
Diversity-Oriented Synthesis: Amino Acetophenones as Building Blocks for the Synthesis of Natural Product Analogs




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
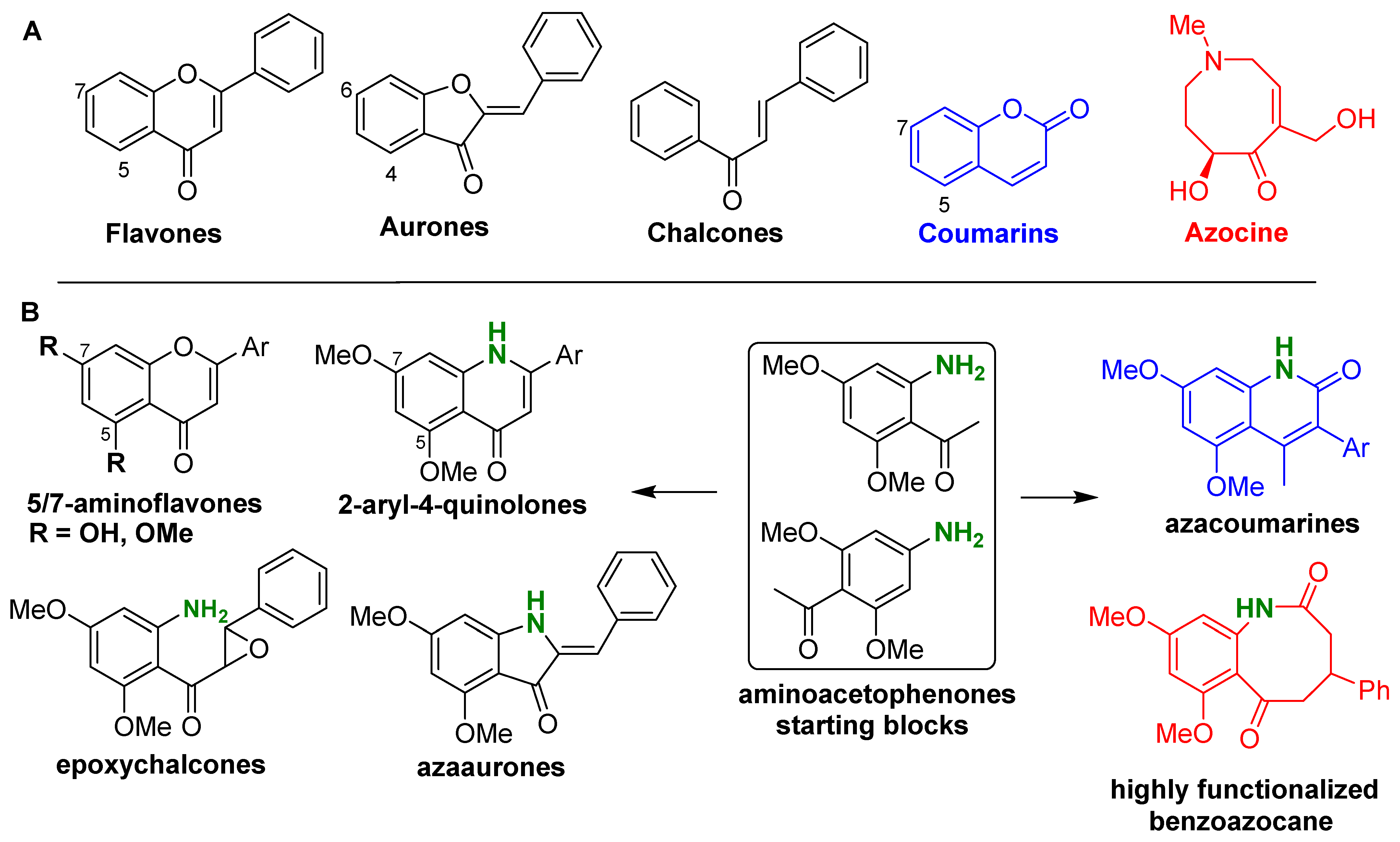
1. Introduction
2. Synthesis of the Starting Blocks
