
Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects


Abstract
:
1. Introduction
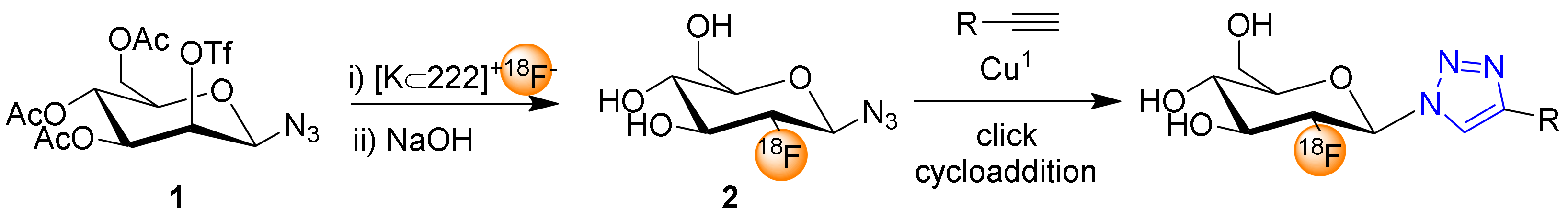
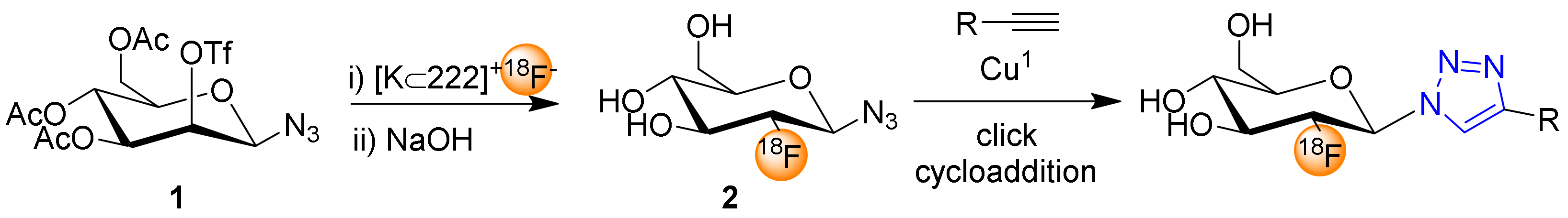
2. 2-Deoxy-2-[18F]fluoro-β-glucosyl Azide for Click Chemistry Based 18F-Fluoroglycosylation
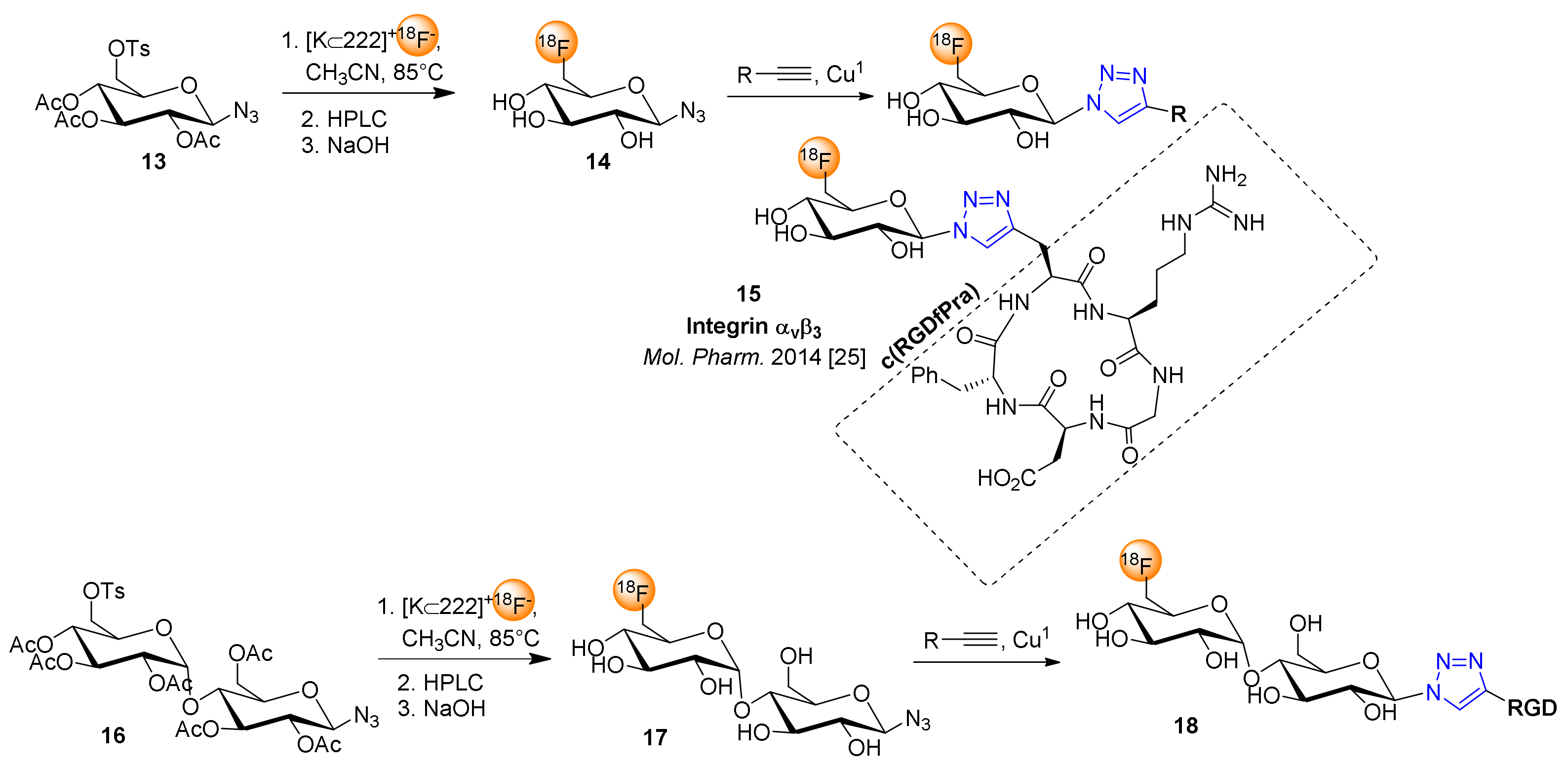
3. 6-Deoxy-6-[18F]fluoro-β-glycosyl Azides for Click Chemistry-Based 18F-Fluoroglycosylation
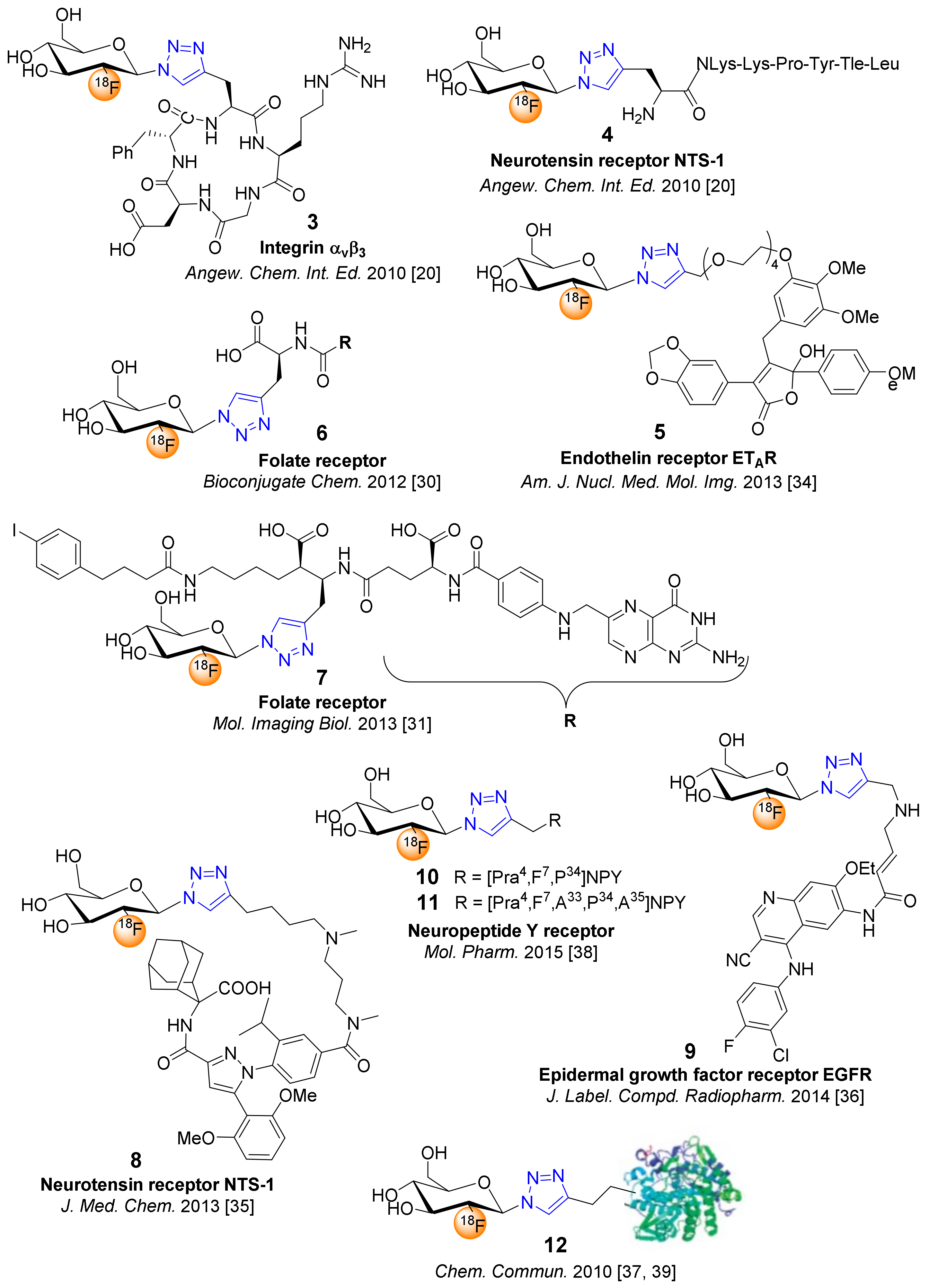
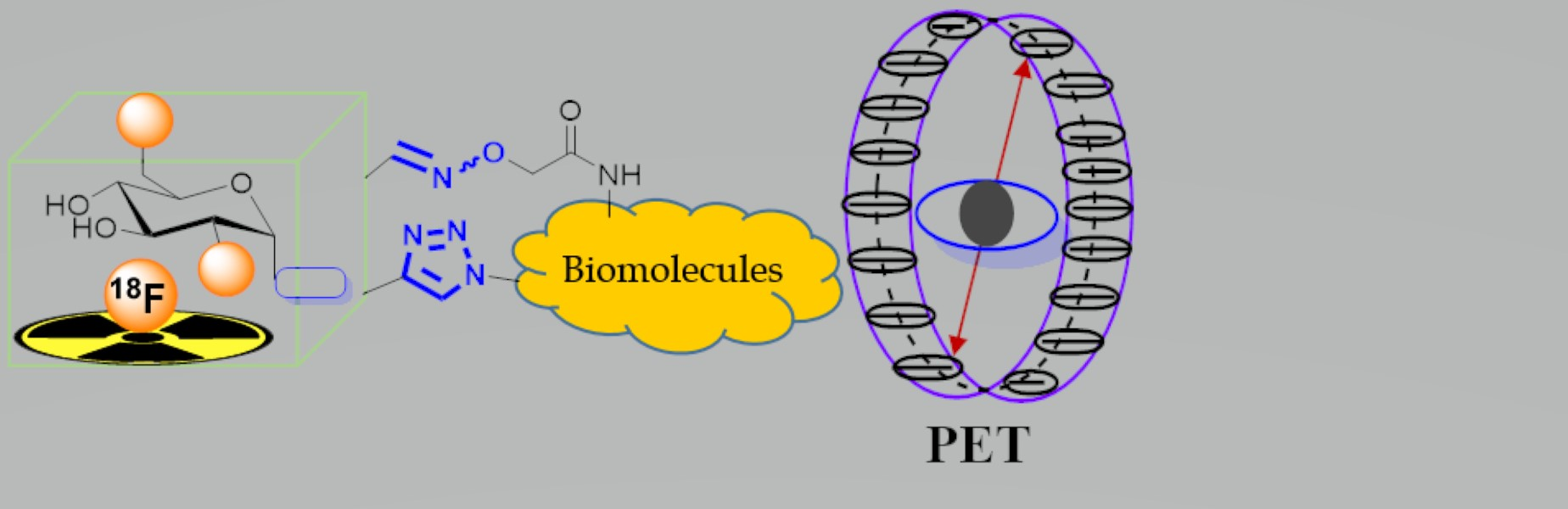
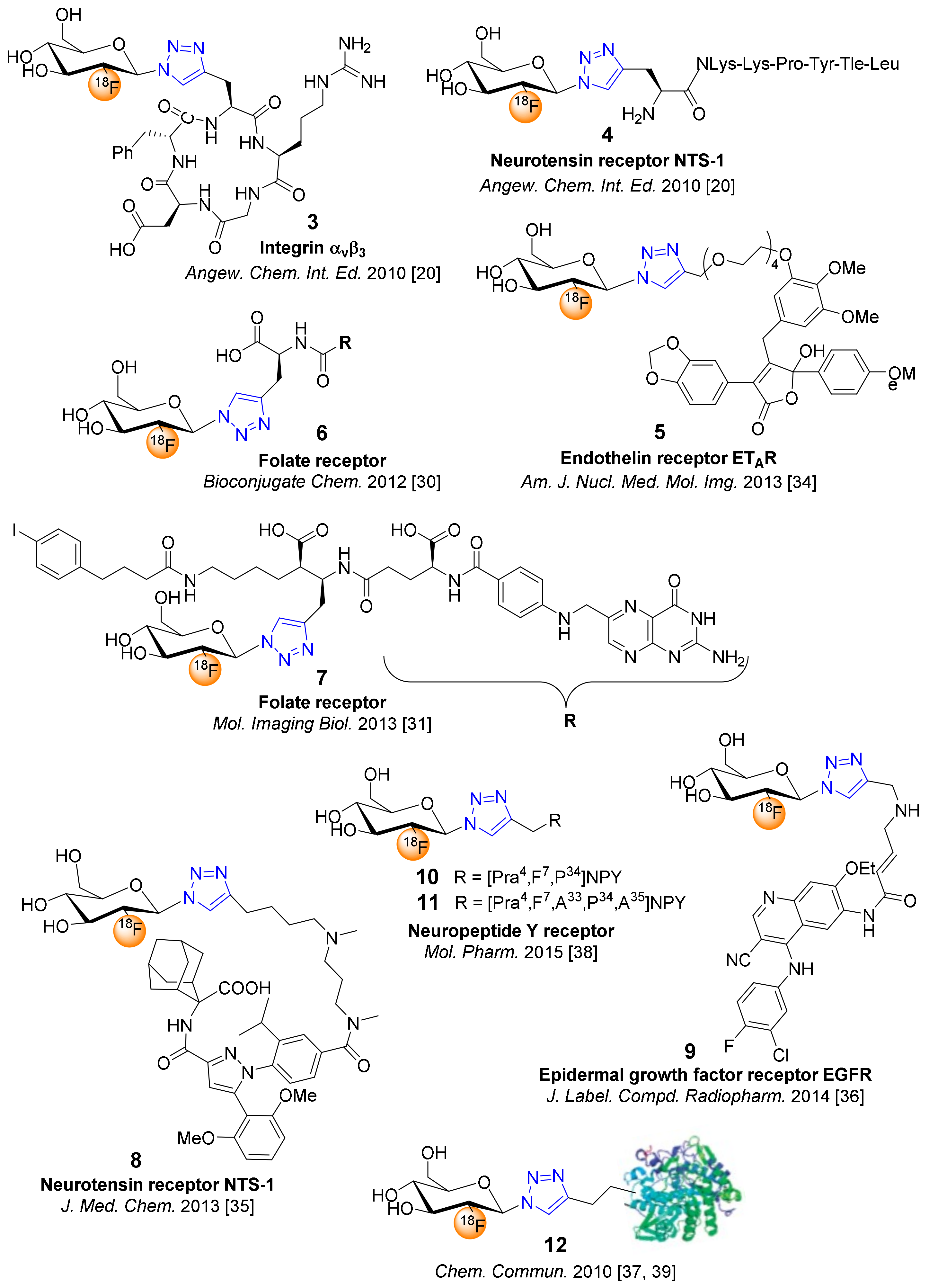
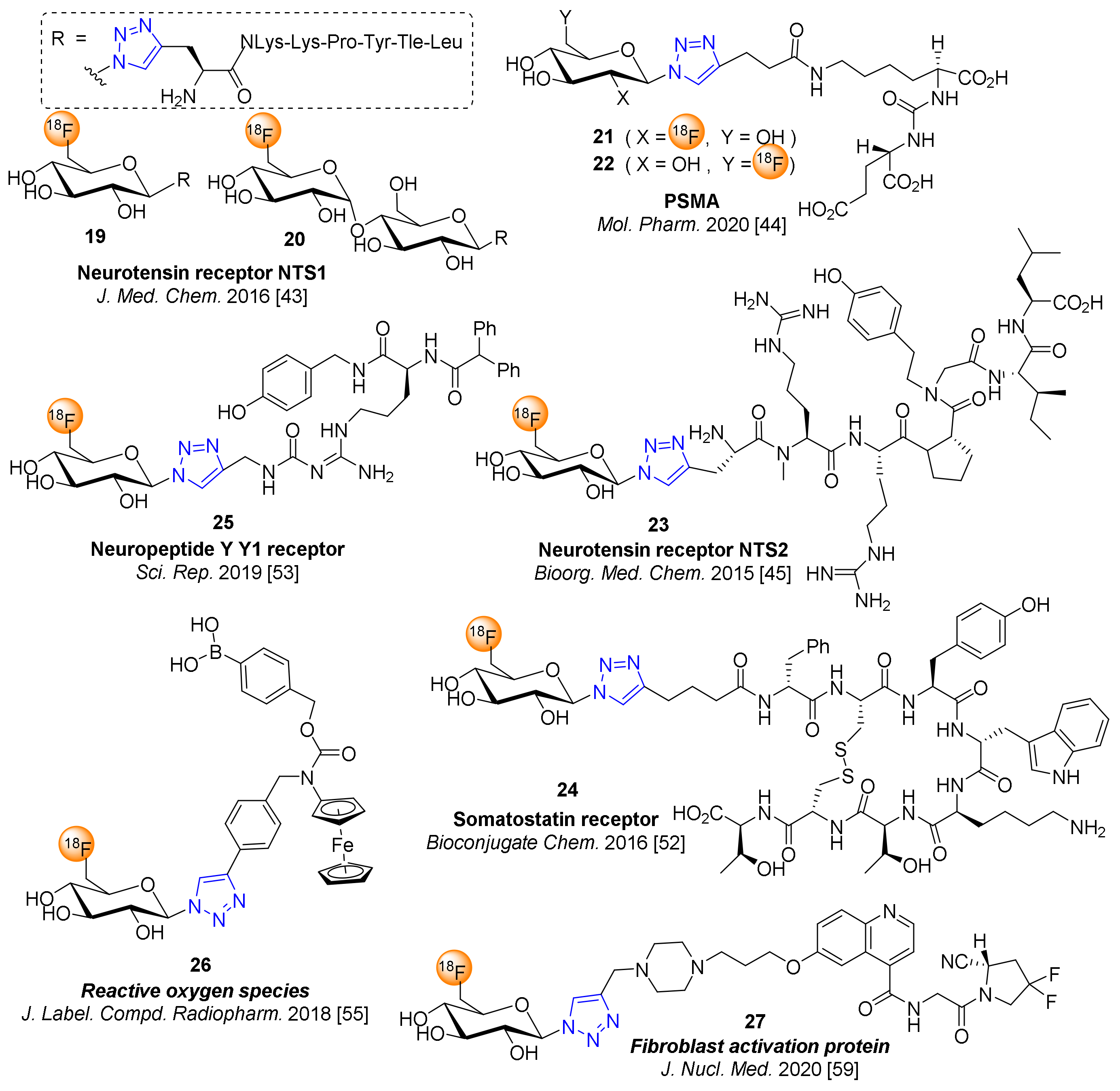
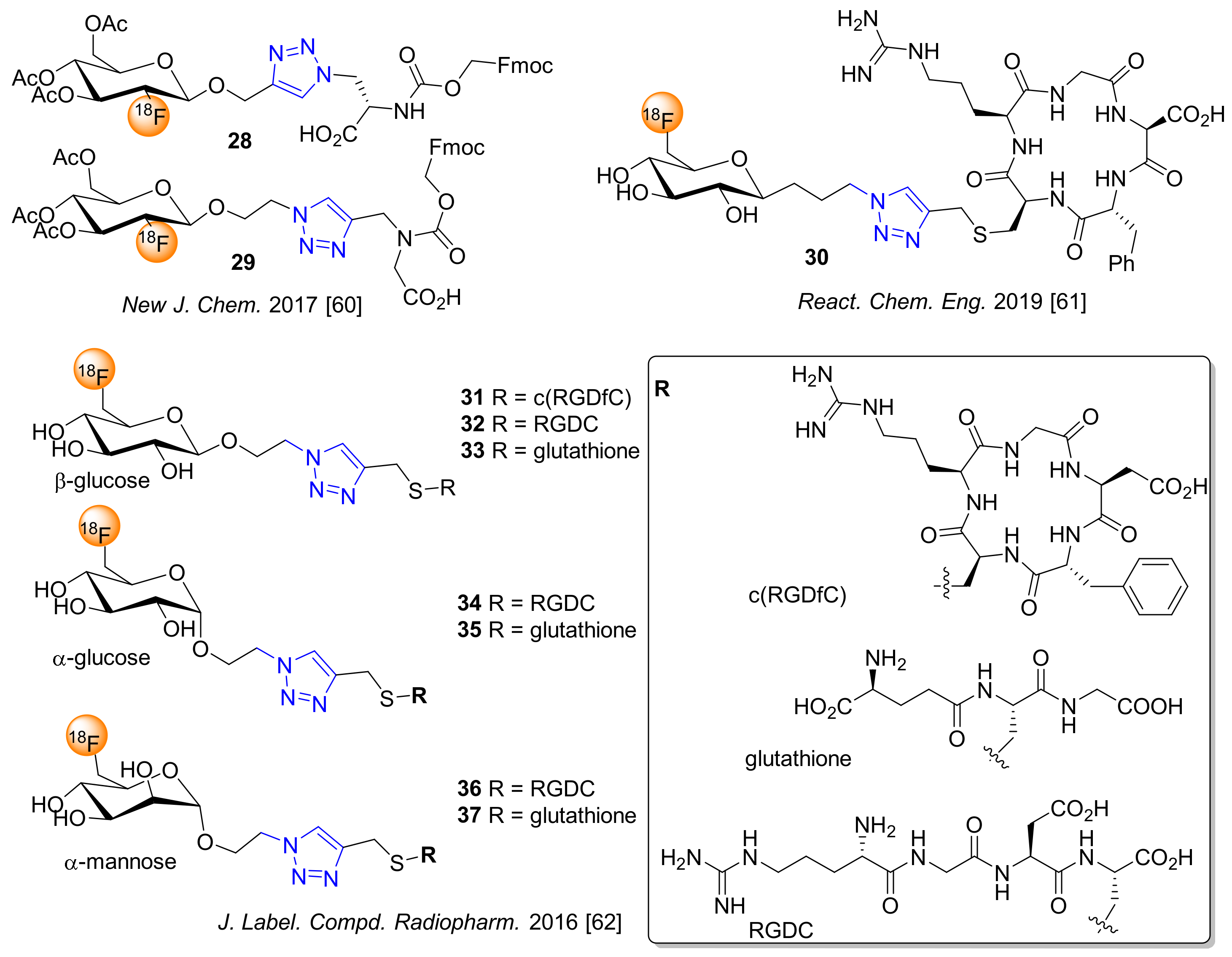
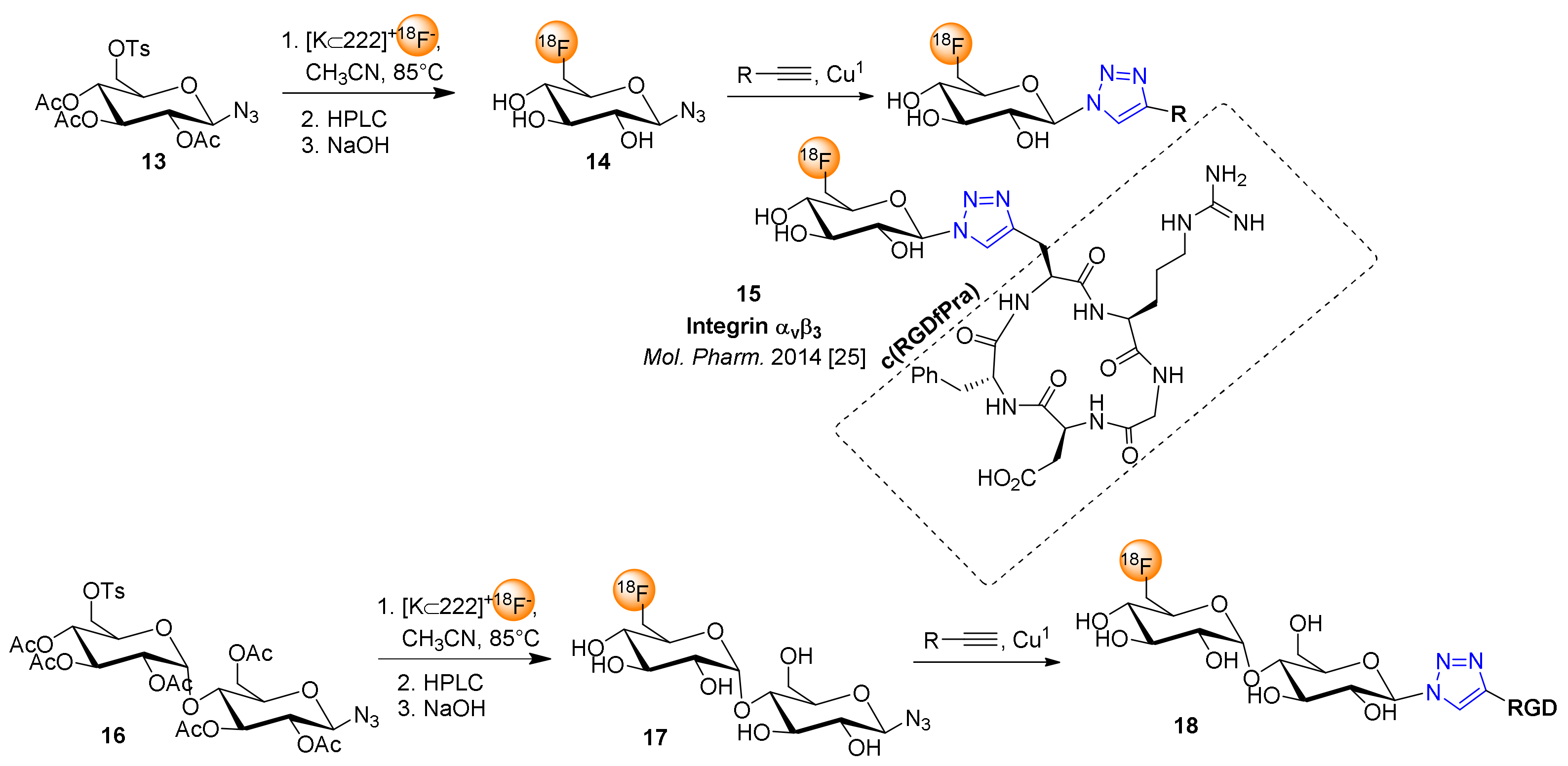
4. 18F-Fluoroglyosylation for the Synthesis of Triazolylalkyl-Linked 18F-Glycoconjugates by CuAAC
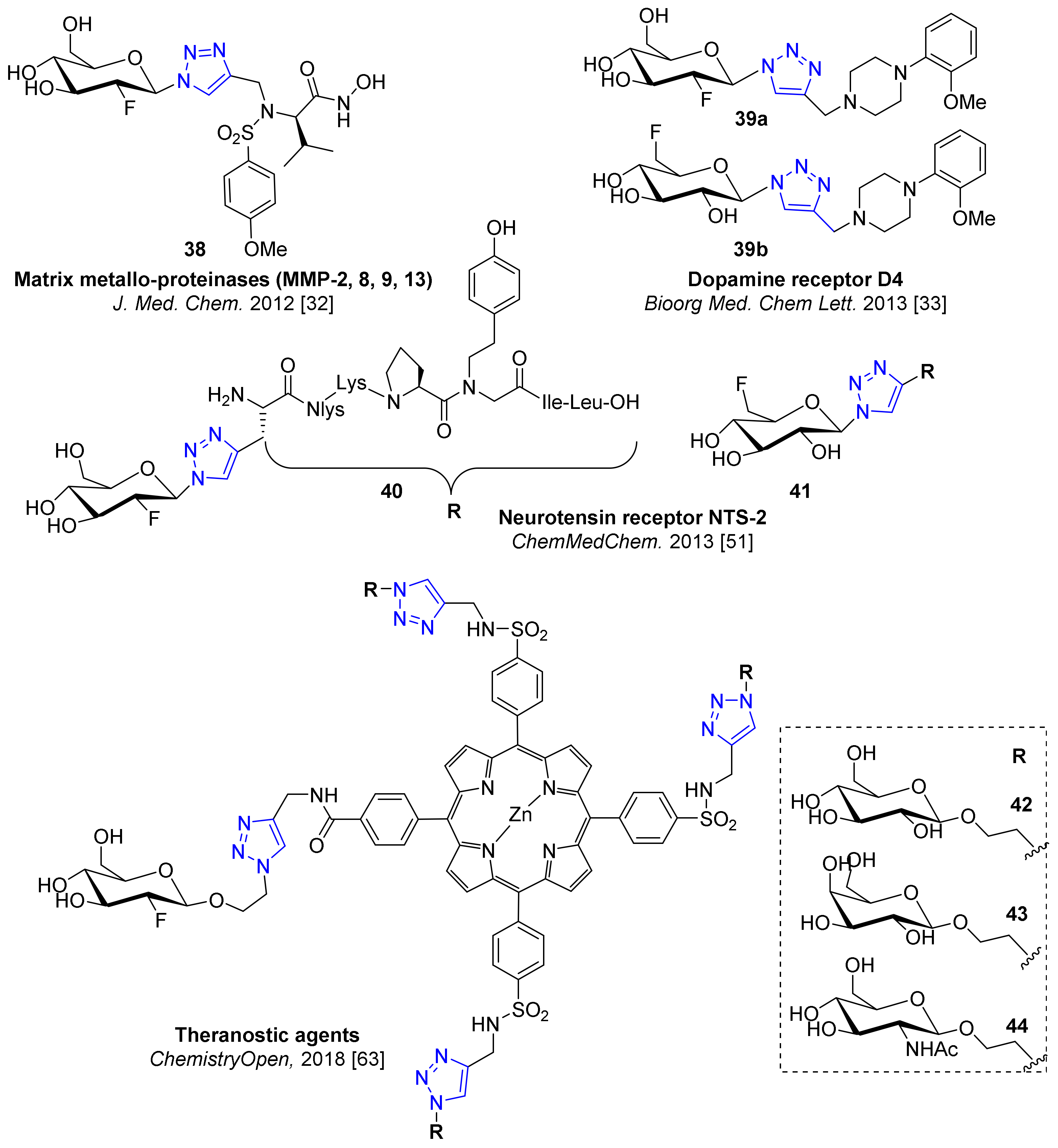
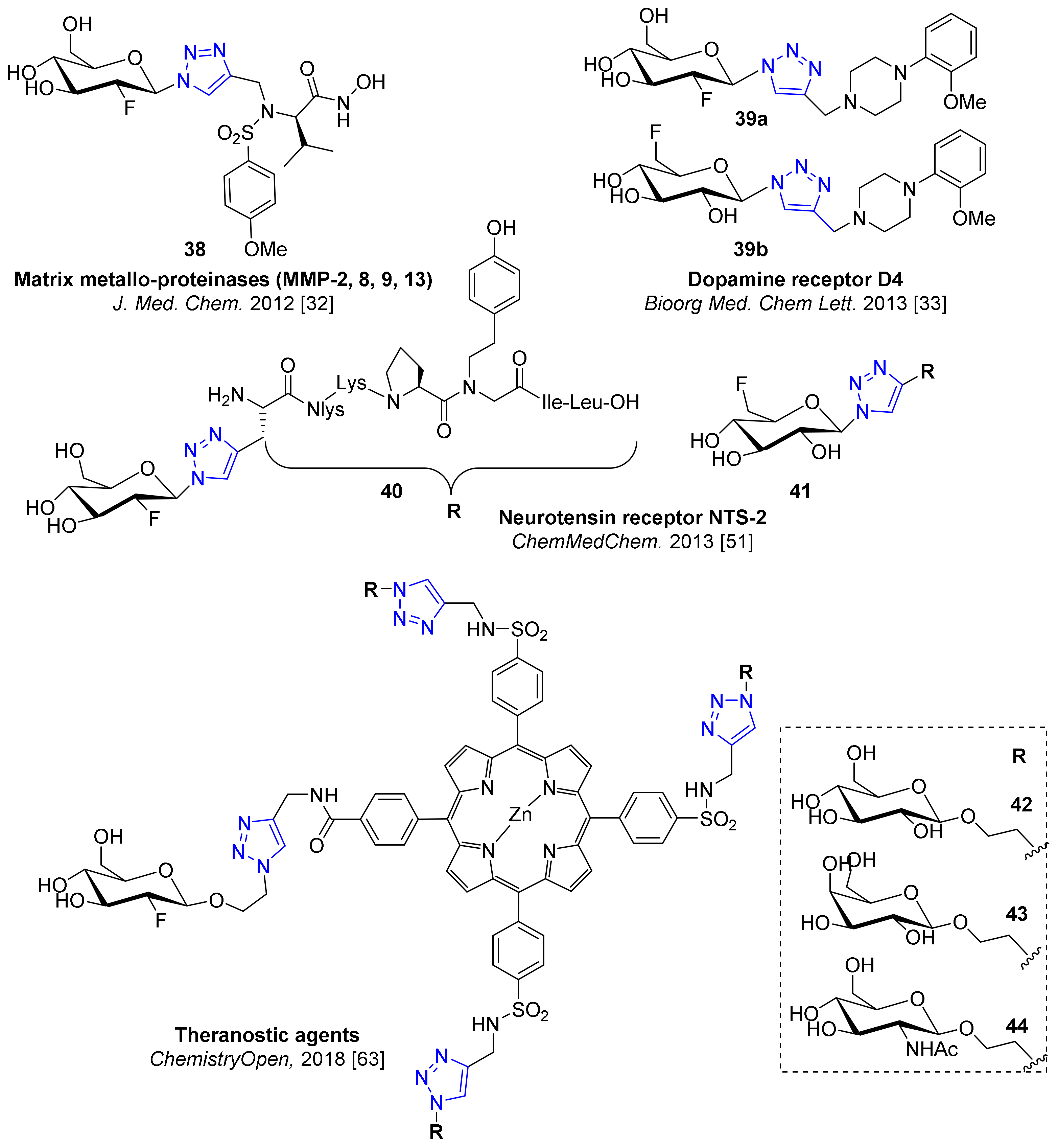
5. Examples of Non-Radioactive Fluoroglycosylation by Click Chemistry and Effects on Inhibitory Potency or Receptor Affinity
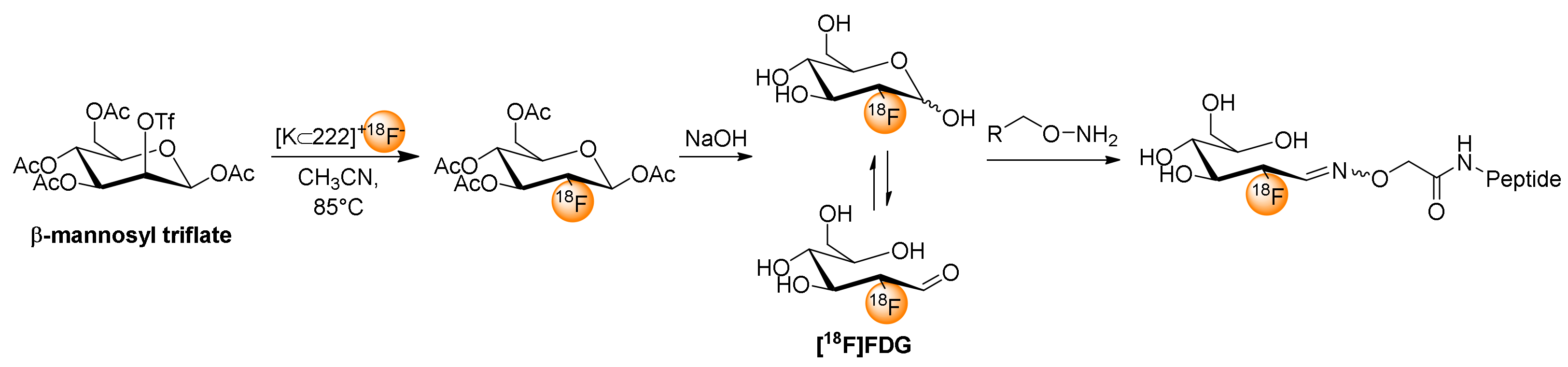
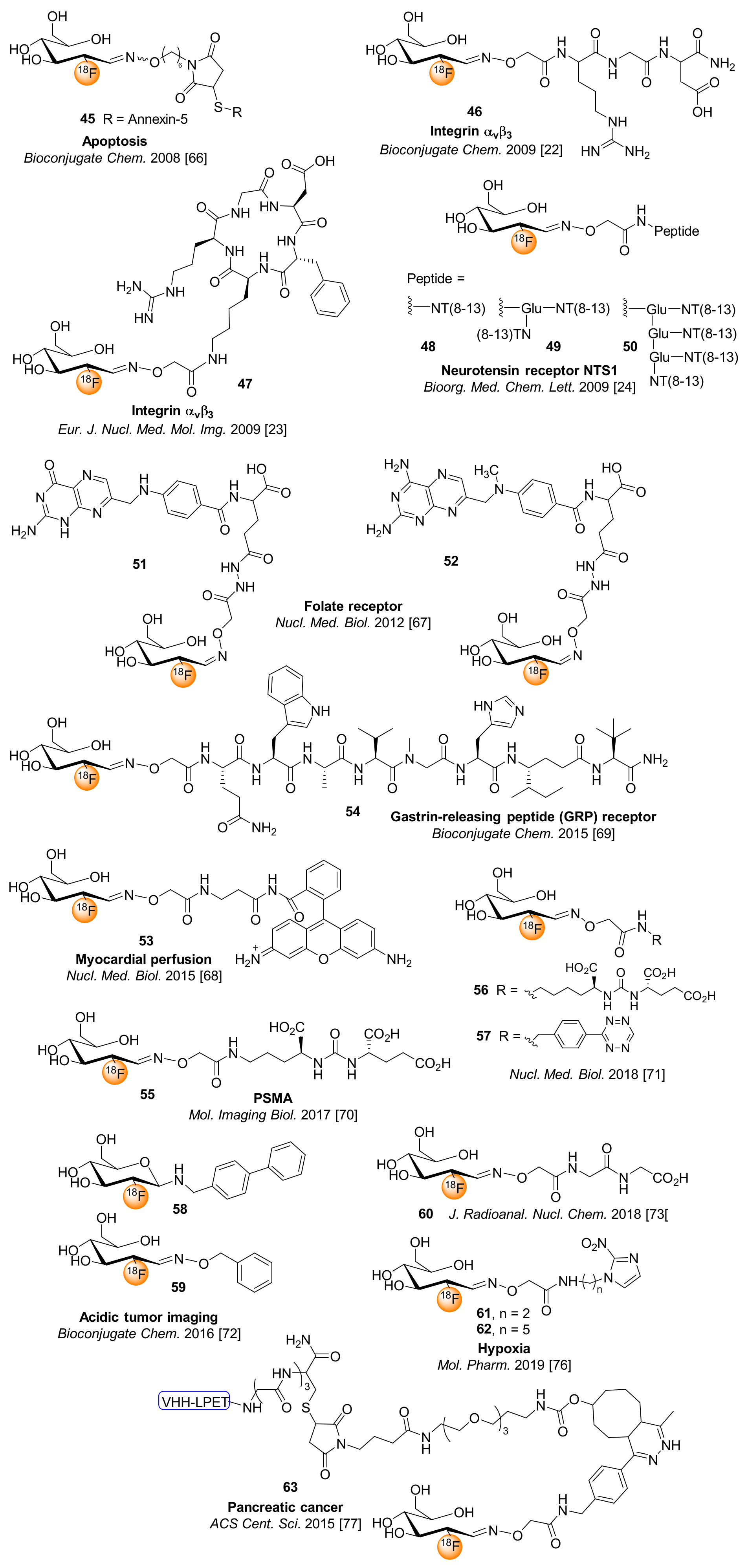
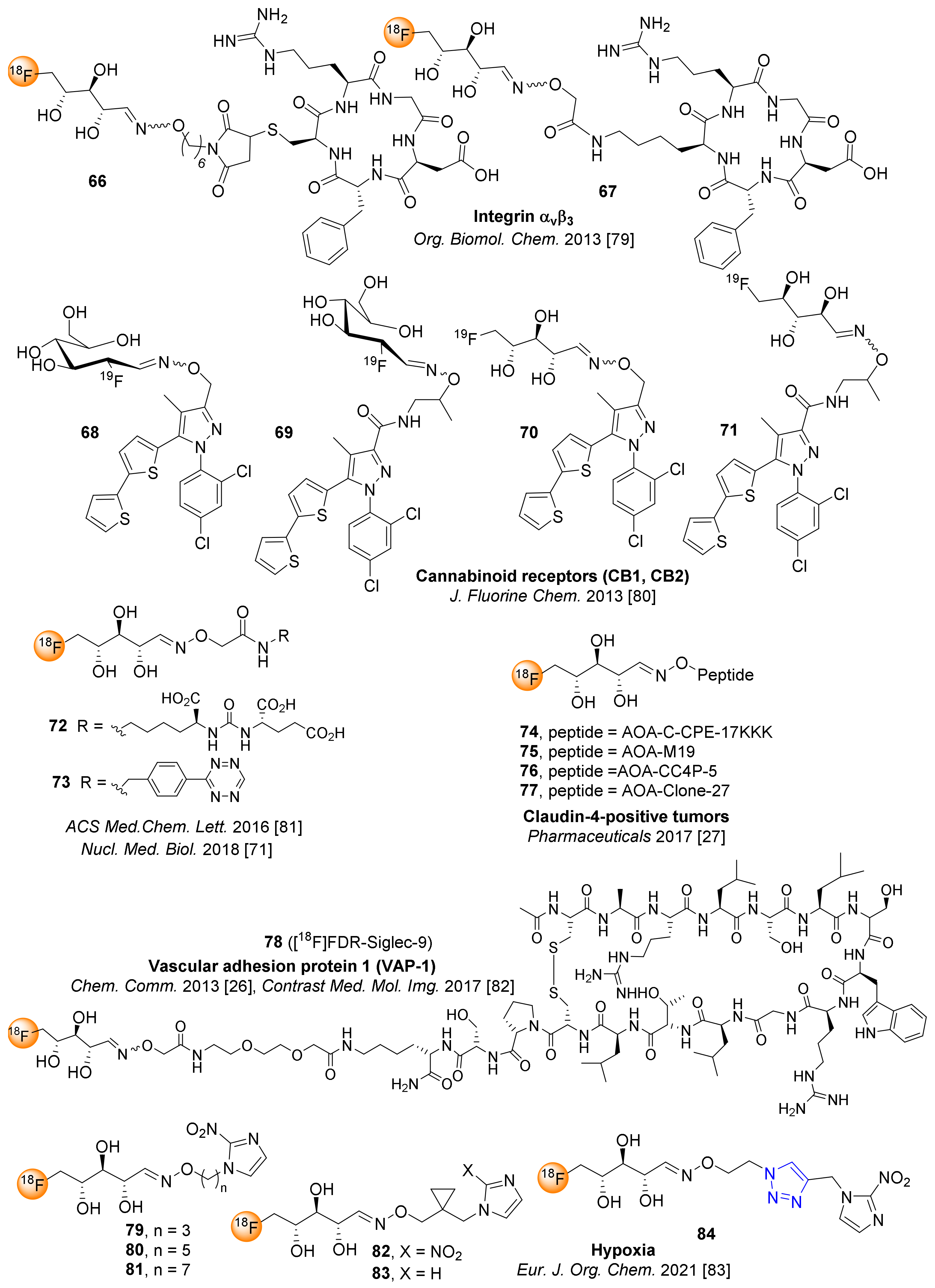
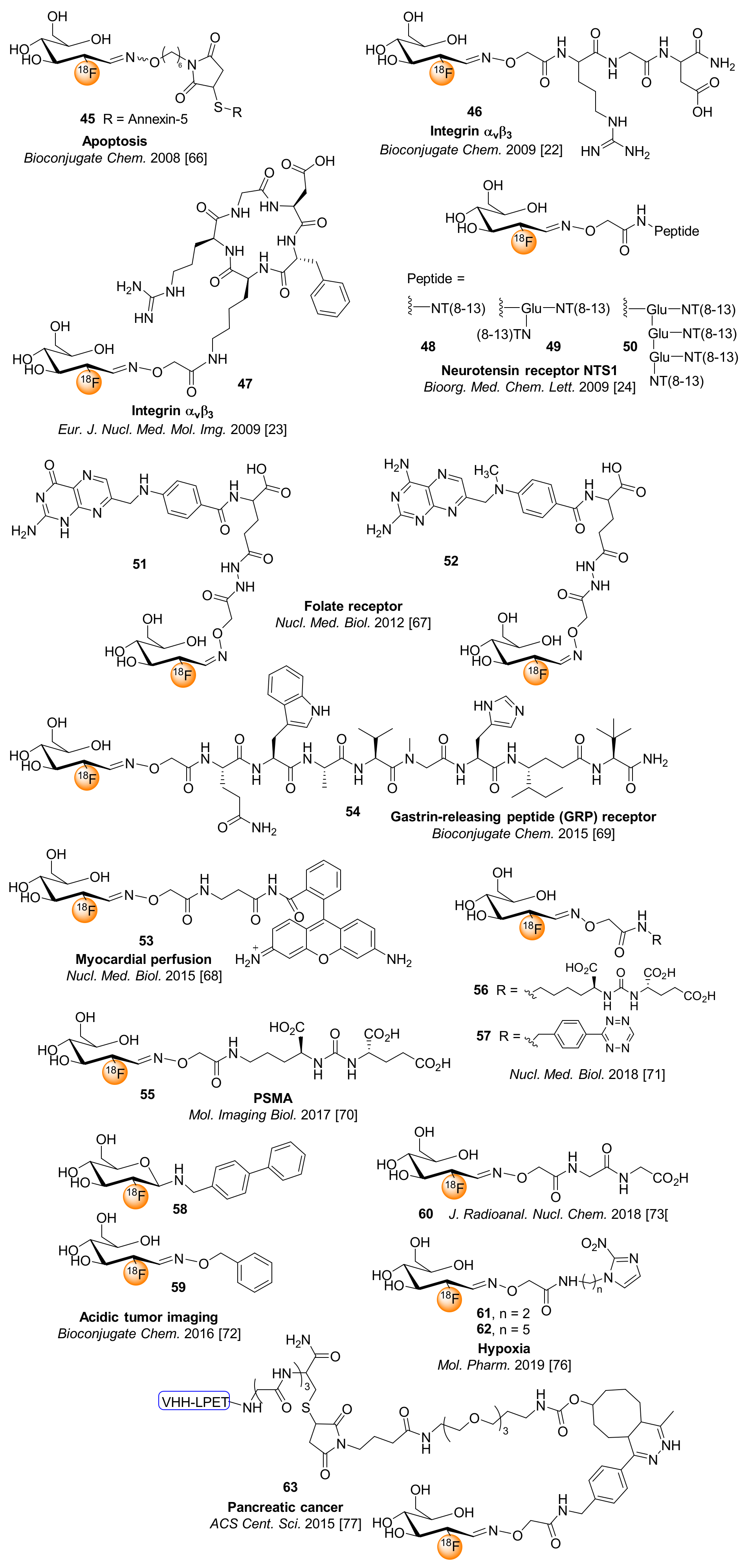
6. [18F]FDG for Chemoselective 18F-Fluoroglycosylation by Oxime Linkage
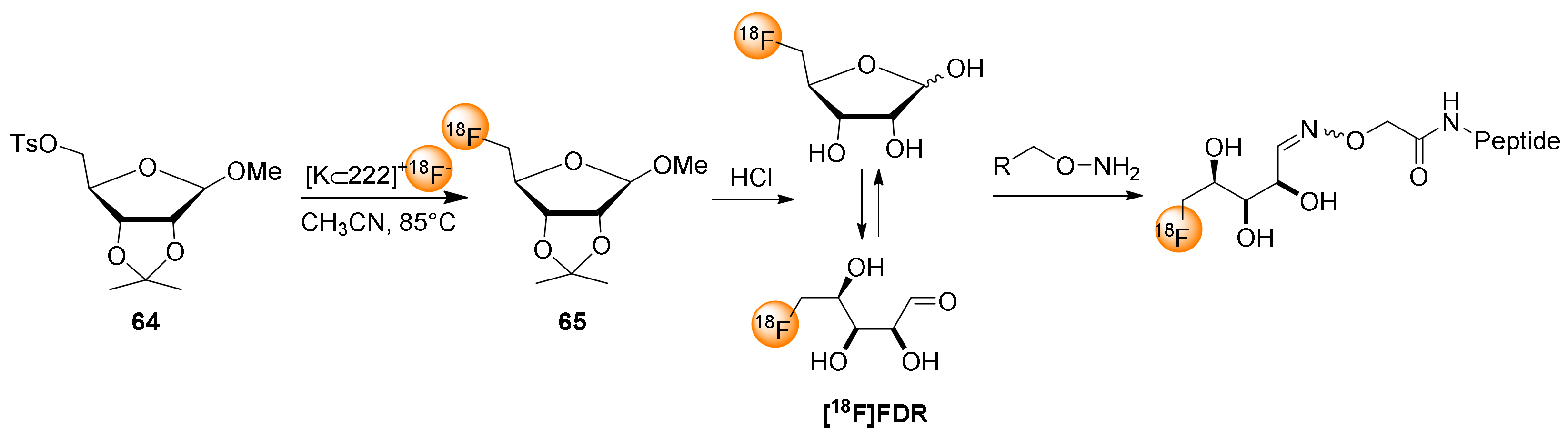
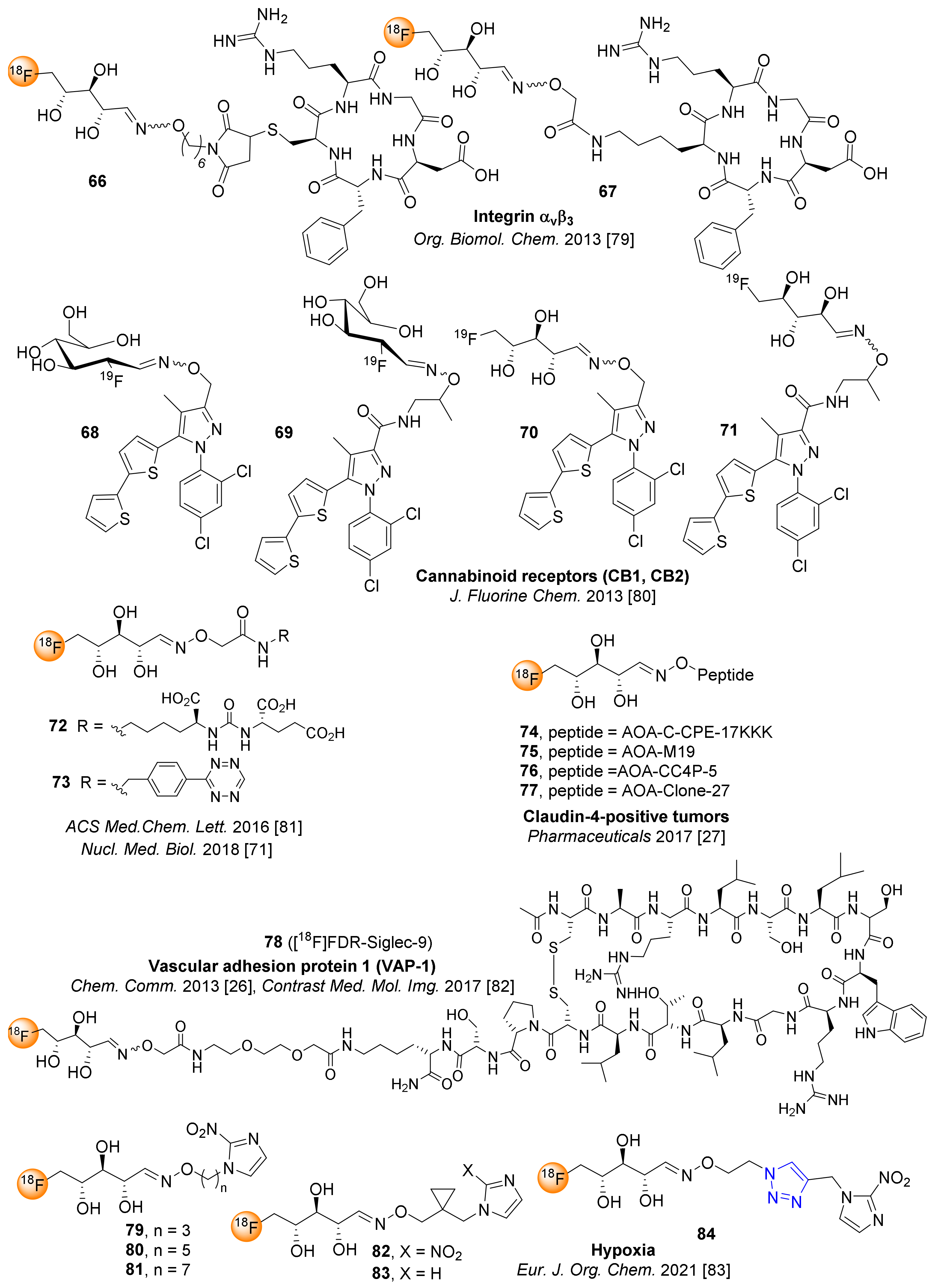
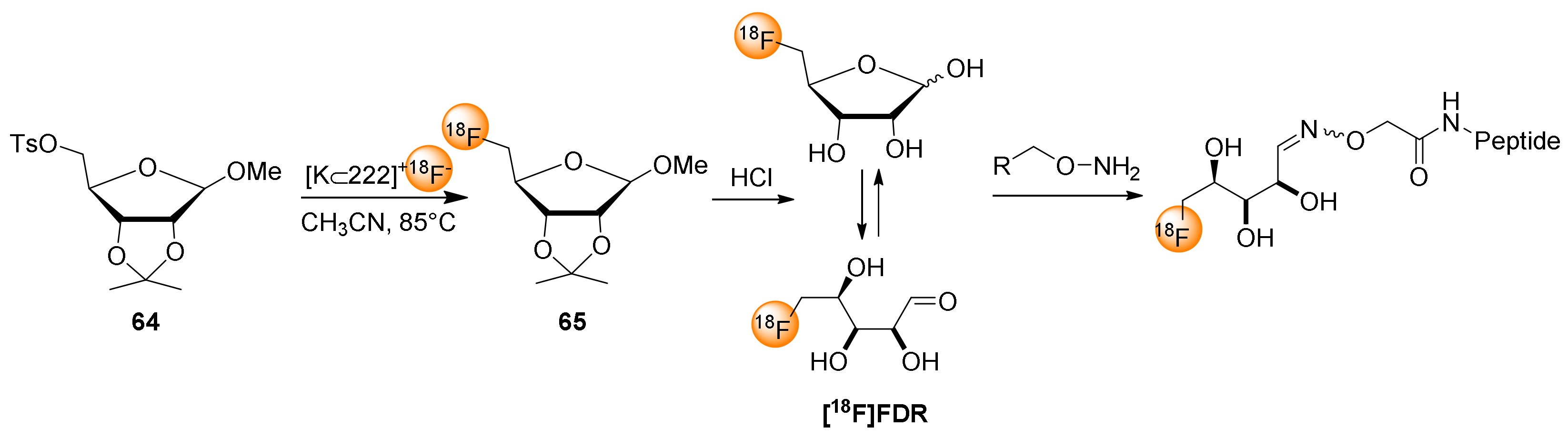
7. 5-[18F]fluoro-5-deoxyribose ([18F]FDR) for Chemoselective 18F-fluoroglycosylation by Oxime Linkage
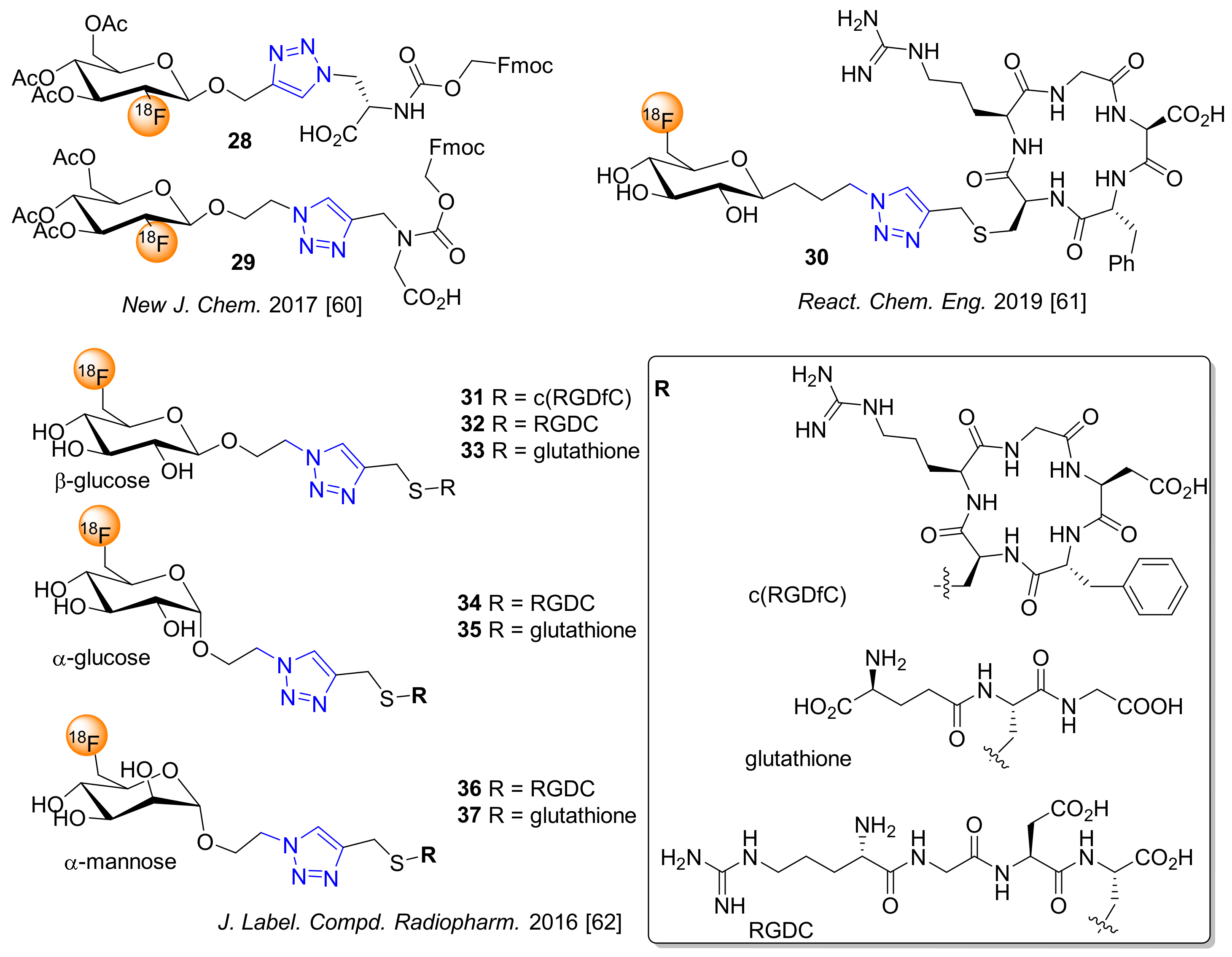
8. Miscellaneous 18F-fluoroglycosylation Reactions
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H. Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions. In PET Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; pp. 15–50. [Google Scholar]
- Littich, R.; Scott, P.J.H. Novel Strategies for Fluorine-18 Radiochemistry. Angew. Chem. Int. Ed. 2012, 51, 1106–1109. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.S.; Patil, S.N. One molecule of ionic liquid and tert-alcohol on a polystyrene-support as catalysts for efficient nucleophilic substitution including fluorination. Org. Biomol. Chem. 2014, 12, 9264–9271. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.S.; Patil, S.N.; Ghatge, A.; Kumar, P. Nucleophilic fluorination using imidazolium based ionic liquid bearing tert-alcohol moiety. New J. Chem. 2015, 39, 4368–4374. [Google Scholar] [CrossRef]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher, K. Phase-transfer catalysed synthesis of 4-β-d-glucopyranosyl-4-thio-d-glucopyranose (thiocellobiose) and 2-β-d-glucopyranosyl-2-thio-d-glucopyranose (thiosophorose). Carbohydr. Res. 1984, 128, 291–295. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H.; Stöcklin, G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar]
