
Acetonitrile Adducts of Tranexamic Acid as Sensitive Ions for Quantification at Residue Levels in Human Plasma by UHPLC-MS/MS


 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
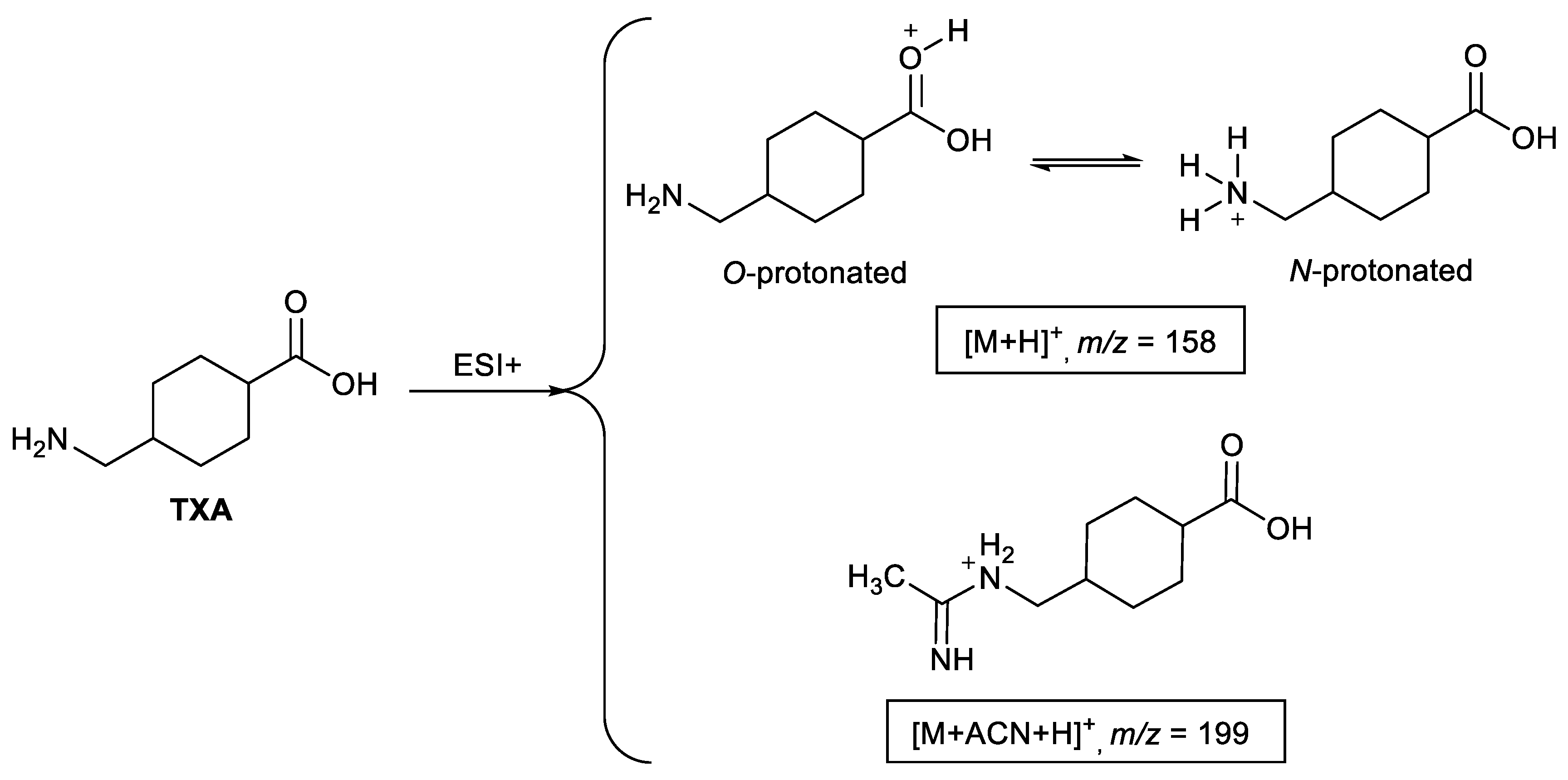
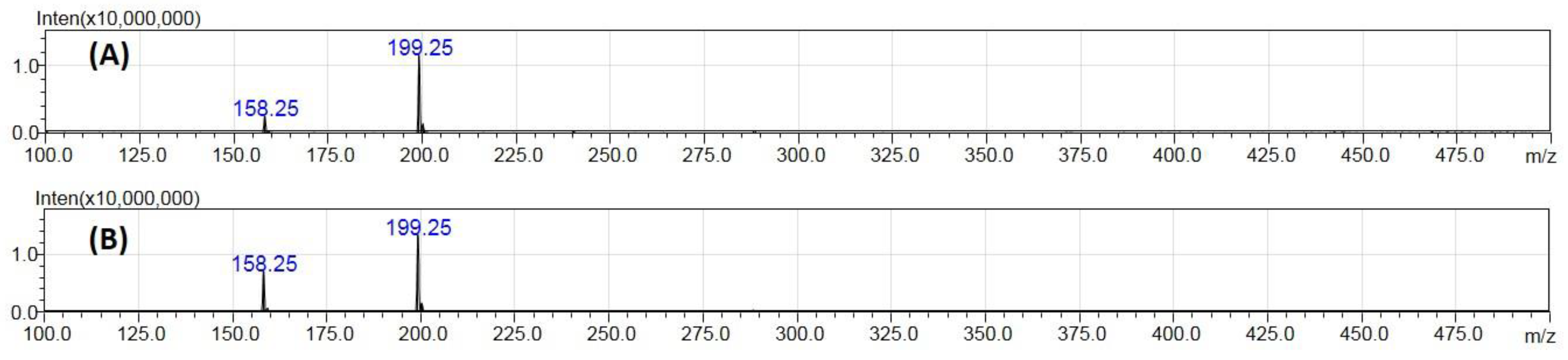
2.1. Ionization Studies
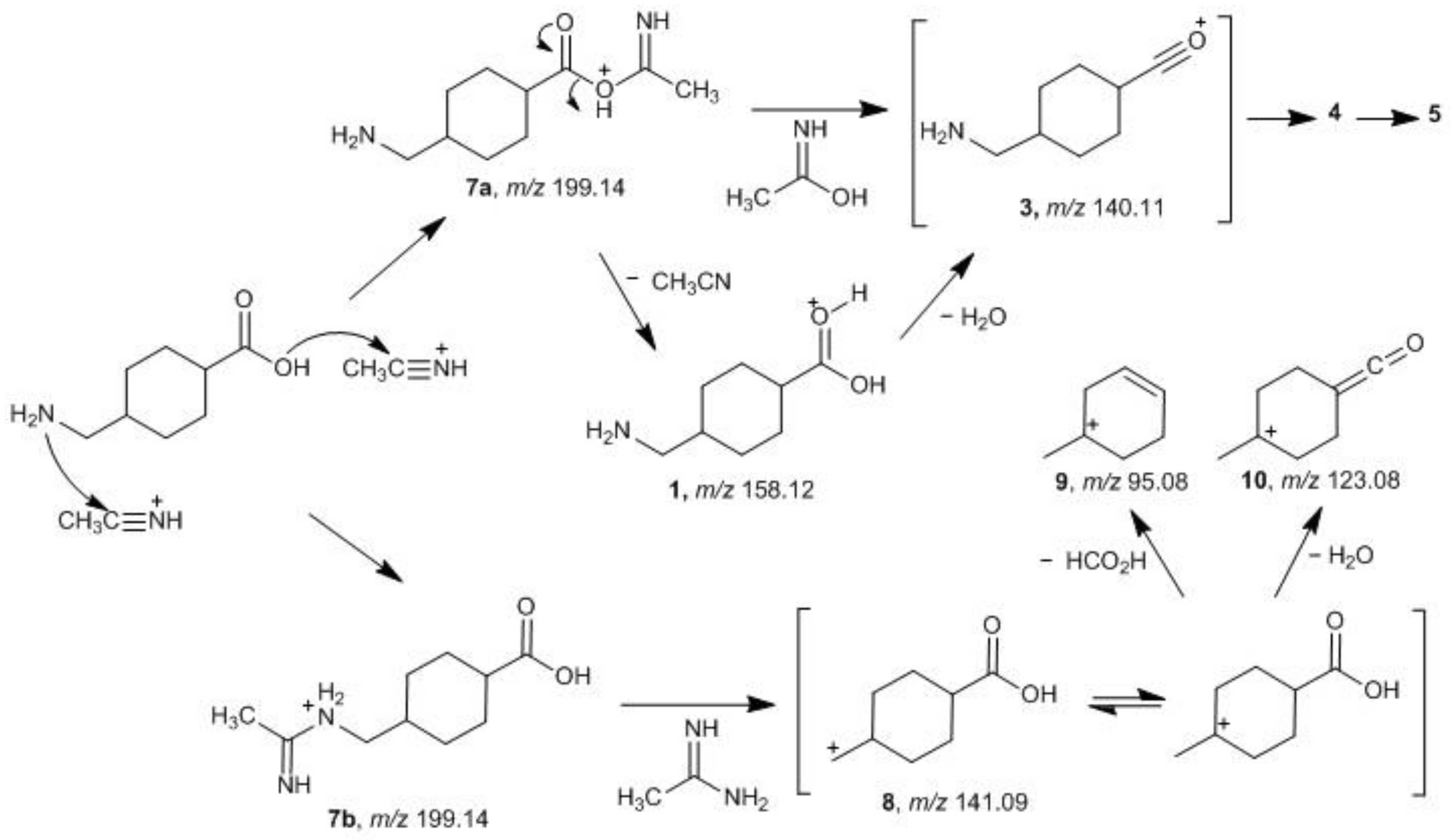
2.2. Fragmentation Mechanisms and DFT Calculations
2.3. Application of the Acetonitrile Adduct for TXA Quantification in Plasma Samples
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. Sample Preparation
3.3. Instrumentation and UHPLC-MS/MS Analysis
3.4. Analytical Figures of Merit of Adduct Ion-Based Quantification Method
3.5. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gross, J.H. Mass Spectrometry, 3rd ed.; Springer Nature: Cham, Switzerland, 2017; Volume 132, pp. 17–18. ISBN 978-3-319-54397-0. [Google Scholar]
- Barreiros, L.; Silva, E.M.P.; Alencastre, I.S.; Lamghari, M.; Segundo, M.A. Determination of neuropeptide Y Y1 receptor antagonist BIBP 3226 and evaluation of receptor expression based on liquid chromatography coupled with tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 6625–6632. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.M.P.; Varandas, P.; Melo, T.; Barros, C.; Alencastre, I.S.; Barreiros, L.; Domingues, P.; Lamghari, M.; Domingues, M.R.M.; Segundo, M.A. Gas-phase structural characterization of neuropeptides Y Y1 receptor antagonists using mass spectrometry: Orbitrap vs triple quadrupole. J. Pharm. Biomed. Anal. 2018, 151, 227–234. [Google Scholar] [CrossRef]
- Barreiros, L.; Cunha-Reis, C.; Silva, E.M.P.; Carvalho, J.R.B.; das Neves, J.; Sarmento, B.; Segundo, M.A. Development and validation of a liquid chromatography-MS/MS method for simultaneous quantification of tenofovir and efavirenz in biological tissues and fluids. J. Pharm. Biomed. Anal. 2017, 136, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.M.P.; Barros, C.; Santos, C.M.M.; Barros, A.S.; Domingues, M.R.M.; Silva, A.M.S. Characterization of 2,3-diarylxanthones by electrospray mass spectrometry: Gas-phase chemistry versus known antioxidant activity properties. Rapid Commun. Mass Spectrom. 2016, 30, 2228–2236. [Google Scholar] [CrossRef]
