
Mechanism of Action of the Sesquiterpene Compound Helenalin in Rhabdomyosarcoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
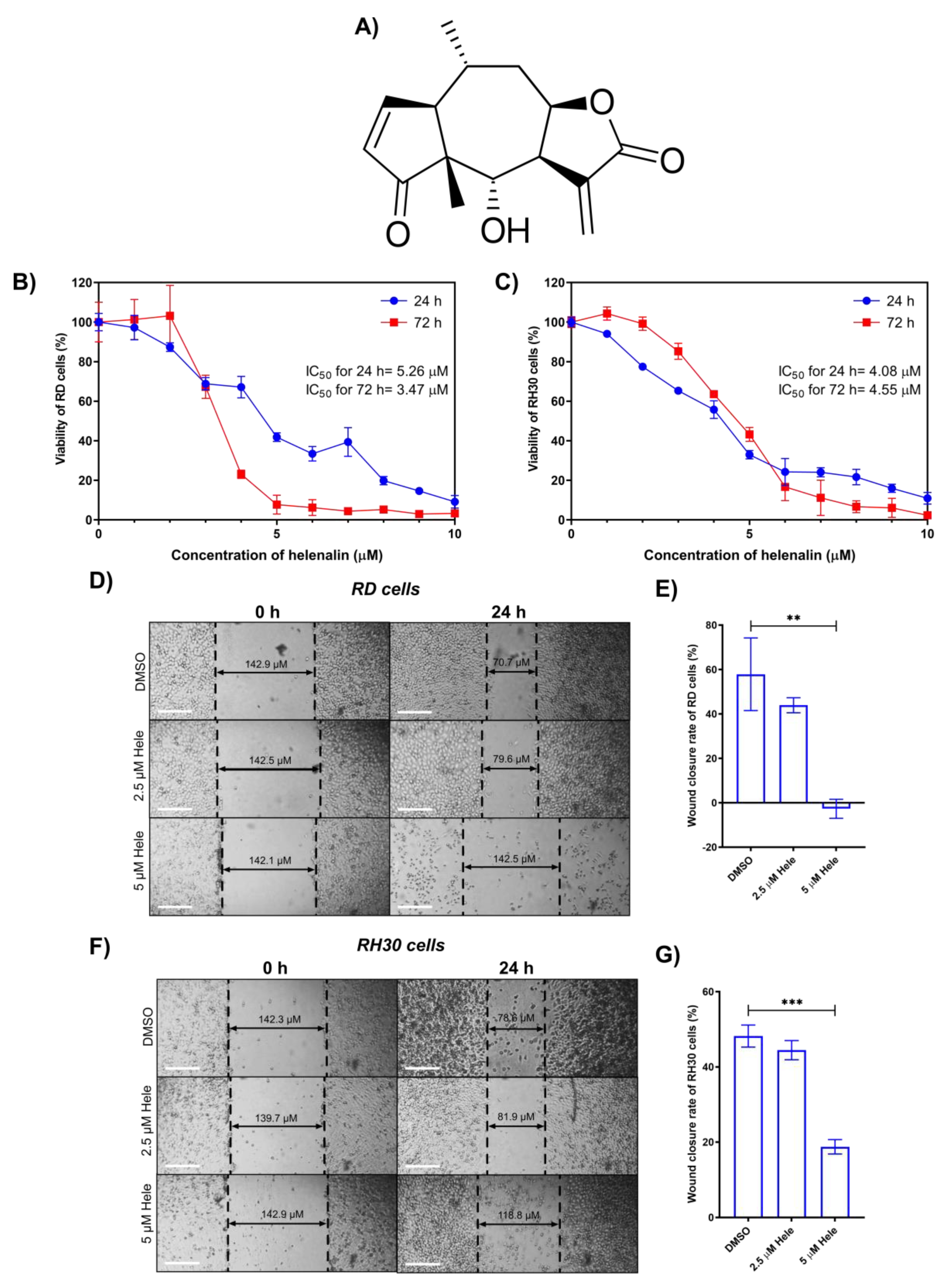
2.1. Cytotoxicity of Helenalin against RMS Cells
2.2. Effect of Helenalin on Migration of RMS Cells
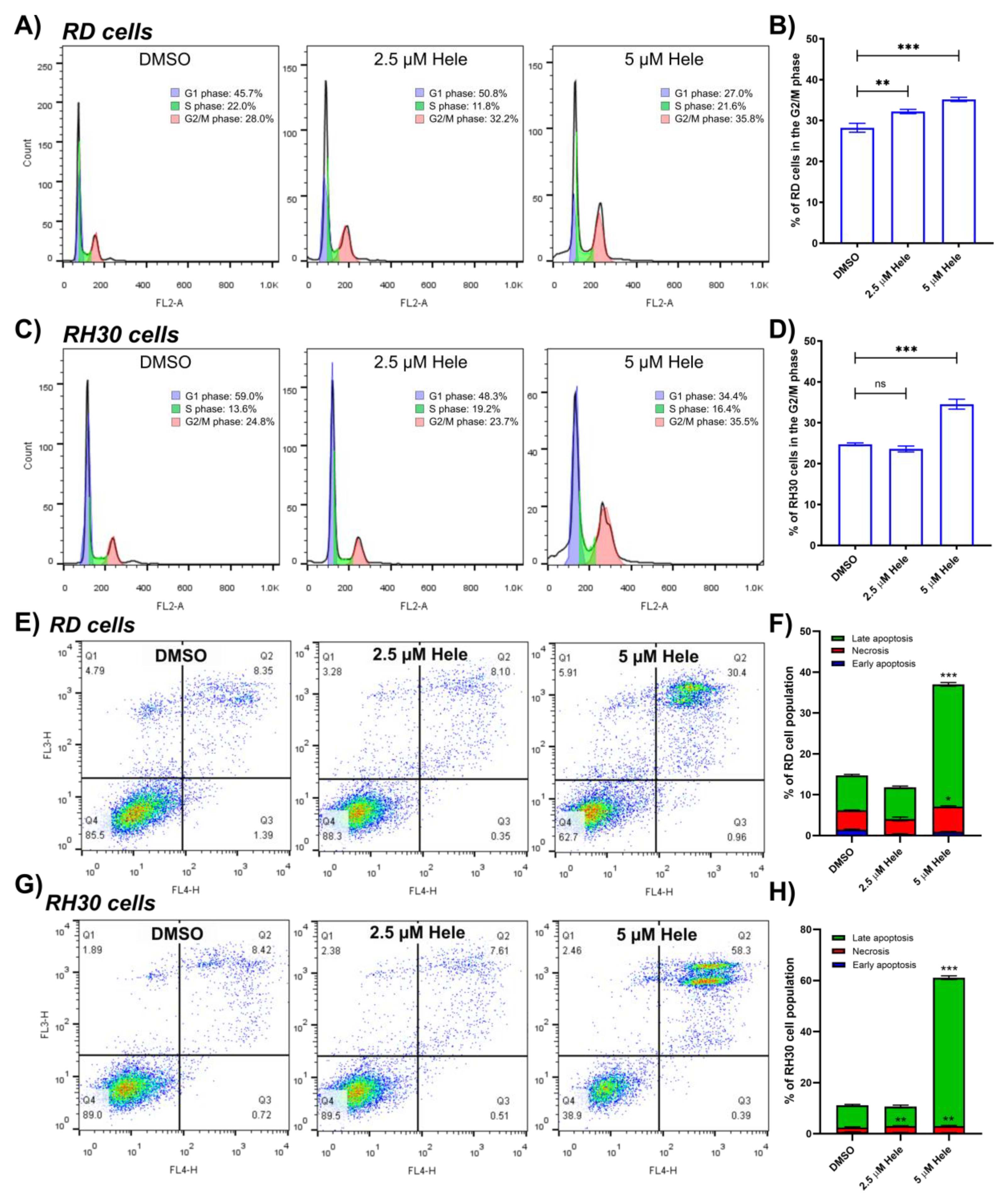
2.3. Assessment of Helenalin-Induced Cell Cycle Arrest and Cell Death
2.4. Evaluation of Action of Helenalin towards RMS Cell Death
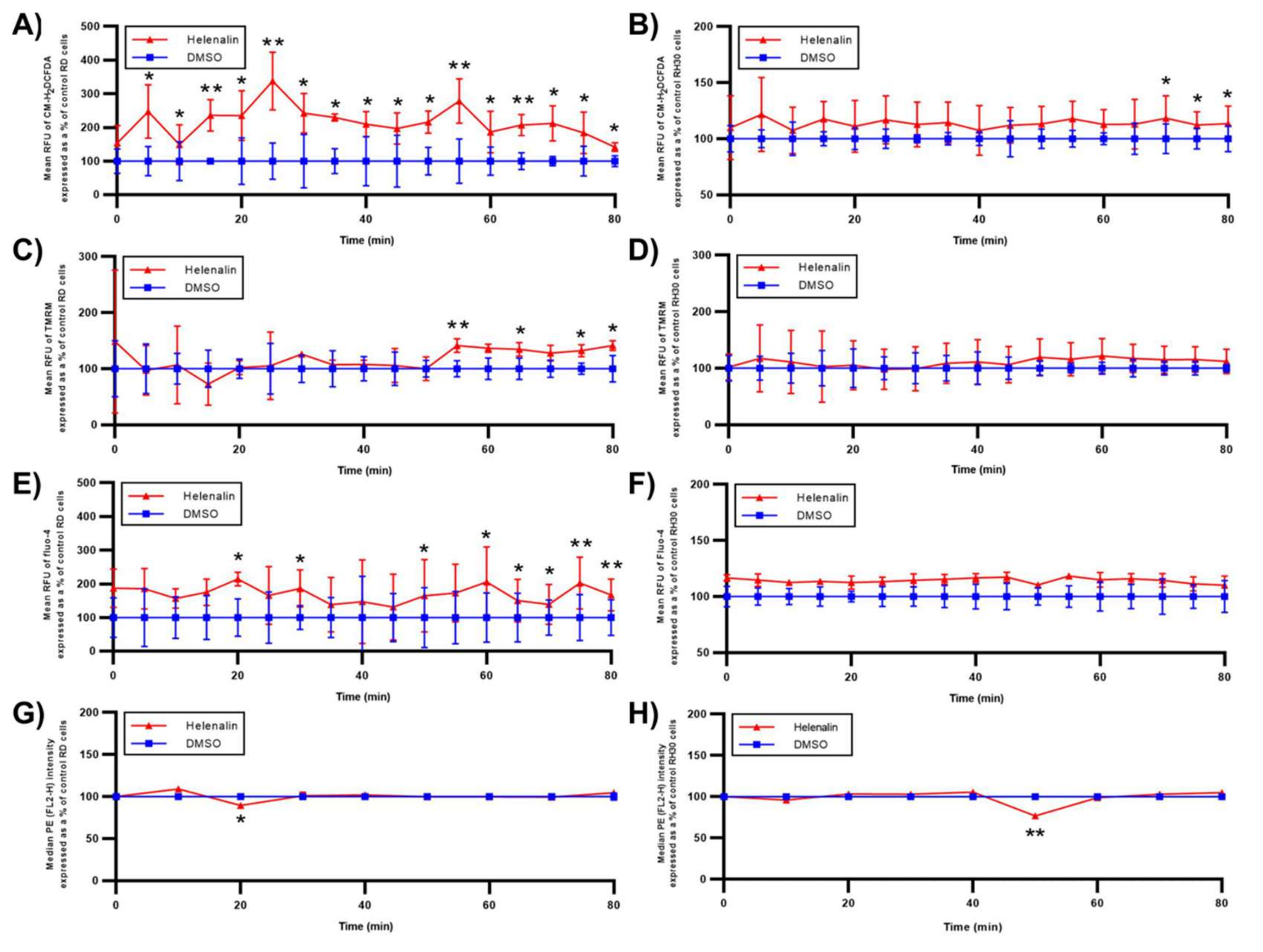
2.4.1. Oxidative Status
2.4.2. Mitochondrial Response
2.4.3. Endoplasmic Reticulum (ER) Response
2.4.4. NF-κB Activation
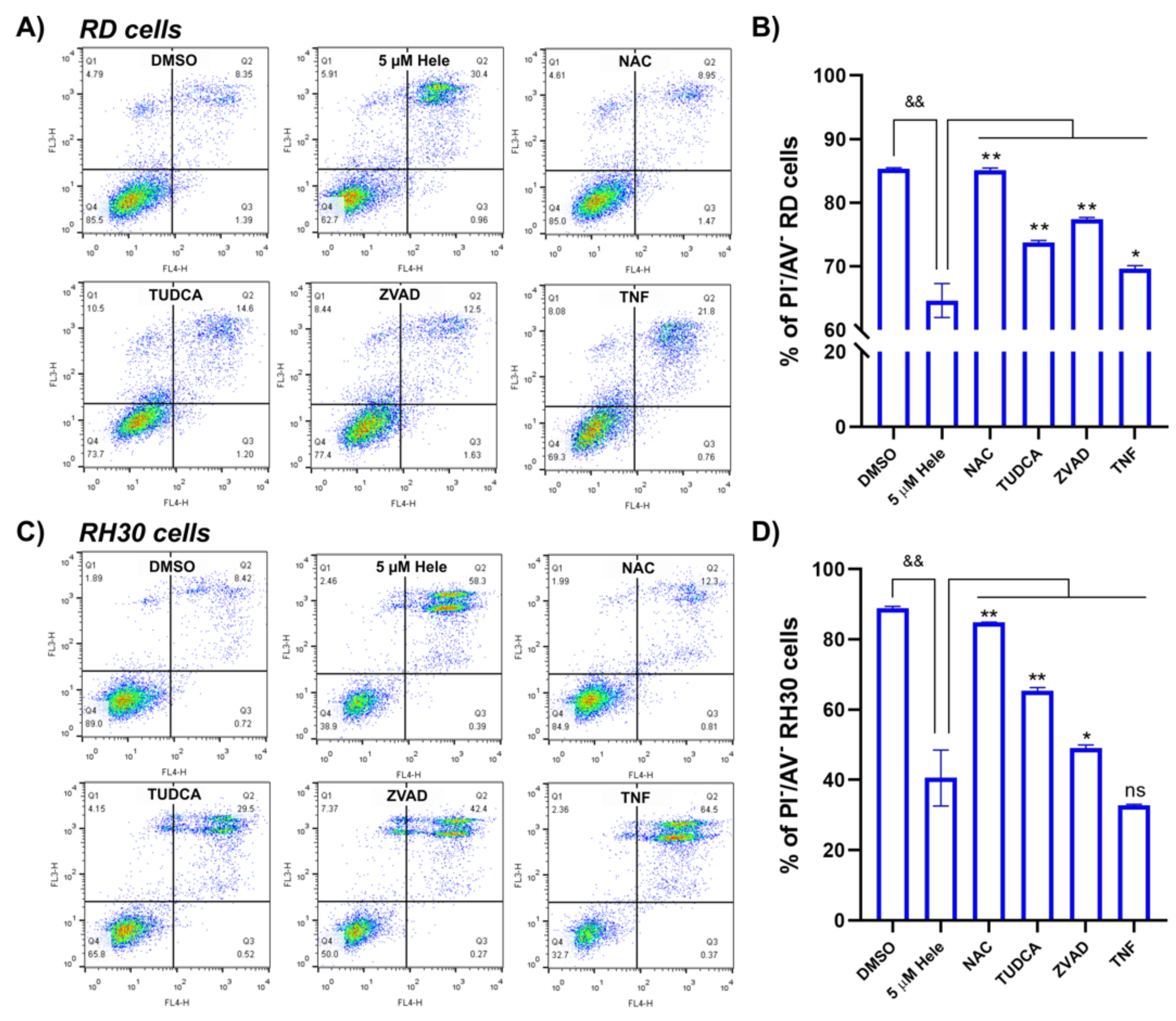
2.4.5. Cell Survival Improvement by Different Antagonists
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Cell Viability Assay
4.3. In Vitro Wound Healing Assay
4.4. Flow Cytometry
4.4.1. Cell Cycle Analysis
4.4.2. Evaluation of Cell Death Status
4.4.3. ROS and Mitochondrial Membrane Potential (MMP) Analysis
4.4.4. Detection of Intracellular Antibody Binding
4.5. Immunoblotting
4.6. Confocal Microscopy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barlow, J.W.; Wiley, J.C.; Mous, M.; Narendran, A.; Gee, M.F.W.; Goldberg, M.; Sexsmith, E.; Malkin, D. Differentiation of Rhabdomyosarcoma Cell Lines Using Retinoic Acid. Pediatric Blood Cancer 2006, 47, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, C. Childhood Rhabdomyosarcoma: Recent Advances and Prospective Views. J. Dent. Res. 2012, 91, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, D.M.; Barr, F.G. Classification of Rhabdomyosarcoma and Its Molecular Basis. Adv. Anat. Pathol. 2013, 20, 387. [Google Scholar] [CrossRef]
- Arden, K.C.; Anderson, M.J.; Finckenstein, F.G.; Czekay, S.; Cavenee, W.K. Detection of the t(2;13) Chromosomal Translocation in Alveolar Rhabdomyosarcoma Using the Reverse Transcriptase-Polymerase Chain Reaction. Genes Chromosomes Cancer 1996, 16, 254–260. [Google Scholar] [CrossRef]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Hinson, A.R.P.; Jones, R.; Crose, L.E.S.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human Rhabdomyosarcoma Cell Lines for Rhabdomyosarcoma Research: Utility and Pitfalls. Front. Oncol. 