- Egleton, R.D.; Davis, T.P. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx 2005, 2, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Kuhnast, B.; Mang, C.; Weber, W.A.; Kessler, H.; Wester, H.-J.; Schwaiger, M. [18F]Galacto-RGD: Synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug. Chem. 2004, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Rau, F.; Reubi, J.C.; Schwaiger, M.; Wester, H.-J. Modulation of pharmacokinetics of radioiodinated sugar-conjugated somatostatin analogues by variation of peptide net charge and carbohydration chemistry. Bioconjug. Chem. 2005, 16, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Wester, H.J.; Weber, W.A.; Mang, C.; Ziegler, S.I.; Goodman, S.L.; Senekowitsch-Schmidtke, R.; Kessler, H.; Schwaiger, M. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001, 61, 1781–1785. [Google Scholar] [PubMed]
- Schottelius, M.; Wester, H.-J.; Reubi, J.C.; Senekowitsch-Schmidtke, R.; Schwaiger, M. Improvement of pharmacokinetics of radioiodinated Tyr3-octreotide by conjugation with carbohydrates. Bioconjug. Chem. 2002, 13, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.; Schottelius, M.; Scheidhauer, K.; Meisetschläger, G.; Herz, M.; Rau, F.; Reubi, J.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Bielski, R. Click Chemistry in Glycoscience: New Developments and Strategies; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Maschauer, S.; Prante, O. A series of 2-O-trifluoromethylsulfonyl-D-mannopyranosides as precursors for concomitant 18F-labeling and glycosylation by click chemistry. Carbohydr. Res. 2009, 344, 753–761. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and Glycosylation of Peptides Using Click Chemistry: A General Approach to 18F-Glycopeptides as Effective Imaging Probes for Positron Emission Tomography. Angew. Chem. Int. Ed. 2010, 49, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Prante, O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: A Short Review. Biomed. Res. Int. 2014, 2014, 214748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenen, H.H.; Gee, A.D.; Adam, M.; Antoni, G.; Cutler, C.S.; Fujibayashi, Y.; Jeong, J.M.; Mach, R.H.; Mindt, T.L.; Pike, V.W.; et al. Consensus nomenclature rules for radiopharmaceutical chemistry-Setting the record straight. Nucl. Med. Biol. 2017, 55, v–xi. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namavari, M.; Cheng, Z.; Zhang, R.; De, A.; Levi, J.; Hoerner, J.K.; Yaghoubi, S.S.; Syud, F.A.; Gambhir, S.S. A Novel Method for Direct Site-Specific Radiolabeling of Peptides Using [18F]FDG. Bioconjug. Chem. 2009, 20, 432–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultsch, C.; Schottelius, M.; Auernheimer, J.; Alke, A.; Wester, H.-J. 18F-Fluoroglucosylation of peptides, exemplified on cyclo(RGDfK). Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Hultsch, C.; Berndt, M.; Bergmann, R. Direct labelling of peptides with 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG). Bioorg. Med. Chem. Lett. 2009, 19, 5426–5428. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-G.; Autio, A.; Ahtinen, H.; Helariutta, K.; Liljenback, H.; Jalkanen, S.; Roivainen, A.; Airaksinen, A.J. Translating the concept of peptide labeling with 5-deoxy-5-[18F]fluororibose into preclinical practice: 18F-labeling of Siglec-9 peptide for PET imaging of inflammation. Chem. Commun. 