- Kebarle, P.; Verkerk, U.H. Electrospray: From ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 2009, 28, 898–917. [Google Scholar] [CrossRef] [PubMed]
- Erngren, I.; Nestor, M.; Pettersson, C.; Hedeland, M. Improved sensitivity in hydrophilic interaction liquid chromatography-electrospray-mass spectrometry after removal of sodium and potassium ions from biological samples. Metabolites 2021, 11, 170. [Google Scholar] [CrossRef]
- Verardo, G.; Baldini, M.; Ferfuia, C.; Gorassini, A. Rapid and selective screening for toxic phorbol esters in Jatropha curcas seed oil using high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. J. Chromatogr. A 2019, 1597, 63–75. [Google Scholar] [CrossRef]
- Hautajarvi, H.; Hukkanen, J.; Turpeinen, M.; Mattila, S.; Tolonen, A. Quantitative analysis of 4 beta- and 4 alpha-hydroxycholesterol in human plasma and serum by UHPLC/ESI-HR-MS. J. Chromatogr. B 2018, 1100, 179–186. [Google Scholar] [CrossRef]
- Kruve, A.; Kaupmees, K.; Liigand, J.; Oss, M.; Leito, I. Sodium adduct formation efficiency in ESI source. J. Mass Spectrom. 2013, 48, 695–702. [Google Scholar] [CrossRef]
- Mortier, K.A.; Zhang, G.F.; Van Peteghem, C.H.; Lambert, W.E. Adduct formation in quantitative bioanalysis: Effect of ionization conditions on paclitaxel. J. Am. Soc. Mass Spectrom. 2004, 15, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.; Chen, X.; Chen, J.F.; Liu, M.P.; Wang, J.Y.; Chen, Z.G.; Xie, Z.Y.; Yao, M.C. Determination of pseudoprotodioscin in rat plasma by UPLC-MS/MS: Assay development and application to pharmacokinetic study. J. Chromatogr. B 2016, 1026, 97–104. [Google Scholar] [CrossRef]
- Bao, Y.W.; Wang, Q.Y.; Tang, P.M. Lithium adduct as precursor ion for sensitive and rapid quantification of 20 (S)-protopanaxadiol in rat plasma by liquid chromatography/quadrupole linear ion trap mass spectrometry and application to rat pharmacokinetic study. J. Mass Spectrom. 2013, 48, 399–405. [Google Scholar] [CrossRef]
