
Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
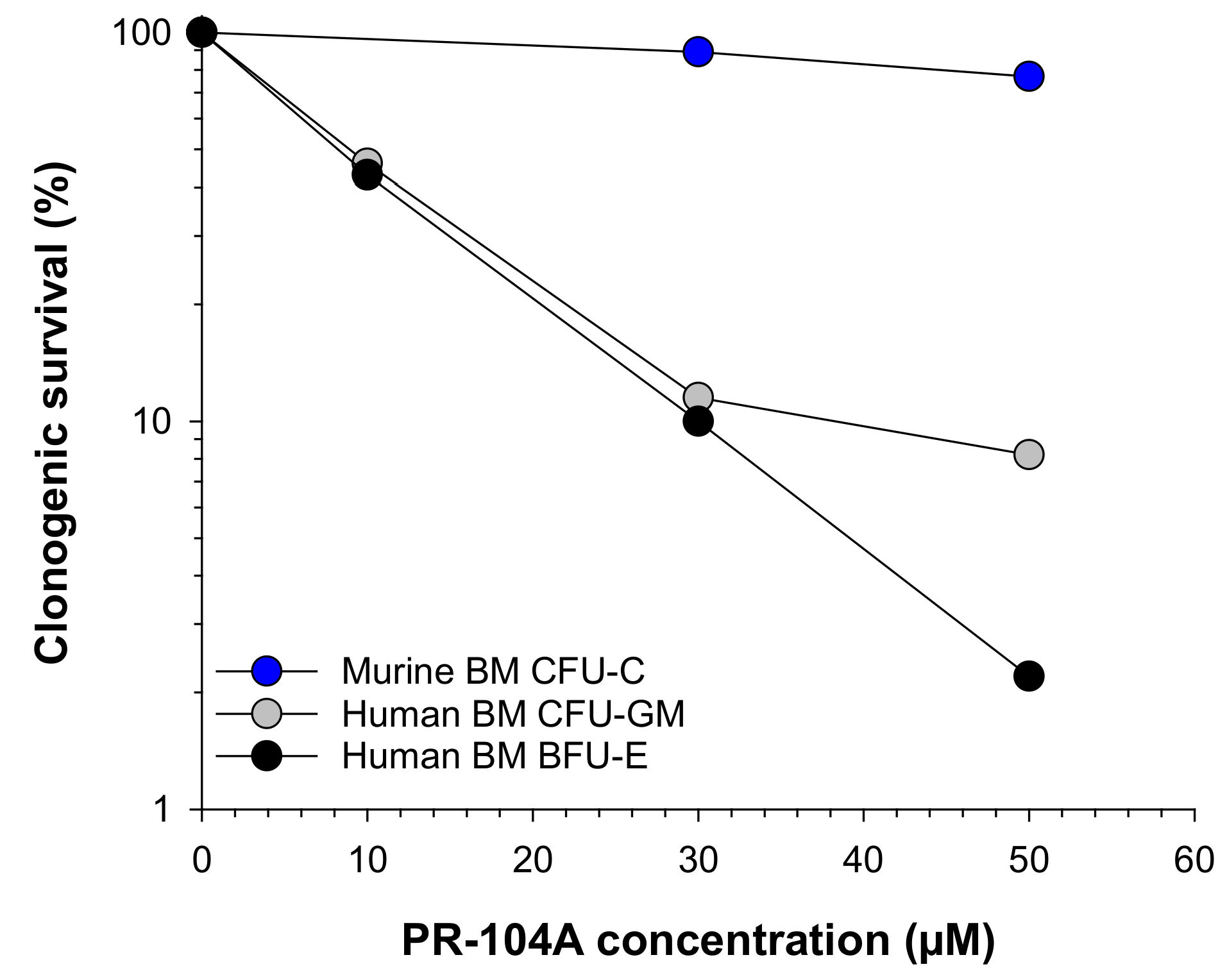
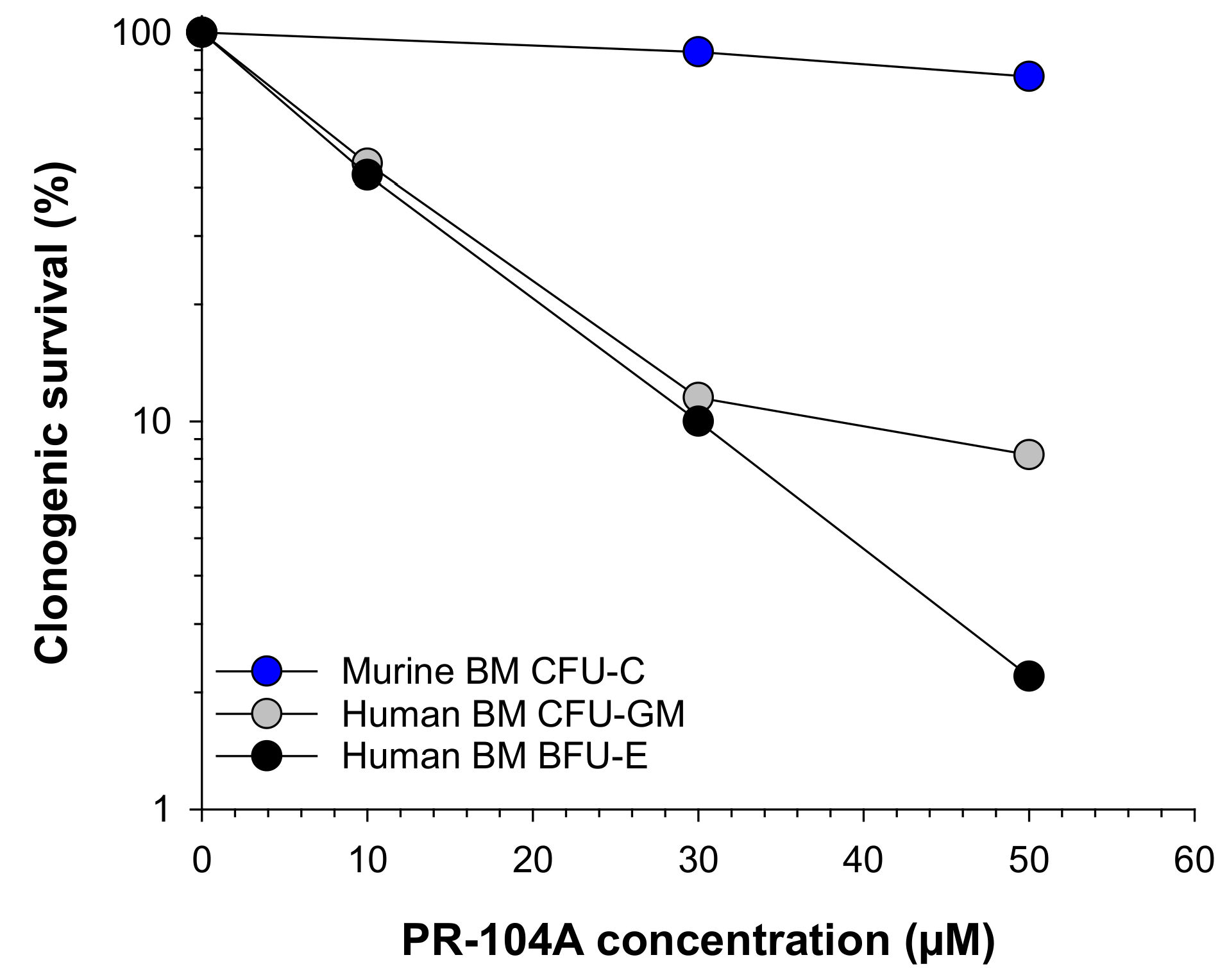
2.1. Human Haematopoietic Cells Are More Sensitive to PR-104A Than Murine Haematopoietic Cells
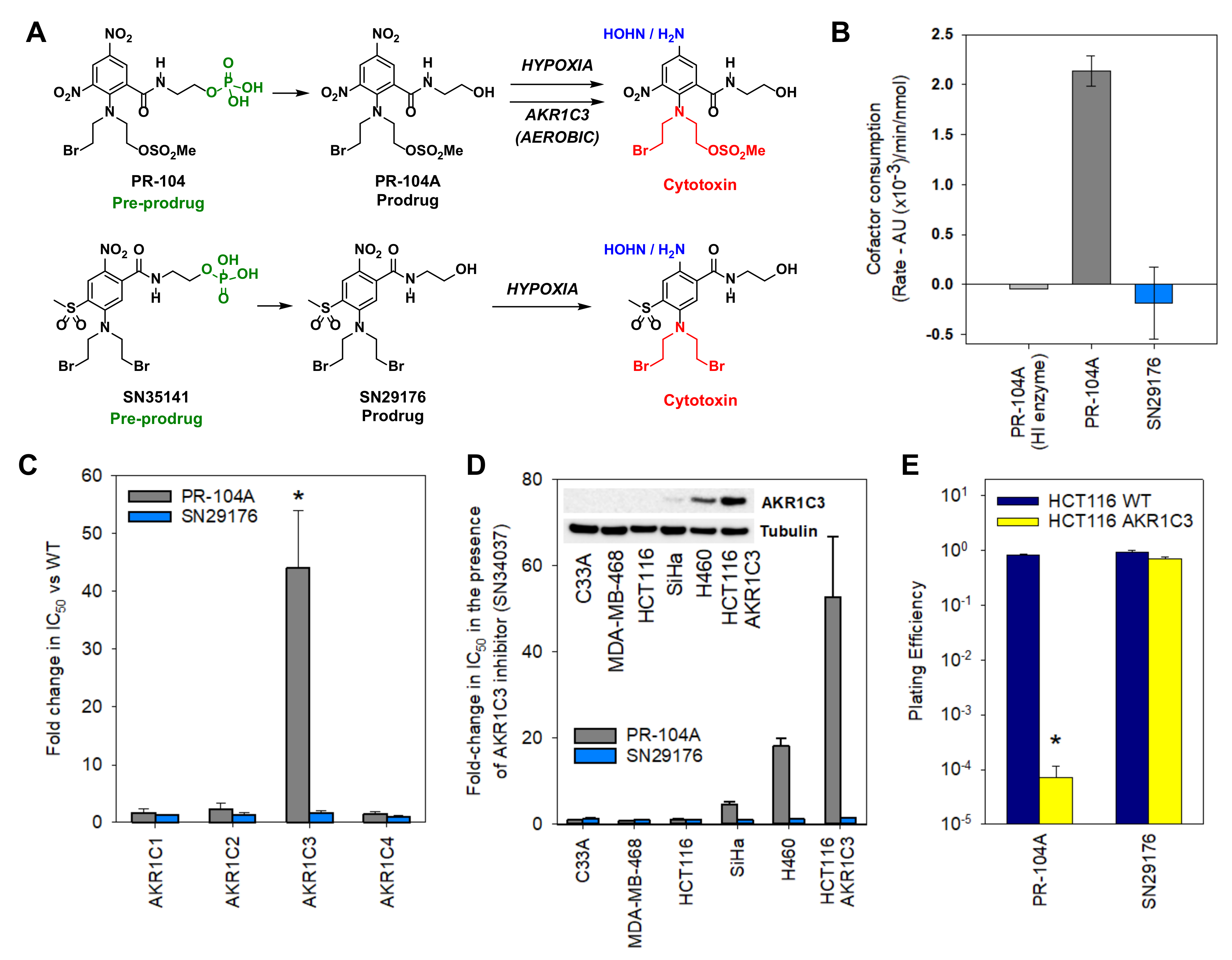
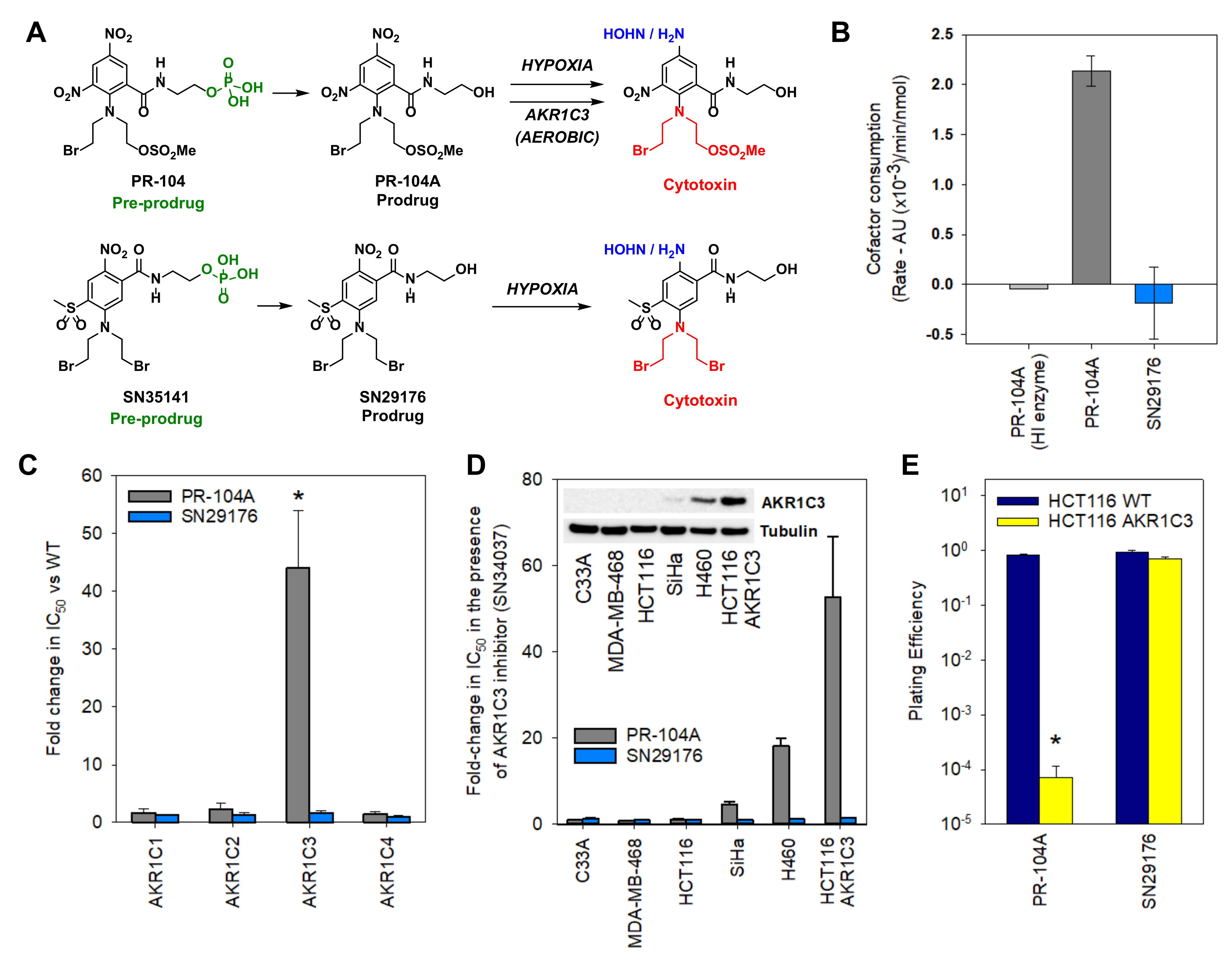
2.2. Design of an AKR1C3-Resistant Nitrobenzamide Mustard
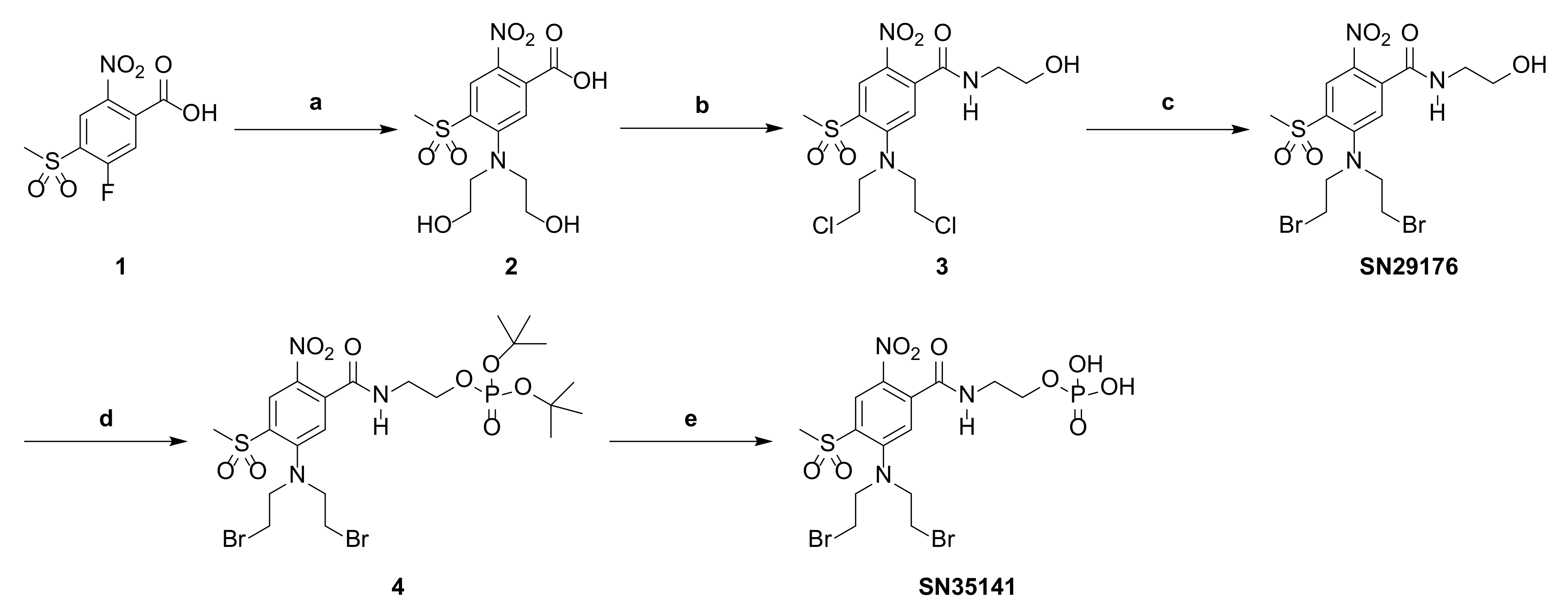
2.3. Chemistry
2.4. SN29176 Is Not a Substrate for Human AKR1C Enzymes
2.5. SN29176 Shows Hypoxia Selectivity In Vitro and Is a Substrate for POR
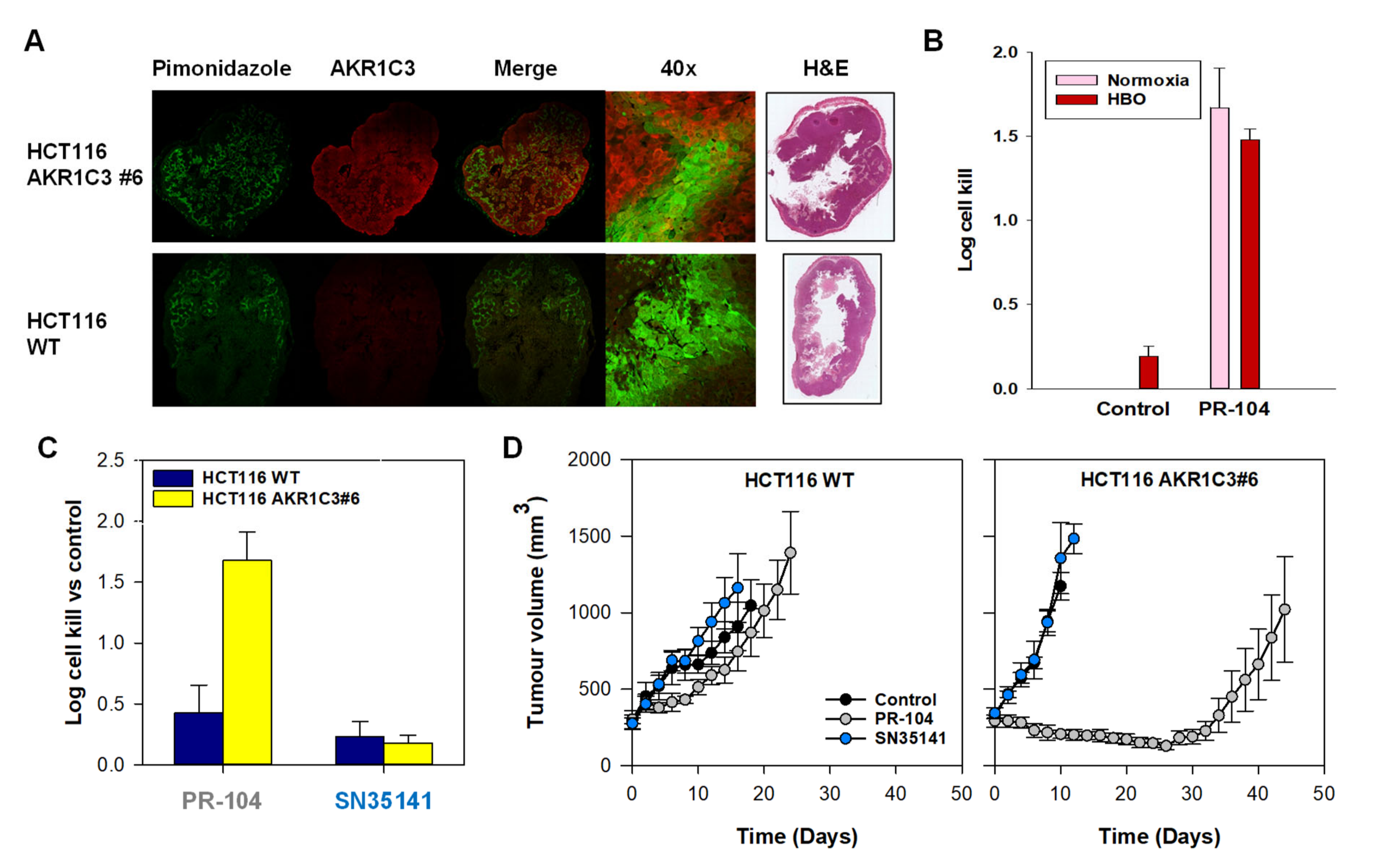
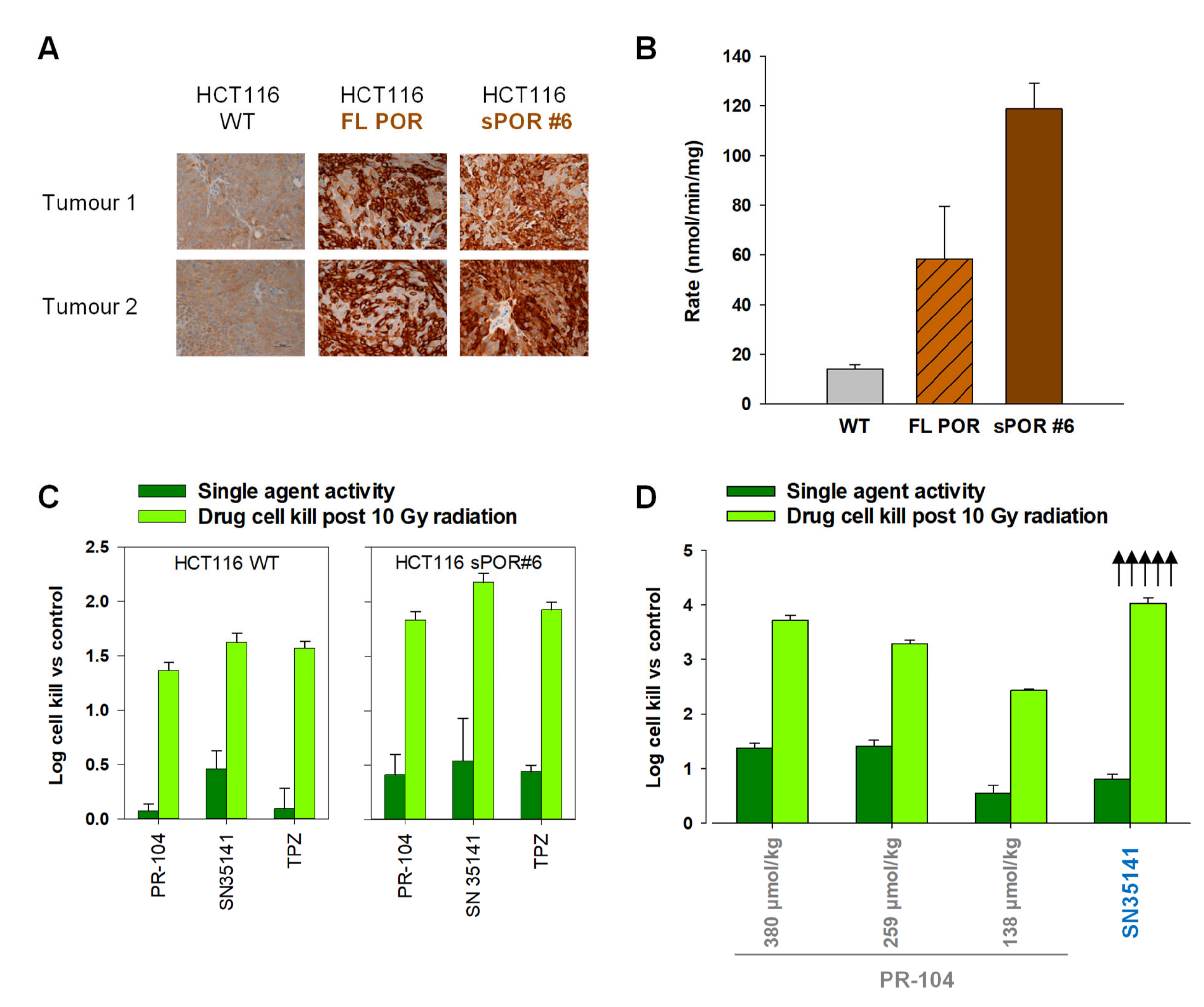
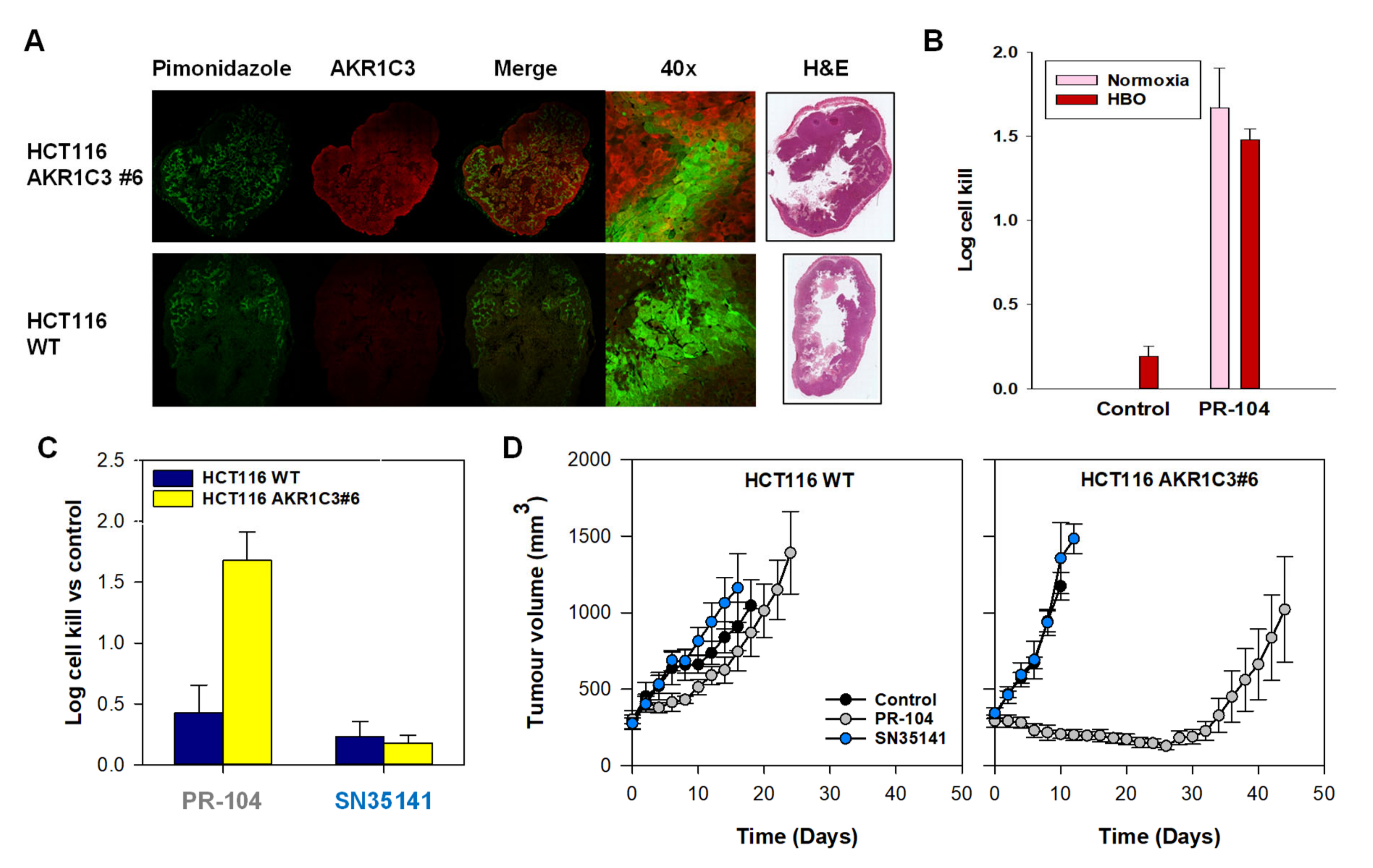
2.6. SN35141, the Phosphate Pre-Prodrug of SN29176, Is Active in Human Tumour Xenografts
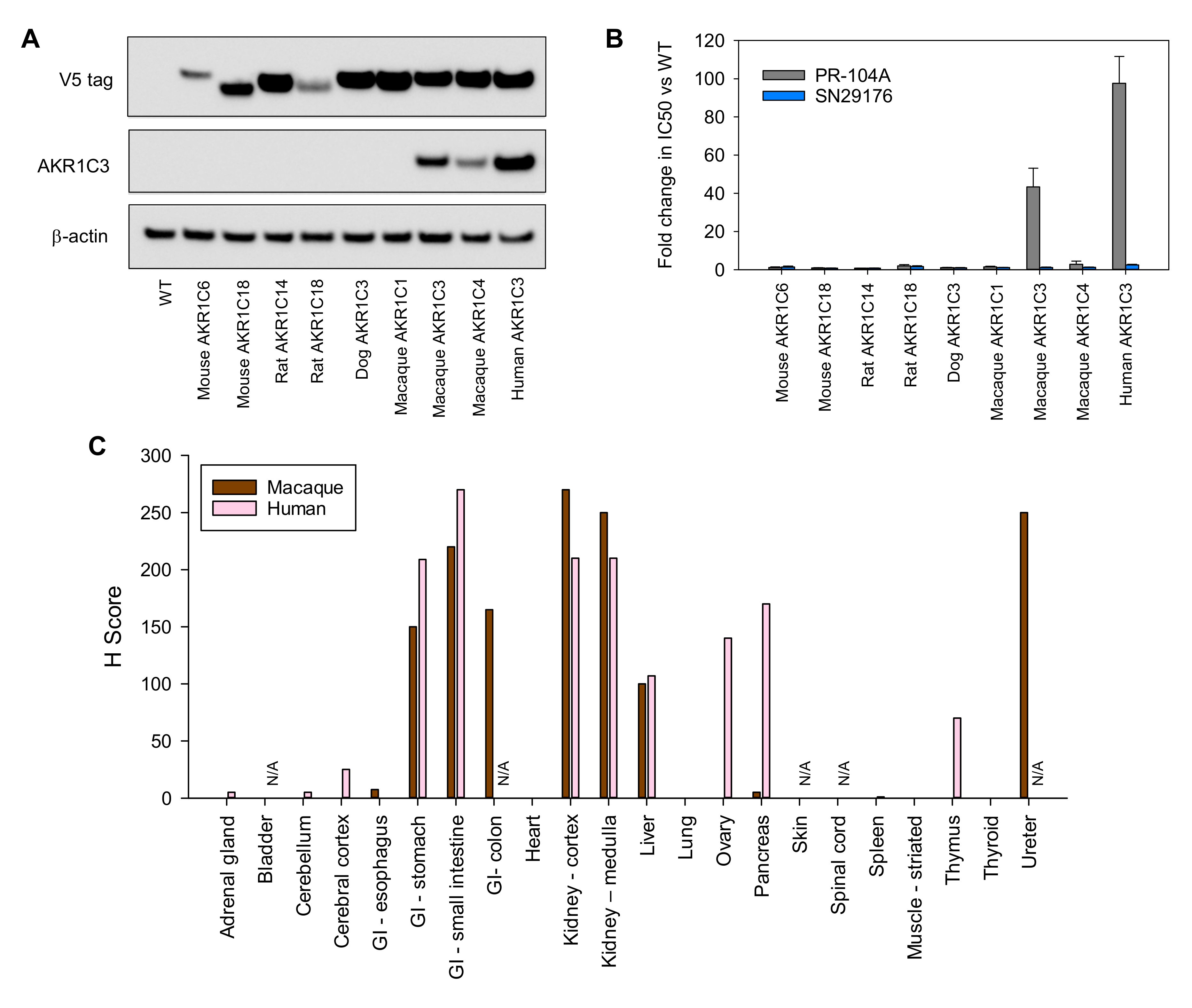
2.7. SN35141 Is Not a Substrate for AKR1C3
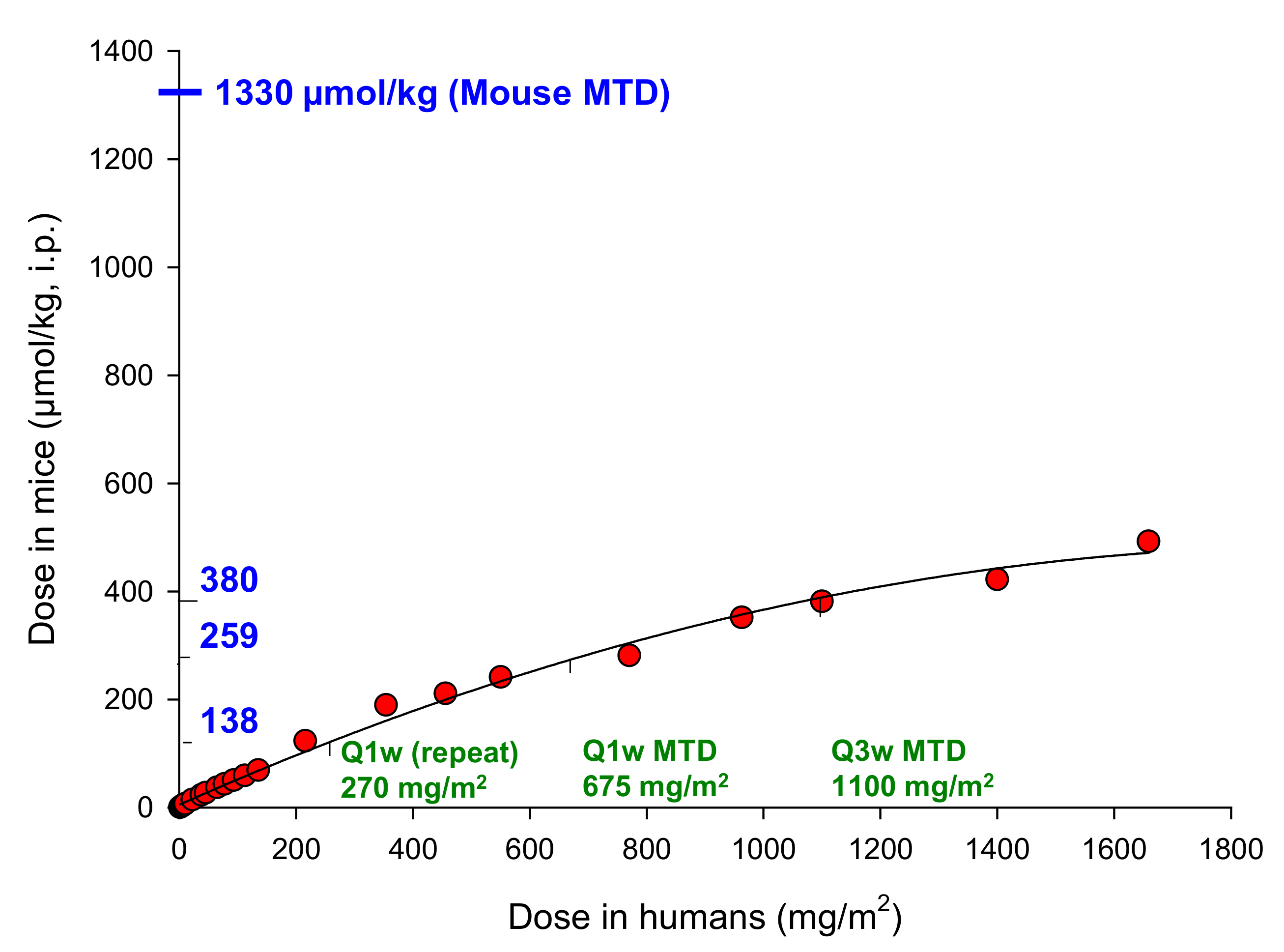
2.8. The Macaque Monkey Is a Suitable Pre-Clinical Animal Model for Evaluation of SN35141
3. Discussion
4. Materials and Methods
4.1. Test Compounds
4.2. Chemistry Experimental
Synthesis of SN29176 and SN35141
4.3. Cell Lines, Cytotoxicity Assays and Multicellular Layer (MCL) Assays
4.4. Clonogenic Survival of Mouse versus Human Bone Marrow Progenitor Cells
4.5. Prodrug Metabolism by Recombinant AKR1C3
4.6. Candidate Gene Expression
4.7. Flow Cytometry
4.8. Western Immunoblotting
4.9. Animals, Excision and Growth Delay Assays
4.10. Immunostaining
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, R.P.; Bristow, R.G.; Fyles, A.; Koritzinsky, M.; Milosevic, M.; Wouters, B.G. Hypoxia and predicting radiation response. Semin. Radiat. Oncol. 2015, 25, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The clinical significance of hypoxia in human cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, P.; Vignali, P.D.A.; Delgoffe, G.M.; Curran, M.A. Hypoxia reduction sensitizes refractory cancers to immunotherapy. Annu. Rev. Med. 2021, 73. [Google Scholar] [CrossRef]