2013, 49, 3682–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feni, L.; Omrane, M.A.; Fischer, M.; Zlatopolskiy, B.D.; Neumaier, B.; Neundorf, I. Convenient Preparation of 18F-Labeled Peptide Probes for Potential Claudin-4 PET Imaging. Pharmaceuticals 2017, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Mirfeizi, L.; Campbell-Verduyn, L.; Dierckx, R.A.; Feringa, B.L.; Elsinga, P.H. Application of Click Chemistry for PET. Curr. Org. Chem. 2013, 17, 2108–2118. [Google Scholar] [CrossRef]
- Fischer, C.R.; Muller, C.; Reber, J.; Muller, A.; Kramer, S.D.; Ametamey, S.M.; Schibli, R. [18F]Fluoro-Deoxy-Glucose Folate: A Novel PET Radiotracer with Improved in Vivo Properties for Folate Receptor Targeting. Bioconjug. Chem. 2012, 23, 805–813. [Google Scholar] [CrossRef]
- Fischer, C.R.; Groehn, V.; Reber, J.; Schibli, R.; Ametamey, S.M.; Muller, C. Improved PET imaging of tumors in mice using a novel 18F-folate conjugate with an albumin-binding entity. Mol. Imaging Biol. 2013, 15, 649–654. [Google Scholar] [CrossRef]
- Hugenberg, V.; Breyholz, H.-J.; Riemann, B.; Hermann, S.; Schober, O.; Schaefers, M.; Gangadharmath, U.; Mocharla, V.; Kolb, H.; Walsh, J.; et al. A New Class of Highly Potent Matrix Metalloproteinase Inhibitors Based on Triazole-Substituted Hydroxamates: (Radio)Synthesis and in Vitro and First In Vivo Evaluation. J. Med. Chem. 2012, 55, 4714–4727. [Google Scholar] [CrossRef]
- Banerjee, A.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Click chemistry based synthesis of dopamine D4 selective receptor ligands for the selection of potential PET tracers. Bioorg. Med. Chem. Lett. 2013, 23, 6079–6082. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Michel, K.; Tripal, P.; Büther, K.; Kuwert, T.; Schober, O.; Kopka, K.; Riemann, B.; Prante, O. Synthesis and In Vivo Evaluation of an 18F-Labeled Glycoconjugate of PD156707 for Imaging ETA Expression in Thyroid Carcinoma by Positron Emission Tomography. Am. J. Nucl. Med. Mol. Imaging 2013, 3, 425–436. [Google Scholar] [PubMed]
- Lang, C.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Synthesis and Evaluation of a 18F-Labeled Diarylpyrazole Glycoconjugate for the Imaging of NTS1-Positive Tumors. J. Med. Chem. 2013, 56, 9361–9365. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Slade, R.L.; Iddon, L.; George, G.P.; Nguyen, Q.D.; Spivey, A.C.; Aboagye, E.O. Synthesis of a new fluorine-18 glycosylated ‘click’ cyanoquinoline for the imaging of epidermal growth factor receptor. J. Labelled Compd. Radiopharm. 2014, 57, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; D’Hooge, F.; Fernandez-Gonzalez, M.; Bernardes, G.J.L.; Sanchez-Navarro, M.; Koeppe, J.R.; Davis, B.G. Fluoroglycoproteins: Ready chemical site-selective incorporation of fluorosugars into proteins. Chem. Commun. 2010, 46, 8142–8144. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Maschauer, S.; Kuwert, T.; Beck-Sickinger, A.G.; Prante, O. Synthesis and in Vitro and in Vivo Evaluation of an 18F-Labeled Neuropeptide Y Analogue for Imaging of Breast Cancer by PET. Mol. Pharm. 