- Kruve, A.; Kaupmees, K. Adduct formation in ESI/MS by mobile phase additives. J. Am. Soc. Mass Spectrom. 2017, 28, 887–894. [Google Scholar] [CrossRef]
- Bogseth, R.; Edgcomb, E.; Jones, C.M.; Chess, E.K.; Hu, P.F. Acetonitrile adduct formation as a sensitive means for simple alcohol detection by LC-MS. J. Am. Soc. Mass Spectrom. 2014, 25, 1987–1990. [Google Scholar] [CrossRef]
- Kolkman, A.; Emke, E.; Bauerlein, P.S.; Carboni, A.; Tran, D.T.; ter Laak, T.L.; van Wezel, A.P.; de Voogt, P. Analysis of (functionalized) fullerenes in water samples by liquid chromatography coupled to high-resolution mass spectrometry. Anal. Chem. 2013, 85, 5867–5874. [Google Scholar] [CrossRef]
- Li, L.; Huhtala, S.; Sillanpaa, M.; Sainio, P. Liquid chromatography-mass spectrometry for C-60 fullerene analysis: Optimisation and comparison of three ionisation techniques. Anal. Bioanal. Chem. 2012, 403, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Maragou, N.C.; Thomaidis, N.S.; Koupparis, M.A. Optimization and comparison of ESI and APCI LC-MS/MS methods: A case study of irgarol 1051, diuron, and their degradation products in environmental samples. J. Am. Soc. Mass Spectrom. 2011, 22, 1826–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grujic, S.; Vasiljevic, T.; Lausevic, M.; Ast, T. Study on the formation of an amoxicillin adduct with methanol using electrospray ion trap tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.Y.; Uboh, C.; Soma, L.; Hess, A.; Luo, Y.; Tsang, D.S. Sensitive liquid chromatographic/tandem mass spectrometric method for the determination of beclomethasone dipropionate and its metabolites in equine plasma and urine. J. Mass Spectrom. 2003, 38, 823–838. [Google Scholar] [CrossRef]
- Silva, E.M.P.; Barreiros, L.; Sa, P.; Afonso, C.; Kozek-Langenecker, S.; Segundo, M.A. Analytical methods for quantification of tranexamic acid in biological fluids: A review. Microchem J. 2017, 134, 333–342. [Google Scholar] [CrossRef]