- Pettersen, E.O.; Ebbesen, P.; Gieling, R.G.; Williams, K.J.; Dubois, L.; Lambin, P.; Ward, C.; Meehan, J.; Kunkler, I.H.; Langdon, S.P.; et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: The METOXIA consortium. J. Enzym. Inhib. Med. Chem 2015, 30, 689–721. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M. SR 4233 (tirapazamine): A new anticancer drug exploiting hypoxia in solid tumours. Br. J. Cancer 1993, 67, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Haffty, B.G.; Wilson, L.D.; Son, Y.H.; Cho, E.I.; Papac, R.J.; Fischer, D.B.; Rockwell, S.; Sartorelli, A.C.; Ross, D.A.; Sasaki, C.T.; et al. Concurrent chemo-radiotherapy with mitomycin C compared with porfiromycin in squamous cell cancer of the head and neck: Final results of a randomized clinical trial. Int. J. Radiat. Oncol. Biol Phys. 2005, 61, 119–128. [Google Scholar] [CrossRef]
- Phillips, R.M.; Hendriks, H.R.; Peters, G.J. EO9 (Apaziquone): From the clinic to the laboratory and back again. Br. J. Pharmacol. 2013, 168, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Patterson, L.H. Rationale for the use of aliphatic N-oxides of cytotoxic anthraquinones as prodrug DNA binding agents: A new class of bioreductive agent. Cancer Metastasis Rev. 1993, 12, 119–134. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. The hypoxia-activated prodrug TH-302: Exploiting hypoxia in cancer therapy. Front. Pharmacol. 2021, 12, 524. [Google Scholar] [CrossRef]
- Estrada-Bernal, A.; Le, A.T.; Doak, A.E.; Tirunagaru, V.G.; Silva, S.; Bull, M.R.; Smaill, J.B.; Patterson, A.V.; Kim, C.; Liu, S.V.; et al. Tarloxotinib is a hypoxia-activated pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin. Cancer Res. 2021, 27, 1463–1475. [Google Scholar] [CrossRef]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Wang, A.T.; Theil, A.; Bridewell, D.J.; Wilson, W.R.; Patterson, A. V Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: A role for NADPH: Cytochrome P450 oxidoreductase under hypoxia. Biochem. Pharmacol. 2007, 74, 810–820. [Google Scholar] [CrossRef]