2013, 3, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyss, G.; Schmidt, T.J.; Merfort, I.; Pahl, H.L. Helenalin, an Anti-Inflammatory Sesquiterpene Lactone from Arnica, Selectively Inhibits Transcription Factor NF-KappaB. Biol. Chem. 1997, 378, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, P.; Schultze, N.; Niehs, S.; Albrecht, D.; Methling, K.; Wurster, M.; Wachlin, G.; Lalk, M.; Lindequist, U.; Haertel, B. Differential Effects of Helenalin, an Anti-Inflammatory Sesquiterpene Lactone, on the Proteome, Metabolome and the Oxidative Stress Response in Several Immune Cell Types. Toxicol. In Vitro 2017, 40, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Hall, I.H.; Mar, E.C.; Starnes, C.O.; ElGebaly, S.A.; Waddell, T.G.; Hadgraft, R.I.; Ruffner, C.G.; Weidner, I. Sesquiterpene Antitumor Agents: Inhibitors of Cellular Metabolism. Science 1977, 196, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-R.; Yeh, Y.-M.; Wang, T.-C.V. Potent Inhibition of Human Telomerase by Helenalin. Cancer Lett. 2005, 227, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, J.; Wasescha, M.R.; Schmidt, T.J. The Influence of Glutathione and Cysteine Levels on the Cytotoxicity of Helenanolide Type Sesquiterpene Lactones against KB Cells. Bioorganic Med. Chem. 2001, 9, 2189–2194. [Google Scholar] [CrossRef]
- Lyß, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The Anti-Inflammatory Sesquiterpene Lactone Helenalin Inhibits the Transcription Factor NF-ΚB by Directly Targeting P65*. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Gupta, D.; Arora, R. NF-KB as a Key Player in Regulation of Cellular Radiation Responses and Identification of Radiation Countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Danilenko, M.; Kim, T.S. Differential Enhancement of Leukaemia Cell Differentiation without Elevation of Intracellular Calcium by Plant-Derived Sesquiterpene Lactone Compounds. Br. J. Pharmacol. 2008, 155, 814–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.B.; Fu, P.Y.; Ky, N.; Zhu, H.S.; Feng, X.; Li, J.; Srinivasan, K.G.; Hamza, M.S.; Zhao, Y. NF-ΚB P65 Repression by the Sesquiterpene Lactone, Helenalin, Contributes to the Induction of Autophagy Cell Death. BMC Complementary Altern. Med. 2012, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Iqbal, T.; Min, K.-J.; Kim, S.; Park, J.-W.; Son, E.-I.; Lee, T.