- Fabresse, N.; Fall, F.; Etting, I.; Devillier, P.; Alvarez, J.C.; Grassin-Delyle, S. LC-MS/MS determination of tranexamic acid in human plasma after phospholipid clean-up. J. Pharm. Biomed. Anal. 2017, 141, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Gorynski, K.; Bojko, B.; Kluger, M.; Jerath, A.; Wasowicz, M.; Pawliszyn, J. Development of SPME method for concomitant sample preparation of rocuronium bromide and tranexamic acid in plasma. J. Pharm. Biomed. Anal. 2014, 92, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Delavenne, X.; Montbel, A.; Hodin, S.; Zufferey, P.; Basset, T. Quantification of total and unbound tranexamic acid in human plasma by ultrafiltration liquid chromatography/tandem mass spectrometry: Application to pharmacokinetic analysis. J. Pharm. Biomed. Anal. 2014, 91, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Bojko, B.; Vuckovic, D.; Mirnaghi, F.; Cudjoe, E.; Wasowicz, M.; Jerath, A.; Pawliszyn, J. Therapeutic monitoring of tranexamic acid concentration: High-throughput analysis with solid-phase microextraction. Ther. Drug Monit. 2012, 34, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bojko, B.; Vuckovic, D.; Cudjoe, E.; Hoque, M.E.; Mirnaghi, F.; Wasowicz, M.; Jerath, A.; Pawliszyn, J. Determination of tranexamic acid concentration by solid phase microextraction and liquid chromatography-tandem mass spectrometry: First step to in vivo analysis. J. Chromatogr. B 2011, 879, 3781–3787. [Google Scholar] [CrossRef]
- Abou-Diwan, C.; Sniecinski, R.M.; Szlam, F.; Ritchie, J.C.; Rhea, J.M.; Tanaka, K.A.; Molinaro, R.J. Plasma and cerebral spinal fluid tranexamic acid quantitation in cardiopulmonary bypass patients. J. Chromatogr. B 2011, 879, 553–556. [Google Scholar] [CrossRef]