- Hunter, F.W.; Young, R.J.; Shalev, Z.; Vellanki, R.N.; Wang, J.; Gu, Y.; Joshi, N.; Sreebavan, S.; Weinreb, I.; Goldstein, D.P.; et al. Identification of P450 oxidoreductase as a major determinant of sensitivity to hypoxia-activated prodrugs. Cancer Res. 2015, 75, 4211–4223. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Abbattista, M.R.; Tipparaju, S.R.; Lambie, N.K.; Su, J.; Li, D.; Wilson, W.R.; Dachs, G.U.; Patterson, A. V Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol. Pharmacol. 2012, 81, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Abbattista, M.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M.; Drury, J.E. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007, 464, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.F.; Lin, H.K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef]
- Jameson, M.B.; Rischin, D.; Pegram, M.; Gutheil, J.; Patterson, A.V.; Denny, W.A.; Wilson, W.R. A phase I trial of PR-104, a nitrogen mustard prodrug activated by both hypoxia and aldo-keto reductase 1C3, in patients with solid tumors. Cancer Chemother. Pharmacol. 2010, 65, 791–801. [Google Scholar] [CrossRef]
- McKeage, M.J.; Gu, Y.; Wilson, W.R.; Hill, A.; Amies, K.; Melink, T.J.; Jameson, M.B. A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumour patients. BMC Cancer 2011, 11, 432. [Google Scholar] [CrossRef] [Green Version]
- McKeage, M.J.; Jameson, M.B.; Ramanathan, R.K.; Rajendran, J.; Gu, Y.; Wilson, W.R.; Melink, T.J.; Tchekmedyian, N.S. PR-104 a bioreductive pre-prodrug combined with gemcitabine or docetaxel in a phase Ib study of patients with advanced solid tumours. BMC Cancer 2012, 12, 496. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Chan, S.L.; Lin, C.C.; Chiorean, E.G.; Holcombe, R.F.; Mulcahy, M.F.; Carter, W.D.; Patel, K.; Wilson, W.R.; Melink, T.J. PR-104 plus sorafenib in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2011, 68, 539–545. [Google Scholar] [CrossRef]
- Patel, K.; Lewiston, D.; Gu, Y.; Hicks, K.O.; Wilson, W.R. Analysis of the hypoxia-activated dinitrobenzamide mustard phosphate prodrug PR-104 and its alcohol metabolite PR-104A in plasma and tissues by liquid chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 856, 302–311. [Google Scholar] [CrossRef]