-J.; Kwon, T.K. Helenalin-Induced Apoptosis Is Dependent on Production of Reactive Oxygen Species and Independent of Induction of Endoplasmic Reticulum Stress in Renal Cell Carcinoma. Toxicol. In Vitro 2013, 27, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Grippo, A.A.; Hall, I.H.; Kiyokawa, H.; Muraoka, O.; Shen, Y.C.; Lee, K.H. The Cytotoxicity of Helenalin, Its Mono and Difunctional Esters, and Related Sesquiterpene Lactones in Murine and Human Tumor Cells. Drug Des. Discov. 1992, 8, 191–206. [Google Scholar] [PubMed]
- Liu, J.; Zhao, Y.; Shi, Z.; Bai, Y. Antitumor Effects of Helenalin in Doxorubicin-Resistant Leukemia Cells Are Mediated via Mitochondrial Mediated Apoptosis, Loss of Mitochondrial Membrane Potential, Inhibition of Cell Migration and Invasion and Downregulation of PI3-Kinase/AKT/m-TOR Signalling Pathway. J. Buon 2019, 24, 2068–2074. [Google Scholar] [PubMed]
- Li, Y.; Zeng, Y.; Huang, Q.; Wen, S.; Wei, Y.; Chen, Y.; Zhang, X.; Bai, F.; Lu, Z.; Wei, J.; et al. Helenalin from Centipeda Minima Ameliorates Acute Hepatic Injury by Protecting Mitochondria Function, Activating Nrf2 Pathway and Inhibiting NF-ΚB Activation. Biomed. Pharmacother. 2019, 119, 109435. [Google Scholar] [CrossRef]
- Kordi, S.; Zarghami, N.; Akbarzadeh, A.; Rahmati, Y.M.; Ghasemali, S.; Barkhordari, A.; Tozihi, M. A Comparison of the Inhibitory Effect of Nano-Encapsulated Helenalin and Free Helenalin on Telomerase Gene Expression in the Breast Cancer Cell Line, by Real-Time PCR. Artif. Cells Nanomed. Biotechnol. 2016, 44, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Potter, A.J.; Gollahon, K.A.; Palanca, B.J.A.; Harbert, M.J.; Choi, Y.M.; Moskovitz, A.H.; Potter, J.D.; Rabinovitch, P.S. Flow Cytometric Analysis of the Cell Cycle Phase Specificity of DNA Damage Induced by Radiation, Hydrogen Peroxide and Doxorubicin. Carcinogenesis 2002, 23, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified Annexin V/Propidium Iodide Apoptosis Assay for Accurate Assessment of Cell Death. J. Vis. Exp. 2011, 50, e2597. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.H.; Roxburgh, C.S.D.; Anderson, J.H.; McKee, R.F.; Foulis, A.K.; Horgan, P.G.; McMillan, D.C. Prognostic Value of Tumour Necrosis and Host Inflammatory Responses in Colorectal Cancer. Br. J. Surg. 2012, 99, 287–294. [Google Scholar] [CrossRef]