- Grassin Delyle, S.; Abe, E.; Batisse, A.; Tremey, B.; Fischler, M.; Devillier, P.; Alvarez, J.C. A validated assay for the quantitative analysis of tranexamic acid in human serum by liquid chromatography coupled with electrospray ionization mass spectrometry. Clin. Chim. Acta 2010, 411, 438–443. [Google Scholar] [CrossRef]
- Chang, Q.; Yin, O.Q.P.; Chow, M.S.S. Liquid chromatography-tandem mass spectrometry method for the determination of tranexamic acid in human plasma. J. Chromatogr. B 2004, 805, 275–280. [Google Scholar] [CrossRef]
- Barreiros, L.; Amoreira, J.L.; Machado, S.; Fernandes, S.R.; Silva, E.M.P.; Sa, P.; Kietaibl, S.; Segundo, M.A. Determination of tranexamic acid in human plasma by UHPLC coupled with tandem mass spectrometry targeting sub-microgram per milliliter levels. Microchem. J. 2019, 144, 144–150. [Google Scholar] [CrossRef]
- Hoff, B.H. Acetonitrile as a building block and reactant. Synthesis 2018, 50, 2824–2852. [Google Scholar] [CrossRef]
- Hegarty, A.F.; Kelly, J.G.; Relihan, C.M. Formation of hemiaminals by N-protonation of ketenimines (etheneimines) sterically hindered at carbon. J. Chem. Soc. Perkin Trans. 1997, 2, 1175–1181. [Google Scholar] [CrossRef]
- Wu, L.M.; Liu, D.Q.; Kord, A.S. Gas-phase Meerwein reaction of epoxides with protonated acetonitrile generated by atmospheric pressure ionizations. J. Am. Soc. Mass Spectrom. 2010, 21, 1802–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.; Alex, A.; Nyaruwata, T.; Parsons, T.; Pullen, F. Using density functional theory to rationalise the mass spectral fragmentation of maraviroc and its metabolites. Rapid Commun. Mass Spectrom. 2010, 24, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Akira, K.; Nohara, A.; Utsunomiya, Y.; Mitome, H.; Watanabe, S.; Tanaka, M.; Tanaka, A. Application of quantitative NMR spectroscopy to the quality evaluation of diclofenac gargles as hospital preparations. Chem. Pharm. Bull. 2021, 69, 721–726. [Google Scholar] [CrossRef]
- Li, Z.H.; Fang, L.; Wang, J.; Dong, L.H.; Guo, Y.N.; Xie, Y.Y. An improved and practical synthesis of tranexamic acid. Org. Process Res. Dev. 2015, 19, 444–448. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline Bioanalytical Method Validation EMEA/CHMP/EWP/192217/2009. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 5 July 2021).