- Patel, K.; Choy, S.F.; Hicks, K.O.; Melink, T.J.; Holford, N.H.G.; Wilson, W.R. A combined pharmacokinetic model for the hypoxia-targeted prodrug PR-104A in humans, dogs, rats and mice predicts species differences in clearance and toxicity. Cancer Chemother. Pharmacol. 2011, 67, 1145–1155. [Google Scholar] [CrossRef]
- Birtwistle, J.; Hayden, R.E.; Khanim, F.L.; Green, R.M.; Pearce, C.; Davies, N.J.; Wake, N.; Schrewe, H.; Ride, J.P.; Chipman, J.K.; et al. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat. Res. 2009, 662, 67–74. [Google Scholar] [CrossRef]
- Desmond, J.C.; Mountford, J.C.; Drayson, M.T.; Walker, E.A.; Hewison, M.; Ride, J.P.; Luong, Q.T.; Hayden, R.E.; Vanin, E.F.; Bunce, C.M. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003, 63, 505–512. [Google Scholar] [PubMed]
- Houghton, P.J.; Lock, R.; Carol, H.; Morton, C.L.; Phelps, D.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; et al. Initial testing of the hypoxia activated prodrug PR-104 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2011, 57, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Mauch, P.; Vergilio, J.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell. Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Atwell, G.J.; Wilson, W.R. Metabolism and excretion of the novel bioreductive prodrug PR-104 in mice, rats, dogs and humans. Drug Metab. Dispos. 2010, 38, 498–508. [Google Scholar] [CrossRef]
- Velica, P.; Davies, N.J.; Rocha, P.P.; Schrewe, H.; Ride, J.P.; Bunce, C.M. Lack of functional and expression homology between human and mouse aldo-keto reductase 1C enzymes: Implications for modelling human cancers. Mol. Cancer 2009, 8, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Singleton, R.S.; Guise, C.P.; Ferry, D.M.; Pullen, S.M.; Dorie, M.J.; Brown, J.M.; Patterson, A.V.; Wilson, W.R. DNA cross-links in human tumor cells exposed to the prodrug PR-104A: Relationships to hypoxia, bioreductive metabolism, and cytotoxicity. Cancer Res. 2009, 69, 3884–3891. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Bellemare, V.; Labrie, F.; Luu-The, V. Molecular characterization of the cynomolgus monkey Macaca fascicularis steroidogenic enzymes belonging to the aldo-keto reductase family. J. Steroid Biochem. Mol. Biol. 2007, 104, 75–80. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Anderson, R.F.; Boyd, M.; Denny, W.A. Hypoxia-selective antitumor agents. Synthesis and hypoxic cell cytotoxicity of regioisomers of the hypoxia-selective cytotoxin 5-[N,N- bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1996, 39, 2518–2528. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Atwell, G.J.; Schultz, D.; Xu, X.Z.; Denny, W.A. Hypoxia-selective antitumor agents. Structure-activity relationships for hypoxia-selective cytotoxicity among analogues of 5-[N,N-bis(2- chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1994, 37, 2175–2184. [Google Scholar] [CrossRef]
- Palmer, B.D.; van Zijl, P.; Denny, W.A.; Wilson, W.R. Reductive chemistry of the novel hypoxia-selective cytotoxin 5-[N,N- bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1995, 38, 1229–1241. [Google Scholar] [CrossRef]
- Atwell, G.J.; Yang, S.; Pruijn, F.B.; Pullen, S.M.; Hogg, A.; Patterson, A.V.; Wilson, W.R.; Denny, W.A. Synthesis and structure-activity relationships for 2,4-dinitrobenzamide-5 mustards as prodrugs for the Escherichia coli nfsB nitroreductase in gene therapy. J. Med. Chem. 2007, 50, 1197–1212. [Google Scholar] [CrossRef]
- Atwell, G.J.; Boyd, M.; Palmer, B.D.; Anderson, R.F.; Pullen, S.M.; Wilson, W.R.; Denny, W.A. Synthesis and evaluation of 4-substituted analogues of 5-[N,N-bis (2- chloroethyl)amino]-2-nitrobenzamide as bioreductively activated prodrugs using an Escherichia coli nitroreductase. Anticancer Drug Des. 1996, 11, 553–567. [Google Scholar]