- Werfel, T.A.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Gonzales-Ericsson, P.I.; Nixon, M.J.; James, J.L.; Balko, J.M.; Scherle, P.A.; et al. Treatment-Induced Tumor Cell Apoptosis and Secondary Necrosis Drive Tumor Progression in the Residual Tumor Microenvironment through MerTK and IDO1. Cancer Res. 2019, 79, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papale, M.; Buccarelli, M.; Mollinari, C.; Russo, M.A.; Pallini, R.; Ricci-Vitiani, L.; Tafani, M. Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression. Int. J. Mol. Sci. 2020, 21, 2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, R.; Verrax, J.; Dejeans, N.; Taper, H.; Calderon, P.B. Menadione Reduction by Pharmacological Doses of Ascorbate Induces an Oxidative Stress That Kills Breast Cancer Cells. Int. J. Toxicol. 2009, 28, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS Inhibitor N-Acetyl-l-Cysteine Antagonizes the Activity of Proteasome Inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Lomeli, N.; Di, K.; Pearre, D.C.; Chung, T.-F.; Bota, D.A. Mitochondrial-Associated Impairments of Temozolomide on Neural Stem/Progenitor Cells and Hippocampal Neurons. Mitochondrion 2020, 52, 56–66. [Google Scholar] [CrossRef]
- Chien, C.-H.; Hsueh, W.-T.; Chuang, J.-Y.; Chang, K.-Y. Dissecting the Mechanism of Temozolomide Resistance and Its Association with the Regulatory Roles of Intracellular Reactive Oxygen Species in Glioblastoma. J. Biomed. Sci. 2021, 28, 18. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Rangel, E.; Olguín-Albuerne, M.; López-Méndez, M.C.; Domínguez-Macouzet, G.; Guerrero-Hernández, A.; Morán, J. Caspase-3 Activation Correlates With the Initial Mitochondrial Membrane Depolarization in Neonatal Cerebellar Granule Neurons. Front. Cell Dev. Biol. 2020, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.-E.; Gottlieb, R.A.; Green, D.R. Caspase-Mediated Loss of Mitochondrial Function and Generation of Reactive Oxygen Species during Apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Huang, H.; Yang, M.; Zhuang, D.; Cheng, X.; Zhang, H.; Fu, X. Microcystin-LR Induces Mitochondria-Mediated Apoptosis in Human Bronchial Epithelial Cells. Exp. Ther. Med. 2016, 12, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Morrison, R.; Lodge, T.; Evidente, A.; Kiss, R.; Townley, H. Ophiobolin A, a Sesterpenoid Fungal Phytotoxin, Displays Different Mechanisms of Cell Death in Mammalian Cells Depending upon the Cancer Cell Origin. Int. J. Oncol. 