2015, 12, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.; D’Hooge, F.; Davis, B.G. Direct radiolabelling of proteins at cysteine using [18F]fluorosugars. Chem. Commun. 2011, 47, 10010–10012. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Z.; Chen, K.; Yan, Y.; Watzlowik, P.; Wester, H.J.; Chin, F.T.; Chen, X. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol. Imaging Biol. 2010, 12, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lang, L.; Hu, S.; Guo, N.; Zhu, L.; Sun, Z.; Ma, Y.; Kiesewetter, D.O.; Niu, G.; Xie, Q. Comparison of three dimeric 18F-AlF-NOTA-RGD tracers. Mol. Imaging Biol. 2014, 16, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Lozada, J.; Wong, M.Q.; Lin, K.-S.; Yapp, D.; Perrin, D.M. Single step 18F-labeling of dimeric cycloRGD for functional PET imaging of tumors in mice. Nucl. Med. Biol. 2013, 40, 959–966. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Prante, O. 18F- and 68Ga-Labeled Neurotensin Peptides for PET Imaging of Neurotensin Receptor 1. J. Med. Chem. 2016, 59, 6480–6492. [Google Scholar] [CrossRef] [PubMed]
- Potemkin, R.; Strauch, B.; Kuwert, T.; Prante, O.; Maschauer, S. Development of 18F-Fluoroglycosylated PSMA-Ligands with Improved Renal Clearance Behavior. Mol. Pharm. 2020, 17, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Greff, C.; Einsiedel, J.; Ott, J.; Tripal, P.; Hübner, H.; Gmeiner, P.; Prante, O. Improved radiosynthesis and preliminary in vivo evaluation of a 18F-labeled glycopeptide–peptoid hybrid for PET imaging of neurotensin receptor 2. Biorg. Med. Chem. 2015, 23, 4026–4033. [Google Scholar] [CrossRef]
- Swift, S.L.; Burns, J.E.; Maitland, N.J. Altered Expression of Neurotensin Receptors Is Associated with the Differentiation State of Prostate Cancer. Cancer Res. 2010, 70, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korner, M.; Waser, B.; Strobel, O.; Buchler, M.; Reubi, J.C. Neurotensin receptors in pancreatic ductal carcinomas. EJNMMI Res. 2015, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.; Nikolaou, S.; Zhu, J.; Jeffery, P.; Goldin, R.; Kinross, J.; Alexander, J.L.; Rasheed, S.; Tekkis, P.; Kontovounisios, C. Characterisation of the Expression of Neurotensin and Its Receptors in Human Colorectal Cancer and Its Clinical Implications. Biomolecules 2020, 10, 1145. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Friess, H.; Buchler, M.; Laissue, J. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut 1998, 42, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Souaze, F.; Dupouy, S.; Viardot-Foucault, V.; Bruyneel, E.; Attoub, S.; Gespach, C.; Gompel, A.; Forgez, P. Expression of neurotensin and NT1 receptor in human breast cancer: A potential role in tumor progression. Cancer Res. 2006, 66, 6243–6249. [Google Scholar] [CrossRef] [Green Version]
- Held, C.; Plomer, M.; Hübner, H.; Meltretter, J.; Pischetsrieder, M.; Gmeiner, P. Development of a Metabolically Stable Neurotensin Receptor 2 (NTS2) Ligand. Chem. Med. Chem. 2013, 8, 75–81. [Google Scholar] [CrossRef]