- Van Der Wiel, A.M.A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.; Lieuwes, N.; Biemans, R.; Lin, X.; et al. Selectively targeting tumor hypoxia with the hypoxia-activated prodrug CP-506. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Denny, W.A.; Atwell, G.J.; Yang, S.; Wilson, W.R.; Patterson, A.V.; Helsby, N.A. Novel Nitrophenyl Mustard and Nitrophenylaziridine Alcohols and Their Corresponding Phosphates and Their Use as Targeted Cytotoxic Agents. Patent WO 2005/042471 A1, 12 May 2005. [Google Scholar]
- Smaill, J.B.; Patterson, A.V.; Ashoorzadeh, A.; Guise, C.P.; Mowday, A.M.; Ackerley, D.F.; Williams, E.; Copp, J.N. Preparation of Nitrobenzamide Mustard Prodrugs Metabolized by Microorganism Nitroreductases or an Hypoxic Environment for Treating Hyperproliferative Disorders. Patent WO 2014/031012 A1, 27 February 2014. [Google Scholar]
- Flanagan, J.U.; Atwell, G.J.; Heinrich, D.M.; Brooke, D.G.; Silva, S.; Rigoreau, L.J.; Trivier, E.; Turnbull, A.P.; Raynham, T.; Jamieson, S.M.; et al. Morpholylureas are a new class of potent and selective inhibitors of the type 5 17-b-hydroxysteroid dehydrogenase (AKR1C3). Bioorg. Med. Chem. 2014, 22, 967–977. [Google Scholar] [CrossRef]
- Manesh, D.M.; Benito, J.; Foehrenbacher, A.; Bohlander, S.K.; Jing, D.; Konopleva, M.; Gu, Y.; Mulaw, M.A.; Smaill, J.B.; El-Hoss, J.; et al. A novel fluorometric assay for aldo-keto reductase 1C3 predicts metabolic activation of the nitrogen mustard prodrug PR-104A in human leukaemia cells. Biochem. Pharmacol. 2014, 88, 36–45. [Google Scholar] [CrossRef]
- Phillips, R.M. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef] [Green Version]
- Denny, W.A. Prospects for hypoxia-activated anticancer drugs. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 395–399. [Google Scholar] [CrossRef]
- Rooseboom, M.; Commandeur, J.N.; Vermeulen, N.P. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol. Rev. 2004, 56, 53–102. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.K.; Crowley, J.J.; Lara, P.N., Jr.; McCoy, J.; Lau, D.H.; Tucker, R.W.; Mills, G.M.; Gandara, D.R. Phase III trial of paclitaxel plus carboplatin with or without tirapazamine in advanced non-small-cell lung cancer: Southwest Oncology Group Trial S0003. J. Clin. Oncol. 2005, 23, 9097–9104. [Google Scholar] [CrossRef]