2017, 50, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Fabian, S.-G.; Jose, L.G.-G.; Mari, C.G.-C.; Federico, V.P. Mitochondrial Biogenesis in Health and Disease. Molecular and Therapeutic Approaches. Curr. Pharm. Des. 2014, 20, 5619–5633. [Google Scholar]
- Doherty, E.; Perl, A. Measurement of Mitochondrial Mass by Flow Cytometry during Oxidative Stress. React. Oxyg. Species 2017, 4, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin Specifically Aggravates ER Stress and Overcomes Chemoresistance in Multidrug-Resistant Gastric Cancer Cells by Inhibiting N-Glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.-S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.-J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic Acid Reduces ER Stress by Regulating of Akt-Dependent Cellular Prion Protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of Tauroursodeoxycholic Acid on Endoplasmic Reticulum Stress-Induced Caspase-12 Activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef]
- Wang, D.; Baldwin, A.S. Activation of Nuclear Factor-KappaB-Dependent Transcription by Tumor Necrosis Factor-Alpha Is Mediated through Phosphorylation of RelA/P65 on Serine 529. J. Biol. Chem. 1998, 273, 29411–29416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.-C.; Lee, Y.-H.; Jung, M.G.; Kwon, S.H.; Kim, M.-J.; Jun, W.J.; Lee, J.; Lee, J.M.; Yoon, H.-G. Gallic Acid Suppresses Lipopolysaccharide-Induced Nuclear Factor-ΚB Signaling by Preventing RelA Acetylation in A549 Lung Cancer Cells. Mol. Cancer Res. 2009, 7, 2011–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. Regulation of NF-ΚB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.; Gergely, P.; Nagy, G.; Koncz, A.; Banki, K. Mitochondrial Hyperpolarization: A Checkpoint of T-Cell Life, Death and Autoimmunity. Trends Immunol. 2004, 25, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Recent Advances and Challenges in the Treatment of Rhabdomyosarcoma. Cancers 2020, 12, 1758. [Google Scholar] [CrossRef]