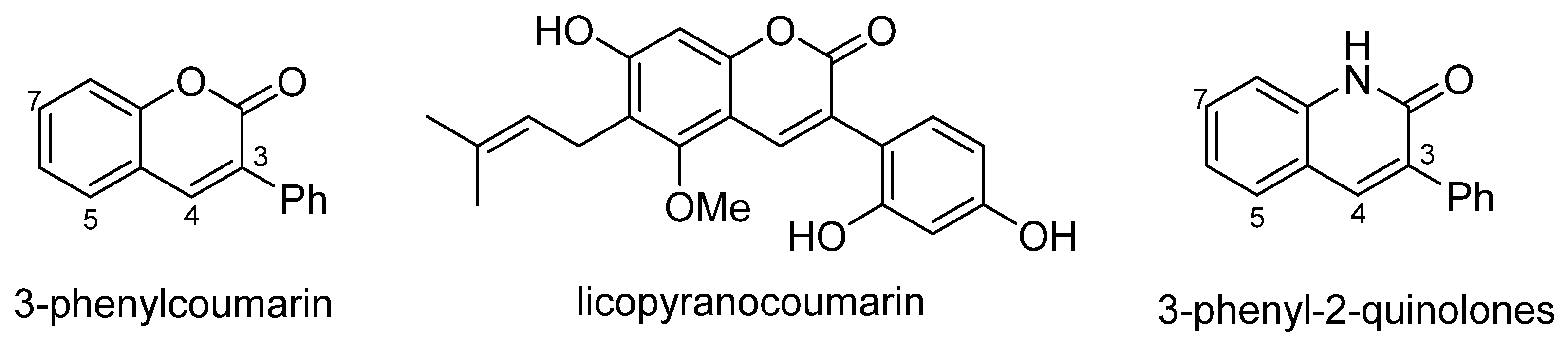
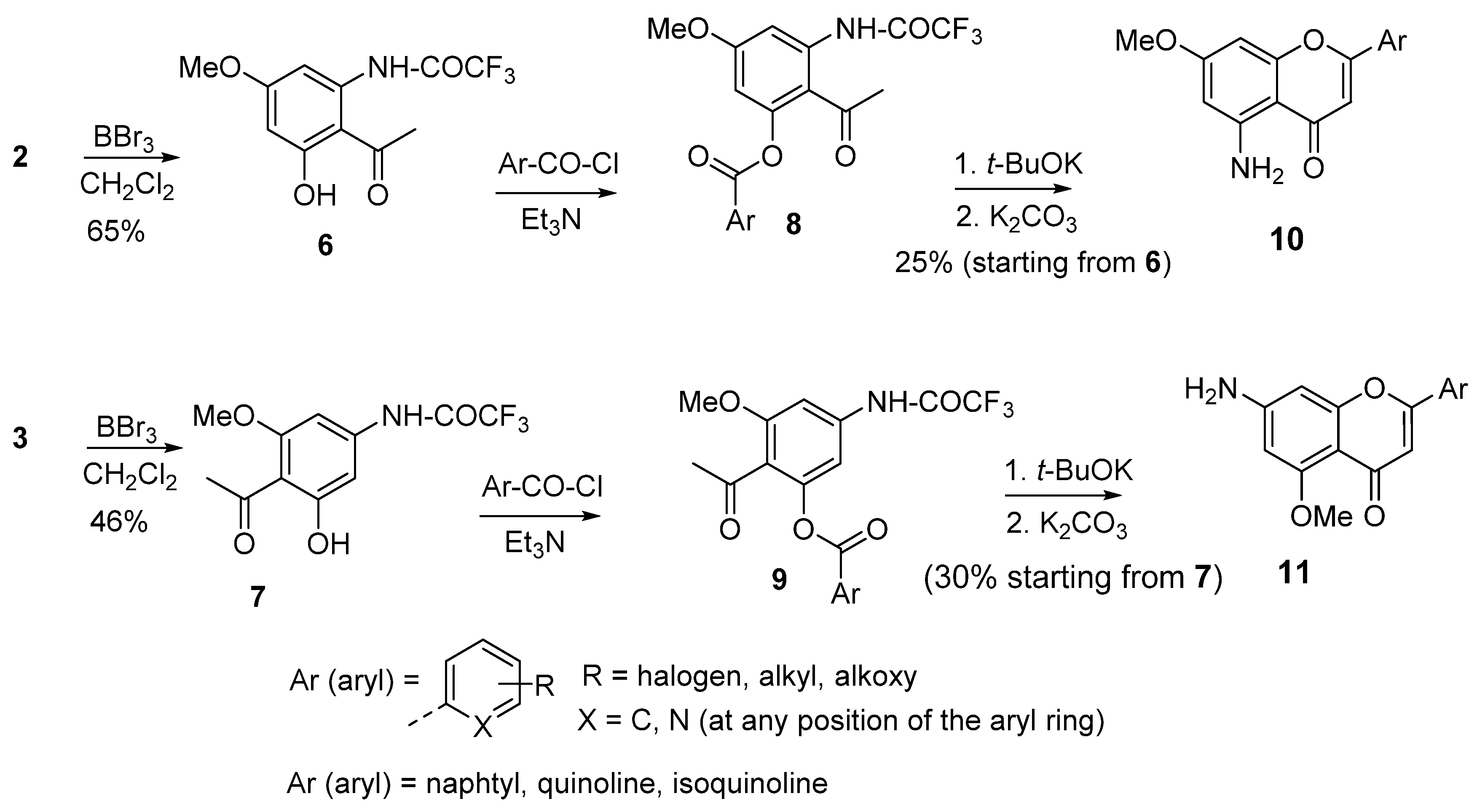
3. Synthesis of 5- and 7-Aminoflavones
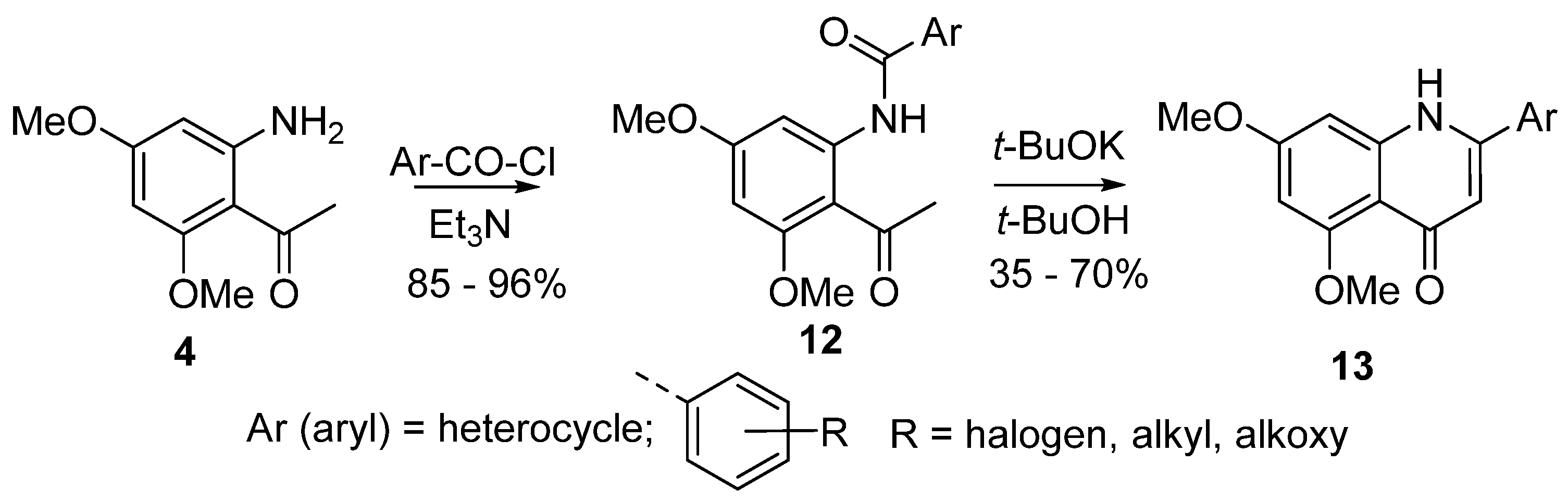
