. Recent progress in silver(I)-, gold(I)/(III)-and palladium(II)-N-heterocyclic carbene complexes: A review towards biological perspectives. J. Organomet. Chem. 2019, 882, 96–111. [Google Scholar] [CrossRef]
- Costello, C.; Karpanen, T.; Lambert, P.A.; Mistry, P.; Parker, K.J.; Rathbone, D.L.; Ren, J.; Wheeldon, L.; Worthington, T. Thiosemicarbazones active against Clostridium Difficile. Bioorg. Med. Chem. Lett. 2008, 18, 1708–1711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Argyrou, A.; Washabaugh, M.W.; Pickart, C.M. Dihydroorotate dehydrogenase from Clostridium oroticum is a class 1B enzyme and utilizes a concerted mechanism of catalysis. Biochemistry 2000, 39, 10373–10384. [Google Scholar] [CrossRef]
- Fraser, W.; Suckling, C.J.; Wood, H.C.S. Latent inhibitors. Part 7. Inhibition of dihydro-orotate dehydrogenase by spirocyclopropanobarbiturates. J. Chem. Soc. Perkin Trans. 1990, 1, 3137–3144. [Google Scholar] [CrossRef]
- Cowden, W.; Jacobsen, N.W.; Stunzi, H. Pyrimidine N-oxides. V. Ionization constants of N-hydroxybarbiturates. Aust. J. Chem. 1982, 35, 1251–1253. [Google Scholar] [CrossRef]
- Golovanov, A.A.; Gusev, D.M.; Odin, I.S.; Zlotskii, S.S. Conjugated 2,4,1-and 1,4,3-enynones as polycentricelectrophiles in synthesis of heterocyclic compounds. Chem. Heterocycl. Comp. 2019, 55, 333–348. [Google Scholar] [CrossRef]
- Grennet, E.; Martinez, J.; Salom-Roig, X.J. Lewis acid induced switch of torquoselectivity in the Nazarov cyclization of activated dienones bearing a chiral sulfoxide. Chem. Eur. J. 2016, 22, 16770–16773. [Google Scholar] [CrossRef] [PubMed]
- Grennet, E.; Martinez, J.; Salom-Roig, X.J. Torquoselective Nazarov cyclization mediated by a chiral sulfoxide: First enantioselective synthesis of two known anticancer agents. Asian J. Org. Chem. 2017, 6, 189–198. [Google Scholar] [CrossRef]
- Leong, W.I.; Saba, J.D. S1P metabolism in cancer and other pathological conditions. Biochimie 2010, 92, 716–723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Truman, J.P.; García-Barros, M.; Obeid, L.M.; Hannun, Y.A. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim. Biophys. Acta 2014, 1841, 1174–1188. [Google Scholar] [CrossRef]
- Heffernan-Stroud, L.A.; Obeid, L.M. Sphingosine kinase 1 in cancer. Adv. Cancer Res. 2013, 117, 201–235. [Google Scholar] [CrossRef]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Shirai, K.; Kaneshiro, T.; Wada, M.; Furuya, H.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M.; Ogretmen, B.; Kawamori, T. A role of sphingosine kinase 1 in head and neck carcinogenesis. Cancer Prev. Res. 2011, 4, 454–462. [Google Scholar] [CrossRef]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298. [Google Scholar] [CrossRef]
- Pesci, E.C.; Milbank, J.B.J.; Pearson, J.P.; McKnight, S.; Kende, A.S.; Greenberg, E.P.; Iglewski, B.H. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Khalilzadeh, P.; Lajoie, B.; El Hage, S.; Furiga, A.; Baziard, G.; Berge, M.; Roques, C. Growth inhibition of adherent Pseudomonas aeruginosa by an N-butanoyl-L-homoserine lactone analog. Can. J. Microbiol. 2010, 56, 317–325. [Google Scholar] [CrossRef]
- Furiga, A.; Lajoie, B.; El Hage, S.; Baziard, G.; Roques, C. Impairment of Pseudomonas aeruginosa biofilm resistance to antibiotics by combining the drugs with a new Quorum-Sensing Inhibitor. Antimicrob. Agents Chemother. 2016, 60, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Nogaret, P.; El Garah, F.; Blanc-Potard, A.-B. A Novel infection protocol in Zebrafish embryo to assess Pseudomonas aeruginosa virulence and validate efficacy of a quorum sensing inhibitor in vivo. Pathogens 2021, 10, 401. [Google Scholar] [CrossRef]