- Hoffmann, R.; von Schwarzenberg, K.; López-Antón, N.; Rudy, A.; Wanner, G.; Dirsch, V.M.; Vollmar, A.M. Helenalin Bypasses Bcl-2-Mediated Cell Death Resistance by Inhibiting NF-ΚB and Promoting Reactive Oxygen Species Generation. Biochem. Pharmacol. 2011, 82, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorg, C.; Schmid, E.; Bortel, N.; Fuchs, J.; Ellerkamp, V. Antitumor Effects of Curcumin in Pediatric Rhabdomyosarcoma in Combination with Chemotherapy and Phototherapy In Vitro. Int. J. Oncol. 2021, 58, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Liu, Y.; Li, L.; Tian, C.; Zhou, H.; Zhang, Q.; Yan, B. The Novel Tubulin Polymerization Inhibitor MHPT Exhibits Selective Anti-Tumor Activity against Rhabdomyosarcoma In Vitro and In Vivo. PLoS ONE 2015, 10, e0121806. [Google Scholar] [CrossRef] [Green Version]
- Yuksel, S.N.; Dikmen, M.; Canturk, Z. Evaluation of Real Time Cell Proliferation, Anti-Inflammatory and Wound Healing Potential of Helenalin on HaCaT Keratinocytes Treated with Lipopolysaccharide Stimulated Monocytes. Indian J. Pharm. Sci. 2021, 83, 219–229. [Google Scholar] [CrossRef]
- DiPaola, R.S. To Arrest or Not to G(2)-M Cell-Cycle Arrest: Commentary Re: A. K. Tyagi et al., Silibinin Strongly Synergizes Human Prostate Carcinoma DU145 Cells to Doxorubicin-Induced Growth Inhibition, G(2)-M Arrest, and Apoptosis. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar] [PubMed]
- Lu, W.-J.; Peng, W.; Sun, Q.-Q.; Li, Y.-H.; Chen, B.; Yu, L.-T.; Xu, Y.-Z.; Wang, S.-Y.; Zhao, Y.-L. #2714, a Novel Active Inhibitor with Potent G2/M Phase Arrest and Antitumor Efficacy in Preclinical Models. Cell Death Discov. 2018, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Łos, M.J. Autophagy, Apoptosis, Mitoptosis and Necrosis: Interdependence Between Those Pathways and Effects on Cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidicelli, G.; Chaigne-Delalande, B.; Dilhuydy, M.-S.; Pinson, B.; Mahfouf, W.; Pasquet, J.-M.; Mahon, F.-X.; Pourquier, P.; Moreau, J.-F.; Legembre, P. The Necrotic Signal Induced by Mycophenolic Acid Overcomes Apoptosis-Resistance in Tumor Cells. PLoS ONE 2009, 4, e5493. [Google Scholar] [CrossRef] [PubMed]