- Wilson, B.M.; Harris, C.R.; Wixted, J.T. Science is not a signal detection problem. Proc. Natl. Acad. Sci. USA 2020, 117, 5559–5567. [Google Scholar] [CrossRef] [PubMed]
- Lewandowsky, S.; Oberauer, K. Low replicability can support robust and efficient science. Nat. Commun. 2020, 11, 12. [Google Scholar] [CrossRef]
- ICH Guideline Q2(R1), Validation of Analytical Procedures: Text and Methodology, 541. 2005. Available online: https://www.ema.europa.eu/en/ich-q2-r1-validation-analytical-procedures-text-methodology (accessed on 5 July 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.-Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Luchini, G.; Alegre-Requena, J.V.; Funes-Ardoiz, I.; Paton, R.S. Goodvibes: Automated thermochemistry for heterogeneous computational chemistry data. F1000Research 2020, 9, 291. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACN Mobile Phase Composition (%, v/v) | [M + H]+/[M + ACN + H]+ Ratio (%) | |
|---|---|---|
| Flow Injection | Chromatography | |
| 80 | 21.4 ± 1.5 I | 10.5 ± 0.1 II |
| 70 | 29.0 ± 1.9 III | 23.3 ± 1.4 IV |
| 60 | 39.4 ± 2.4 V | 29.5 ± 1.7 VI |
| 50 | 54.7 ± 4.2 VII | 33.8 ± 0.1 VIII |
| Parameters | Precursor ion [M + ACN + H]+ (This Work) | Precursor ion [M + H]+ [30] |
|---|---|---|
| Linear range (ng mL−1) | 30–600 (180–3600 in plasma) | 30–600 (180–3600 in plasma) |
| Calibration curve | y = 0.00291 (±0.00003) x + 0.026 (±0.009) | y = 0.00296 (±0.00002) x + 0.035 (±0.007) |
| Correlation coefficient (r2) | ≥0.9983 | ≥0.9974 |
| LOD (ng mL−1) a | 3 | 18 |
| LOQ (ng mL−1) a | 6 | 36 |
| Precision (CV, %) b | ||
| Intra-day | 0.6 | 1.7 |
| Inter-day | 2.4 | 1.7 |
| Accuracy (%) b | ||
| Intra-day | 94.7 | 96.3 |
| Inter-day | 92.2 | 94.7 |
| Recovery (%) c | ||
| TXA | 98.8 ± 0.5 I | 96.3 ± 3.6 I |
| TXA-IS | 100.6 ± 1.0 II | 94.4 ± 1.0 III |
| Matrix factor (%) d | ||