- Nguyen, T.H.D.; Tam, J.; Wu, R.A.; Greber, B.J.; Toso, D.; Nogales, E.; Collins, K. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 2018, 557, 190–198. [Google Scholar] [CrossRef]
- Beekman, A.M.; Cominetti, M.M.D.; Walpole, S.J.; Prabhu, S.; O’Connell, M.A.; Angulo, J.; Searcey, M. Identification of selective protein–protein interaction inhibitors using efficient in silico peptide-directed ligand design. Chem. Sci. 2019, 10, 4502–4508. [Google Scholar] [CrossRef]
- Zhang, C.; Fortin, P.Y.; Barnoin, G.; Qin, X.; Wang, X.; Fernandez Alvarez, A.; Bijani, C.; Maddelein, M.L.; Hemmert, C.; Cuvillier, O.; et al. An artemisinin-derivative–(NHC) Gold (I) hybrid with enhanced cytotoxicity through inhibition of NRF2 transcriptional activity. Angew. Chem. Int. Ed. 2020, 59, 12062–12068. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; de la Torre, B.G.; Albericio, F. Hydroxamate siderophores: Natural occurrence, chemical synthesis, iron binding affinity and use as trojan horses against pathogens. Eur. J. Med. Chem. 2020, 208, 112791–112819. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, R.; Meijler, M.M. Total synthesis of pyoverdin D. Org. Lett. 2013, 15, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C.Q.; Wells, N.; Weymouth-Wilson, A. Investigating the influence of (deoxy)fluorination on the lipophilicity of non-UV-active fluorinated alkanols and carbohydrates by a new log P determination method. Angew. Chem. Int. Ed. 2016, 55, 674–6781. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, B.; Wang, Z.; Troup, R.I.; Goupille, A.; Le Questel, J.-Y.; Fallan, C.; Scott, J.S.; Chiarparin, E.; Graton, J.; Linclau, B. Lipophilicity trends upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl groups. Beilstein J. Org. Chem. 2020, 16, 2141–2150. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, B.; Wang, Z.; Felstead, H.R.; Le Questel, J.-Y.; Scott, J.S.; Chiarparin, E.; Graton, J.; Linclau, B. Systematic investigation of lipophilicity modulation by aliphatic fluorination motifs. J. Med. Chem. 2020, 63, 1002–1031. [Google Scholar] [CrossRef]













































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helesbeux, J.-J.; Carro, L.; McCarthy, F.O.; Moreira, V.M.; Giuntini, F.; O’Boyle, N.; Matthews, S.E.; Bayraktar, G.; Bertrand, S.; Rochais, C.; et al. 29th Annual GP2A Medicinal Chemistry Conference. Pharmaceuticals 2021, 14, 1278. https://doi.org/10.3390/ph14121278
Helesbeux J-J, Carro L, McCarthy FO, Moreira VM, Giuntini F, O’Boyle N, Matthews SE, Bayraktar G, Bertrand S, Rochais C, et al. 29th Annual GP2A Medicinal Chemistry Conference. Pharmaceuticals. 2021; 14(12):1278. https://doi.org/10.3390/ph14121278
Chicago/Turabian StyleHelesbeux, Jean-Jacques, Laura Carro, Florence O. McCarthy, Vânia M. Moreira, Francesca Giuntini, Niamh O’Boyle, Susan E. Matthews, Gülşah Bayraktar, Samuel Bertrand, Christophe Rochais, and et al. 2021. "29th Annual GP2A Medicinal Chemistry Conference" Pharmaceuticals 14, no. 12: 1278. https://doi.org/10.3390/ph14121278
APA StyleHelesbeux, J.-J., Carro, L., McCarthy, F. O., Moreira, V. M., Giuntini, F., O’Boyle, N., Matthews, S. E., Bayraktar, G., Bertrand, S., Rochais, C., & Marchand, P. (2021). 29th Annual GP2A Medicinal Chemistry Conference. Pharmaceuticals, 14(12), 1278. https://doi.org/10.3390/ph14121278