- Maschauer, S.; Heilmann, M.; Wängler, C.; Schirrmacher, R.; Prante, O. Radiosynthesis and Preclinical Evaluation of 18F-Fluoroglycosylated Octreotate for Somatostatin Receptor Imaging. Bioconjug. Chem. 2016, 27, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Ott, J.J.; Bernhardt, G.; Kuwert, T.; Keller, M.; Prante, O. 18F-labelled triazolyl-linked argininamides targeting the neuropeptide Y Y1R for PET imaging of mammary carcinoma. Sci. Rep. 2019, 9, 12990. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, E.M.; Roise, J.J.; Li, I.C.; Das, R.; Murthy, N. Advances in Imaging Reactive Oxygen Species. J. Nucl. Med. 2021, 62, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Toms, J.; Reshetnikov, V.; Maschauer, S.; Mokhir, A.; Prante, O. Radiosynthesis of an 18F-fluoroglycosylated aminoferrocene for in-vivo imaging of reactive oxygen species activity by PET. J. Label. Compd. Radiopharm. 2018, 61, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Toms, J.; Reshetnikov, V.; Ozkan, H.G.; Hampel, F.; Maschauer, S.; Hakimioun, A.; Beierlein, F.; Sellner, L.; Schmitt, M.; et al. Identification of Boronic Acid Derivatives as an Active Form of N-Alkylaminoferrocene-Based Anticancer Prodrugs and Their Radiolabeling with 18F. Bioconjug. Chem. 2019, 30, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. 2019, 60, 801. [Google Scholar] [CrossRef] [Green Version]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers. J. Nucl. Med. 2019, 60, 386. [Google Scholar] [CrossRef] [Green Version]
- Toms, J.; Kogler, J.; Maschauer, S.; Daniel, C.; Schmidkonz, C.; Kuwert, T.; Prante, O. Targeting Fibroblast Activation Protein: Radiosynthesis and Preclinical Evaluation of an 18F-Labeled FAP Inhibitor. J. Nucl. Med. 2020, 61, 1806. [Google Scholar] [CrossRef]
- Elgland, M.; Nordeman, P.; Fyrner, T.; Antoni, G.; Nilsson, K.P.R.; Konradsson, P. β-Configured clickable [18F]FDGs as novel 18F-fluoroglycosylation tools for PET. New J. Chem. 2017, 41, 10231–10236. [Google Scholar] [CrossRef] [Green Version]
- Collet, C.; Vucko, T.; Ariztia, J.; Karcher, G.; Pellegrini-Moïse, N.; Lamandé-Langle, S. Fully automated radiosynthesis of [18F]fluoro-C-glyco-c(RGDfC): Exploiting all the abilities of the AllInOne synthesizer. React. Chem. Eng. 2019, 4, 2088–2098. [Google Scholar] [CrossRef]
- Collet, C.; Maskali, F.; Clément, A.; Chrétien, F.; Poussier, S.; Karcher, G.; Marie, P.-Y.; Chapleur, Y.; Lamandé-Langle, S. Development of 6-[18F]fluoro-carbohydrate-based prosthetic groups and their conjugation to peptides via click chemistry. J. Label. Compd. Radiopharm. 2016, 59, 54–62. [Google Scholar] [CrossRef]
- Arja, K.; Elgland, M.; Appelqvist, H.; Konradsson, P.; Lindgren, M.; Nilsson, K.P.R. Synthesis and Characterization of Novel Fluoro-glycosylated Porphyrins that can be Utilized as Theranostic Agents. ChemistryOpen 2018, 7, 495–503. [Google Scholar] [CrossRef]