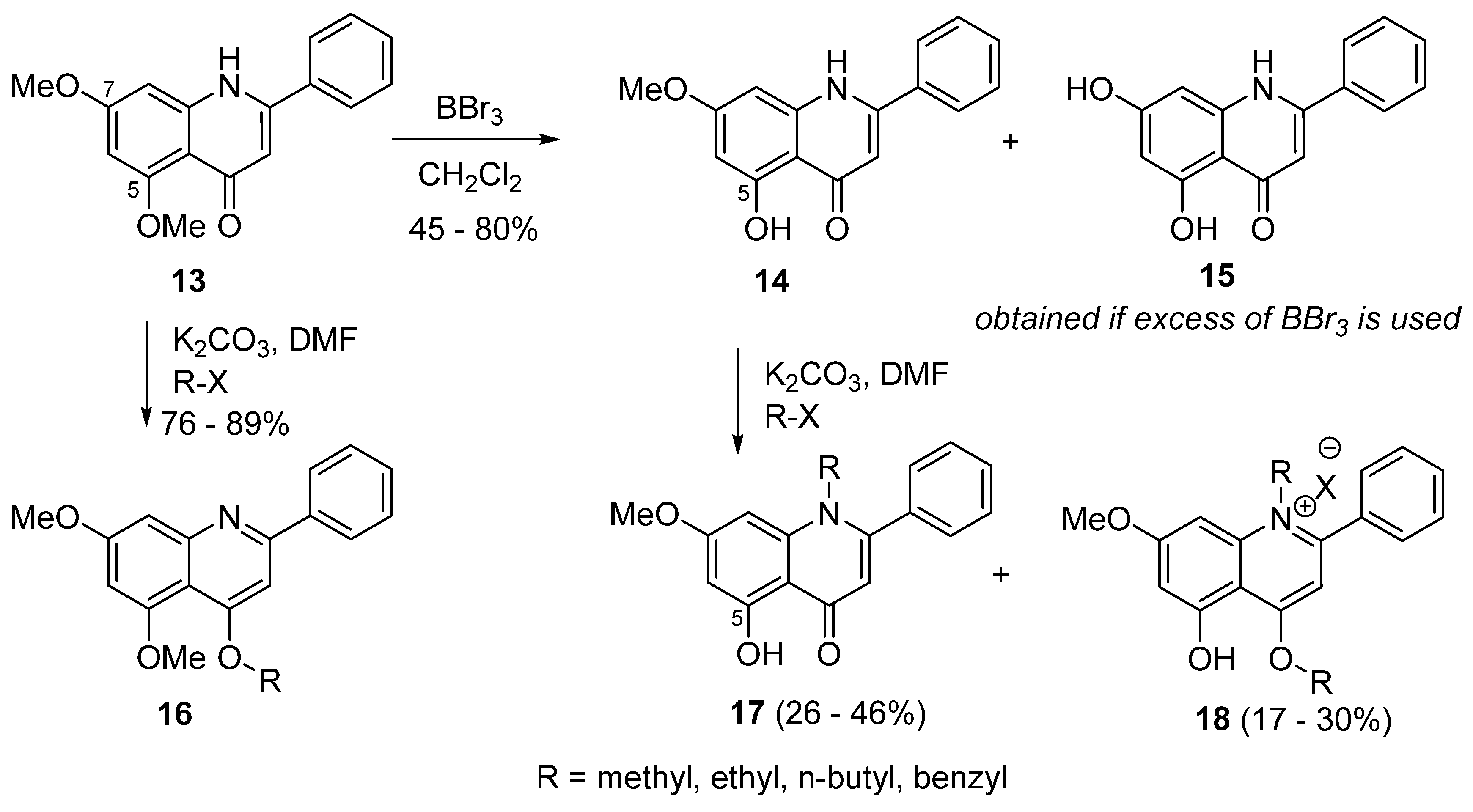
4. Synthesis of 2-Aryl-4-quinolones (Azaflavones)
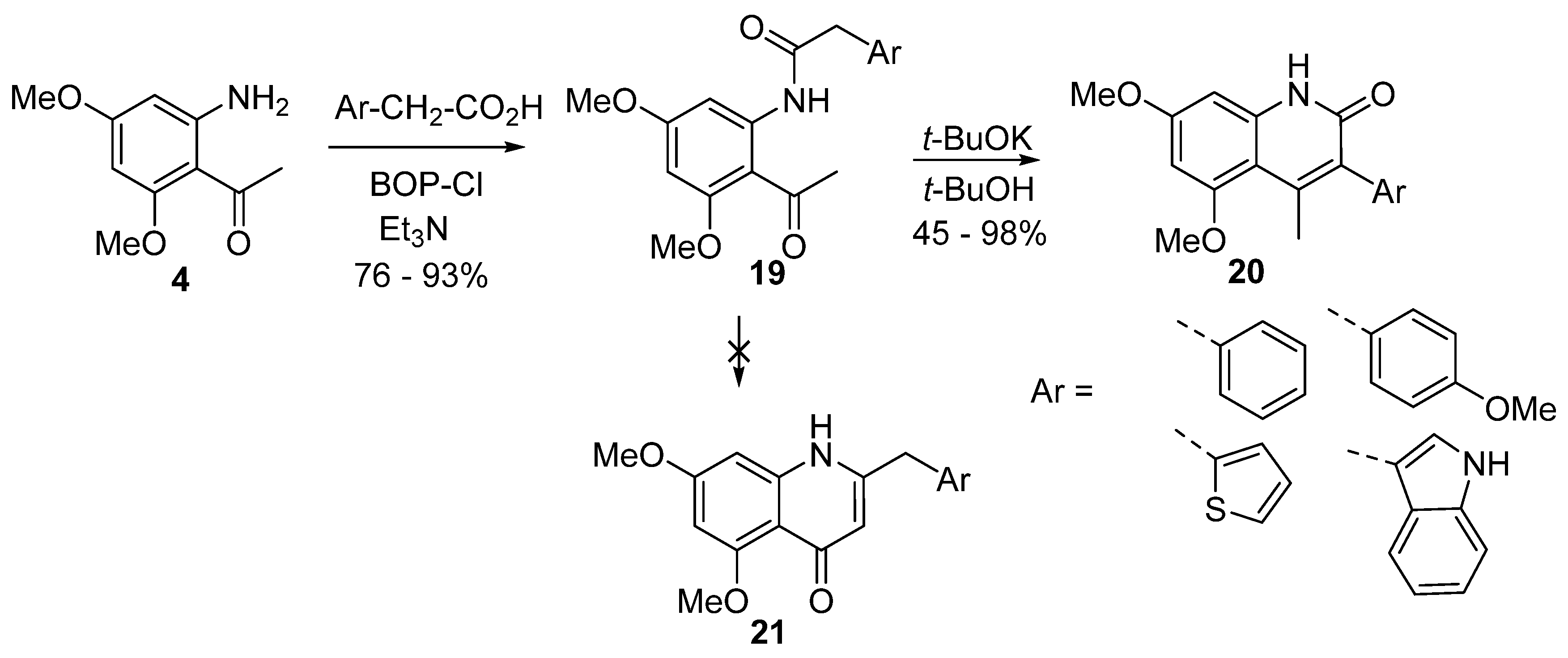