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin Suppresses Essential Immune Functions of Activated CD4+ T Cells by Multiple Mechanisms. Mol. Immunol. 2009, 46, 2892–2901. [Google Scholar] [CrossRef] [PubMed]
- Gach, K.; Długosz, A.; Janecka, A. The Role of Oxidative Stress in Anticancer Activity of Sesquiterpene Lactones. Naunyn-Schmiedeberg’s Arch Pharm. 2015, 388, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Morán, J. Reactive Oxygen Species Induce Different Cell Death Mechanisms in Cultured Neurons. Free. Radic. Biol. Med. 2004, 36, 1112–1125. [Google Scholar] [CrossRef]
- Joanna, D.; Anna, J. Helenalin—A Sesquiterpene Lactone with Multidirectional Activity. Curr. Drug Targets 2019, 20, 444–452. [Google Scholar]
- Tsujimoto, Y.; Shimizu, S. Role of the Mitochondrial Membrane Permeability Transition in Cell Death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasul, A.; Khan, M.; Yu, B.; Ali, M.; Bo, Y.J.; Yang, H.; Ma, T. Isoalantolactone, a Sesquiterpene Lactone, Induces Apoptosis in SGC-7901 Cells via Mitochondrial and Phosphatidylinositol 3-Kinase/Akt Signaling Pathways. Arch. Pharm. Res. 2013, 36, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Hamzeloo-Moghadam, M.; Aghaei, M.; Fallahian, F.; Jafari, S.M.; Dolati, M.; Abdolmohammadi, M.H.; Hajiahmadi, S.; Esmaeili, S. Britannin, a Sesquiterpene Lactone, Inhibits Proliferation and Induces Apoptosis through the Mitochondrial Signaling Pathway in Human Breast Cancer Cells. Tumor Biol. 2015, 36, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-N.; Huang, H.-H.; Wu, C.-L.; Lin, C.P.C.; Hsu, J.T.A.; Hsieh, H.-P.; Chuang, S.-E.; Lai, G.-M. Isocostunolide, a Sesquiterpene Lactone, Induces Mitochondrial Membrane Depolarization and Caspase-Dependent Apoptosis in Human Melanoma Cells. Cancer Lett. 2007, 246, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Normington, K.; Sambrook, J.; Gething, M.J.; Mori, K. The Promoter Region of the Yeast KAR2 (BiP) Gene Contains a Regulatory Domain That Responds to the Presence of Unfolded Proteins in the Endoplasmic Reticulum. Mol. Cell. Biol. 1993, 13, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Ruddock, L.W. The Human Protein Disulphide Isomerase Family: Substrate Interactions and Functional Properties. EMBO Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Jäättelä, M. Connecting Endoplasmic Reticulum Stress to