| TXA | 105.4 ± 3.5 IV | 110.0 ± 7.6 IV |
| TXA-IS | 103.6 ± 6.9 V | 110.0 ± 6.3 V |
| IS normalized matrix factor e | 102.0 ± 5.4 VI | 100.0 ± 1.4 VI |
| Plasma Samples a | C[M + ACN + H]+ (This Work) | C[M + H]+ [30] |
|---|---|---|
| End of surgery | ||
| Patient 1 | 10.9 ± 0.1 I | 11.2 ± 0.1 I |
| Patient 2 | 15.7 ± 0.1 II | 16.0 ± 0.1 II |
| Patient 3 | 17.1 ± 0.5 III | 17.1 ± 0.2 III |
| Patient 4 | 14.0 ± 0.3 IV | 14.2 ± 0.1 IV |
| Patient 5 | 9.8 ± 0.1 V | 10.0 ± 0.1 V |
| 24 h after surgery | ||
| Patient 1 | 0.213 ± 0.001 VI | 0.219 ± 0.002 VI |
| Patient 2 | 0.226 ± 0.001 VII | 0.229 ± 0.001 VII |
| Patient 3 | 0.325 ± 0.002 VIII | 0.332 ± 0.002 VIII |
| Patient 4 | 0.214 ±0.004 IX | 0.226 ± 0.006 IX |
| Patient 5 | 0.269 ± 0.004 X | 0.275 ± 0.001 X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, E.M.P.; Barreiros, L.; Fernandes, S.R.; Sá, P.; Ramalho, J.P.P.; Segundo, M.A. Acetonitrile Adducts of Tranexamic Acid as Sensitive Ions for Quantification at Residue Levels in Human Plasma by UHPLC-MS/MS. Pharmaceuticals 2021, 14, 1205. https://doi.org/10.3390/ph14121205
Silva EMP, Barreiros L, Fernandes SR, Sá P, Ramalho JPP, Segundo MA. Acetonitrile Adducts of Tranexamic Acid as Sensitive Ions for Quantification at Residue Levels in Human Plasma by UHPLC-MS/MS. Pharmaceuticals. 2021; 14(12):1205. https://doi.org/10.3390/ph14121205
Chicago/Turabian StyleSilva, Eduarda M. P., Luisa Barreiros, Sara R. Fernandes, Paula Sá, João P. Prates Ramalho, and Marcela A. Segundo. 2021. "Acetonitrile Adducts of Tranexamic Acid as Sensitive Ions for Quantification at Residue Levels in Human Plasma by UHPLC-MS/MS" Pharmaceuticals 14, no. 12: 1205. https://doi.org/10.3390/ph14121205
APA StyleSilva, E. M. P., Barreiros, L., Fernandes, S. R., Sá, P., Ramalho, J. P. P., & Segundo, M. A. (2021). Acetonitrile Adducts of Tranexamic Acid as Sensitive Ions for Quantification at Residue Levels in Human Plasma by UHPLC-MS/MS. Pharmaceuticals, 14(12), 1205. https://doi.org/10.3390/ph14121205