- DiSilvestro, P.A.; Ali, S.; Craighead, P.S.; Lucci, J.A.; Lee, Y.C.; Cohn, D.E.; Spirtos, N.M.; Tewari, K.S.; Muller, C.; Gajewski, W.H.; et al. Phase III randomized trial of weekly cisplatin and irradiation versus cisplatin and tirapazamine and irradiation in stages IB2, IIA, IIB, IIIB, and IVA cervical carcinoma limited to the pelvis: A Gynecologic Oncology Group study. J. Clin. Oncol. 2014, 32, 458–464. [Google Scholar] [CrossRef]
- Chawla, S.P.; Cranmer, L.D.; Van Tine, B.A.; Reed, D.R.; Okuno, S.H.; Butrynski, J.E.; Adkins, D.R.; Hendifar, A.E.; Kroll, S.; Ganjoo, K.N. Phase II Study of the Safety and Antitumor Activity of the Hypoxia-Activated Prodrug TH-302 in Combination with Doxorubicin in Patients with Advanced Soft Tissue Sarcoma. J. Clin. Oncol. 2014, 32, 3299–3306. [Google Scholar] [CrossRef]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J., Jr.; Chiorean, E.G.; Rosen, P.J.; et al. Randomized Phase II Trial of Gemcitabine plus TH-302 versus Gemcitabine in Patients with Advanced Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.K.; Houghton, P.J. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur. J. Cancer 2004, 40, 837–844. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.K.; McMahon, M.; Plummer, S.M.; Higgins, L.G.; Penning, T.M.; Igarashi, K.; Hayes, J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: Demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009, 30, 1571–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Atwell, G.J.; Denny, W.A. Synthesis of asymmetric halomesylate mustards with aziridineethanol/alkali metal halides: Application to an improved synthesis of the hypoxia prodrug PR-104. Tetrahedron 2007, 63, 5470–5476. [Google Scholar] [CrossRef]
- Atwell, G.J.; Denny, W.A. Synthesis of 3H- and 2H4-labelled versions of the hypoxia-activated pre-prodrug 2-[(2-bromoethyl)-2,4-dinitro-6-[[[2-(phosphonooxy)ethyl]amino]carbonyl]anilino]ethyl methanesulfonate (PR-104). J. Label. Comp. Radiopharm. 2007, 50, 7–12. [Google Scholar] [CrossRef]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar]