Autophagy by Unfolded Protein Response and Calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, P.; Ma, X.; Qiao, F.; Ma, Y.; Qing, S.; Zhang, Y.; Wang, Y.; Cui, W. Tauroursodeoxycholic Acid (TUDCA) Alleviates Endoplasmic Reticulum Stress of Nuclear Donor Cells under Serum Starvation. PLoS ONE 2018, 13, e0196785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, B.; Wen, S.; Li, Y.; Bai, F.; Wei, Y.; Xiong, Y.; Huang, Q.; Lin, X. Prediction and Verification of Target of Helenalin against Hepatic Stellate Cell Activation Based on MiR-200a-Mediated PI3K/Akt and NF-ΚB Pathways. Int. Immunopharmacol. 2021, 92, 107208. [Google Scholar] [CrossRef] [PubMed]
- Mitomo, K.; Nakayama, K.; Fujimoto, K.; Sun, X.; Seki, S.; Yamamoto, K. Two Different Cellular Redox Systems Regulate the DNA-Binding Activity of the P50 Subunit of NF-Kappa B In Vitro. Gene 1994, 145, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Rüngeler, P.; Castro, V.; Mora, G.; Gören, N.; Vichnewski, W.; Pahl, H.L.; Merfort, I.; Schmidt, T.J. Inhibition of Transcription Factor NF-ΚB by Sesquiterpene Lactones: A Proposed Molecular Mechanism of Action. Bioorganic Med. Chem. 1999, 7, 2343–2352. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sulen, A.; Gullaksen, S.-E.; Bader, L.; McClymont, D.W.; Skavland, J.; Gavasso, S.; Gjertsen, B.T. Signaling Effects of Sodium Hydrosulfide in Healthy Donor Peripheral Blood Mononuclear Cells. Pharmacol. Res. 2016, 113, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mun, H.; Townley, H.E. Mechanism of Action of the Sesquiterpene Compound Helenalin in Rhabdomyosarcoma Cells. Pharmaceuticals 2021, 14, 1258. https://doi.org/10.3390/ph14121258
Mun H, Townley HE. Mechanism of Action of the Sesquiterpene Compound Helenalin in Rhabdomyosarcoma Cells. Pharmaceuticals. 2021; 14(12):1258. https://doi.org/10.3390/ph14121258
Chicago/Turabian StyleMun, Hakmin, and Helen Elizabeth Townley. 2021. "Mechanism of Action of the Sesquiterpene Compound Helenalin in Rhabdomyosarcoma Cells" Pharmaceuticals 14, no. 12: 1258. https://doi.org/10.3390/ph14121258
APA StyleMun, H., & Townley, H. E. (2021). Mechanism of Action of the Sesquiterpene Compound Helenalin in Rhabdomyosarcoma Cells. Pharmaceuticals, 14(12), 1258. https://doi.org/10.3390/ph14121258