- Ulrich, S.; Boturyn, D.; Marra, A.; Renaudet, O.; Dumy, P. Oxime ligation: A chemoselective click-type reaction for accessing multifunctional biomolecular constructs. Chemistry 2014, 20, 34–41. [Google Scholar] [CrossRef]
- Li, X.G.; Haaparanta, M.; Solin, O. Oxime formation for fluorine-18 labeling of peptides and proteins for positron emission tomography (PET) imaging: A review. J. Fluor. Chem. 2012, 143, 49–56. [Google Scholar] [CrossRef]
- Wuest, F.; Berndt, M.; Bergmann, R.; van, d.H.J.; Pietzsch, J. Synthesis and Application of [18F]FDG-Maleimidehexyloxime ([18F]FDG-MHO): A [18F]FDG-Based Prosthetic Group for the Chemoselective 18F-Labeling of Peptides and Proteins. Bioconjug. Chem. 2008, 19, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Al Jammaz, I.; Al-Otaibi, B.; Amer, S.; Al-Hokbany, N.; Okarvi, S. Novel synthesis and preclinical evaluation of folic acid derivatives labeled with 18F-FDG for PET imaging of folate receptor-positive tumors. Nucl. Med. Biol. 2012, 39, 864–870. [Google Scholar] [CrossRef]
- AlJammaz, I.; Al-Otaibi, B.; AlHindas, H.; Okarvi, S.M. Novel synthesis and initial preclinical evaluation of 18F[FDG] labeled rhodamine: A potential PET myocardial perfusion imaging agent. Nucl. Med. Biol. 2015, 42, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, M.; Bergman, C.N.; Way, J.D.; Krieger, S.; Rogers, B.E.; Wuest, F. Rerouting the Metabolic Pathway of 18F-Labeled Peptides: The Influence of Prosthetic Groups. Bioconjug. Chem. 2015, 26, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, V.; Wuest, M.; Bailey, J.J.; Bergman, C.; Janzen, N.; Valliant, J.F.; Wuest, F. Targeting Prostate-Specific Membrane Antigen (PSMA) with F-18-Labeled Compounds: The Influence of Prosthetic Groups on Tumor Uptake and Clearance Profile. Mol. Imaging Biol. 2017, 19, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Keinänen, O.; Partelová, D.; Alanen, O.; Antopolsky, M.; Sarparanta, M.; Airaksinen, A.J. Efficient cartridge purification for producing high molar activity [18F]fluoro-glycoconjugates via oxime formation. Nucl. Med. Biol. 2018, 67, 27–35. [Google Scholar] [CrossRef]
- Flavell, R.R.; Truillet, C.; Regan, M.K.; Ganguly, T.; Blecha, J.E.; Kurhanewicz, J.; VanBrocklin, H.F.; Keshari, K.R.; Chang, C.J.; Evans, M.J.; et al. Caged [18F]FDG Glycosylamines for Imaging Acidic Tumor Microenvironments Using Positron Emission Tomography. Bioconjug. Chem. 2016, 27, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Şenışık, A.M.; İçhedef, Ç.; Kılçar, A.Y.; Uçar, E.; Arı, K.; Göksoy, D.; Parlak, Y.; Sayıt Bilgin, B.E.; Teksöz, S. One-step conjugation of glycylglycine with [18F]FDG and a pilot PET imaging study. J. Radioanal. Nucl. Chem. 2018, 316, 457–463. [Google Scholar] [CrossRef]
- Lopci, E.; Grassi, I.; Chiti, A.; Nanni, C.; Cicoria, G.; Toschi, L.; Fonti, C.; Lodi, F.; Mattioli, S.; Fanti, S. PET radiopharmaceuticals for imaging of tumor hypoxia: A review of the evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384. [Google Scholar] [PubMed]