- Ahn, G.O.; Botting, K.J.; Patterson, A.V.; Ware, D.C.; Tercel, M.; Wilson, W.R. Radiolytic and cellular reduction of a novel hypoxia-activated cobalt(III) prodrug of a chloromethylbenzindoline DNA minor groove alkylator. Biochem. Pharmacol. 2006, 71, 1683–1694. [Google Scholar] [CrossRef]
- Halim, M.; Yee, D.J.; Sames, D. Imaging induction of cytoprotective enzymes in intact human cells: Coumberone, a metabolic reporter for human AKR1C enzymes reveals activation by panaxytriol, an active component of red ginseng. J. Am. Chem. Soc. 2008, 130, 14123–14128. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, M.; Marks, D.F.; DeMaria, M.A.; Connole, M.; Johnson, R.P. Identification of primitive hematopoietic progenitor cells in the rhesus macaque. J. Med. Primatol. 2001, 30, 36–45. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbattista, M.R.; Ashoorzadeh, A.; Guise, C.P.; Mowday, A.M.; Mittra, R.; Silva, S.; Hicks, K.O.; Bull, M.R.; Jackson-Patel, V.; Lin, X.; et al. Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3. Pharmaceuticals 2021, 14, 1231. https://doi.org/10.3390/ph14121231
Abbattista MR, Ashoorzadeh A, Guise CP, Mowday AM, Mittra R, Silva S, Hicks KO, Bull MR, Jackson-Patel V, Lin X, et al. Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3. Pharmaceuticals. 2021; 14(12):1231. https://doi.org/10.3390/ph14121231
Chicago/Turabian StyleAbbattista, Maria R., Amir Ashoorzadeh, Christopher P. Guise, Alexandra M. Mowday, Rituparna Mittra, Shevan Silva, Kevin O. Hicks, Matthew R. Bull, Victoria Jackson-Patel, Xiaojing Lin, and et al. 2021. "Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3" Pharmaceuticals 14, no. 12: 1231. https://doi.org/10.3390/ph14121231
APA StyleAbbattista, M. R., Ashoorzadeh, A., Guise, C. P., Mowday, A. M., Mittra, R., Silva, S., Hicks, K. O., Bull, M. R., Jackson-Patel, V., Lin, X., Prosser, G. A., Lambie, N. K., Dachs, G. U., Ackerley, D. F., Smaill, J. B., & Patterson, A. V. (2021). Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3. Pharmaceuticals, 14(12), 1231. https://doi.org/10.3390/ph14121231