- Patt, M.; Sorger, D.; Scheunemann, M.; Stöcklin, G. Adduct of 2-[18F]FDG and 2-nitroimidazole as a putative radiotracer for the detection of hypoxia with PET: Synthesis, in vitro- and in vivo-characterization. Appl. Radiat. Isot. 2002, 57, 705–712. [Google Scholar] [CrossRef]
- Yang, X.; Wang, F.; Zhu, H.; Yang, Z.; Chu, T. Synthesis and Bioevaluation of Novel [18F]FDG-Conjugated 2-Nitroimidazole Derivatives for Tumor Hypoxia Imaging. Mol. Pharm. 2019, 16, 2118–2128. [Google Scholar] [CrossRef]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-Fluorodeoxyglucose to Label Antibody Fragments for Immuno-Positron Emission Tomography of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Li, X.G.; Dall’Angelo, S.; Schweiger, L.F.; Zanda, M.; O’Hagan, D. [18F]-5-Fluoro-5-deoxyribose, an efficient peptide bioconjugation ligand for positron emission tomography (PET) imaging. Chem. Commun. 2012, 48, 5247–5249. [Google Scholar] [CrossRef]
- Dall’Angelo, S.; Zhang, Q.; Fleming, I.N.; Piras, M.; Schweiger, L.F.; O’Hagan, D.; Zanda, M. Efficient bioconjugation of 5-fluoro-5-deoxy-ribose (FDR) to RGD peptides for positron emission tomography (PET) imaging of αvβ3 integrin receptor. Org. Biomol. Chem. 2013, 11, 4551–4558. [Google Scholar] [CrossRef] [PubMed]
- Frau, S.; Dall’Angelo, S.; Baillie, G.L.; Ross, R.A.; Pira, M.; Tseng, C.-C.; Lazzari, P.; Zanda, M. Pyrazole-type cannabinoid ligands conjugated with fluoro-deoxy-carbohydrates as potential PET-imaging agents: Synthesis and CB1/CB2 receptor affinity evaluation. J. Fluor. Chem. 2013, 152, 166–172. [Google Scholar] [CrossRef]
- Keinänen, O.; Li, X.-G.; Chenna, N.K.; Lumen, D.; Ott, J.; Molthoff, C.F.M.; Sarparanta, M.; Helariutta, K.; Vuorinen, T.; Windhorst, A.D.; et al. A New Highly Reactive and Low Lipophilicity Fluorine-18 Labeled Tetrazine Derivative for Pretargeted PET Imaging. ACS Med. Chem. Lett. 2016, 7, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, H.; Silvola, J.M.U.; Autio, A.; Li, X.-G.; Liljenbäck, H.; Hellberg, S.; Siitonen, R.; Ståhle, M.; Käkelä, M.; Airaksinen, A.J.; et al. Comparison of 68Ga-DOTA-Siglec-9 and 18F-Fluorodeoxyribose-Siglec-9: Inflammation Imaging and Radiation Dosimetry. Contrast Media Mol. Imaging 2017, 2017, 7645070. [Google Scholar] [CrossRef] [Green Version]
- Musolino, M.; Fleming, I.N.; Schweiger, L.F.; O’Hagan, D.; Dall’Angelo, S.; Zanda, M. Synthesis, Radiosynthesis, and in vitro Studies on Novel Hypoxia PET Tracers Incorporating [18F]FDR. Eur. J. Org. Chem. 2021, 2021, 1429–1439. [Google Scholar] [CrossRef]
- Maschauer, S.; Kuwert, T.; Prante, O. 18F-glycosylation using Koenigs-Knorr conditions: A comparative study. J. Label. Compd. Radiopharm. 2006, 49, 101–108. [Google Scholar] [CrossRef]
- Maschauer, S.; Pischetsrieder, M.; Kuwert, T.; Prante, O. Utility of 1,3,4,6-tetra-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranoside for no-carrier-added 18F-glycosylation of amino acids. J. Label. Compd. Radiopharm. 2005, 48, 701–719. [Google Scholar] [CrossRef]
- Prante, O.; Einsiedel, J.; Haubner, R.; Gmeiner, P.; Wester, H.J.; Kuwert, T.; Maschauer, S. 3,4,6-tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl phenylthiosulfonate: A thiol-reactive agent for the chemoselective 18F-glycosylation of peptides. Bioconjug. Chem. 2007, 18, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Ozkaya, F.; Unak, P.; Medine, E.I.; Sakarya, S.; Unak, G.; Timur, S. 18FDG conjugated magnetic nanoparticle probes: Synthesis and in vitro investigations on MCF-7 breast cancer cells. J. Radioanal. Nucl. Chem. 2013, 295, 1789–1796. [Google Scholar] [CrossRef]
- Unak, G.; Ozkaya, F.; Ilker Medine, E.; Kozgus, O.; Sakarya, S.; Bekis, R.; Unak, P.; Timur, S. Gold nanoparticle probes: Design and in vitro applications in cancer cell culture. Colloids Surf. B. Biointerfaces 2012, 90, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bormans, G.; Verbruggen, A. Enzymatic synthesis and biodistribution in mice of beta-O-D-galactopyranosyl-(1,4′)-2 ‘-[18F]fluoro-2′-deoxy-D-glucopyranose (2’-[18F]fluorodeoxylactose). J. Label. Compd. Radiopharm. 2001, 44, 417–423. [Google Scholar] [CrossRef]
- Prante, O.; Hamacher, K.; Coenen, H.H. Chemoenzymatic n.c.a synthesis of the coenzyme UDP-2-deoxy-2-[18F]fluoro-α-D-glucopyranose as substrate of glycosyltransferases. J. Label. Compd. Radiopharm. 2007, 50, 55–63. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, J.; Brownell, A.-L.; Wang, H.; Li, C.; Meng, X.; Yuan, L.; Liu, H.; Li, S.; Xie, J. Synthesis and evaluation of an N-[18F]fluorodeoxyglycosyl amino acid for PET imaging of tumor metabolism. Nucl. Med. Biol. 2018, 66, 40–48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Labeling Precursor | Prosthetic Group | Reaction Conditions | Ref. |
|---|---|---|---|
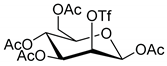 |  | 1. K222, K2CO3 2. NaOH | [23,24,25] |
 | 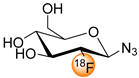 | 1. K222, K2CO3 2. NaOH | [19] |
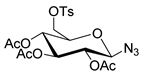 |  | 1. K222, K2CO3, KH2PO4 2. NaOH | [26] |
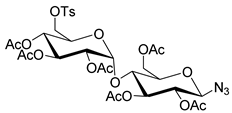 |  | 1. K222, K2CO3, KH2PO4 2. NaOH | [26] |
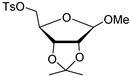 | 1. K222, K2CO3, 2. DMT-Cl, pyridine 3. HCl | [27] | |
 | 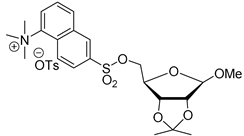 | 1. MeCN, 120 °C 2. 1M HCl, 110 °C | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinde, S.S.; Maschauer, S.; Prante, O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects. Pharmaceuticals 2021, 14, 1175. https://doi.org/10.3390/ph14111175
Shinde SS, Maschauer S, Prante O. Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects. Pharmaceuticals. 2021; 14(11):1175. https://doi.org/10.3390/ph14111175
Chicago/Turabian StyleShinde, Sandip S., Simone Maschauer, and Olaf Prante. 2021. "Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects" Pharmaceuticals 14, no. 11: 1175. https://doi.org/10.3390/ph14111175
APA StyleShinde, S. S., Maschauer, S., & Prante, O. (2021). Sweetening Pharmaceutical Radiochemistry by 18F-Fluoroglycosylation: Recent Progress and Future Prospects. Pharmaceuticals, 14(11), 1175. https://doi.org/10.3390/ph14111175