5. Synthesis of 3-Aryl-2-quinolones
6. Synthesis of Epoxychalcones
7. Synthesis of Azaaurones
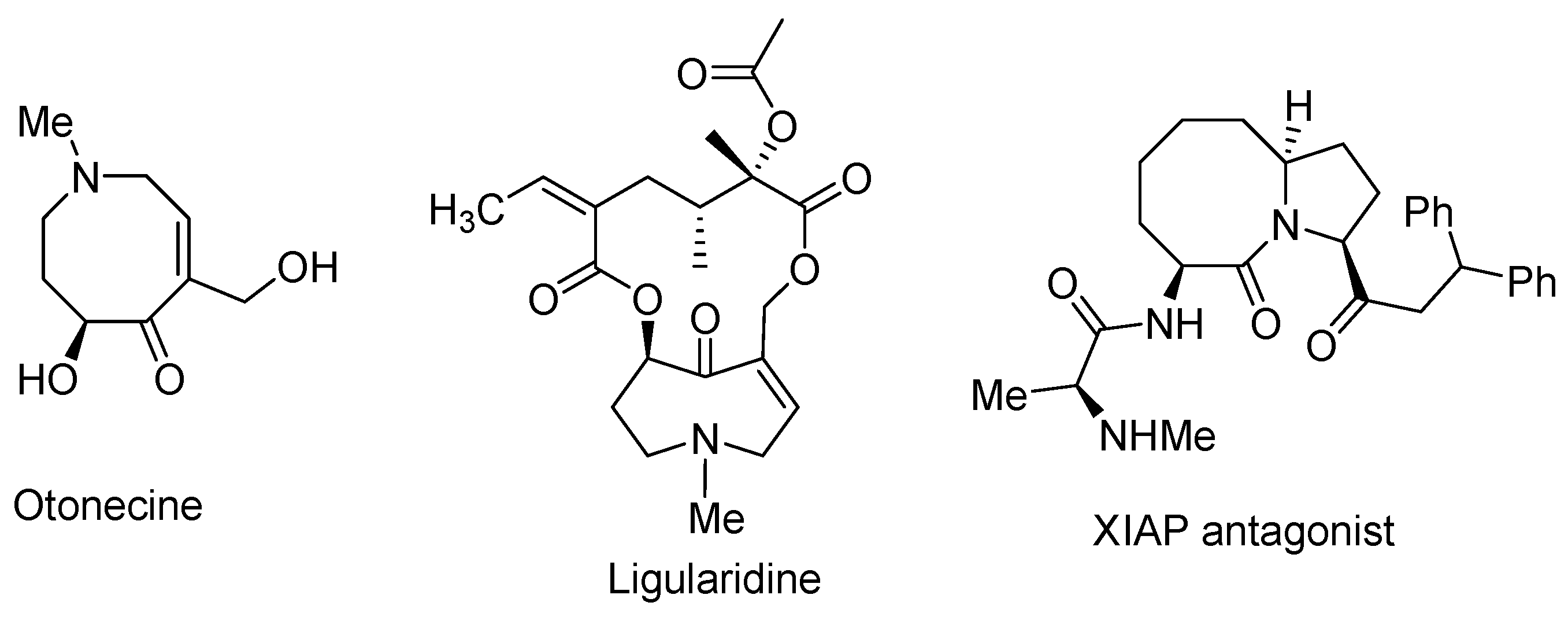
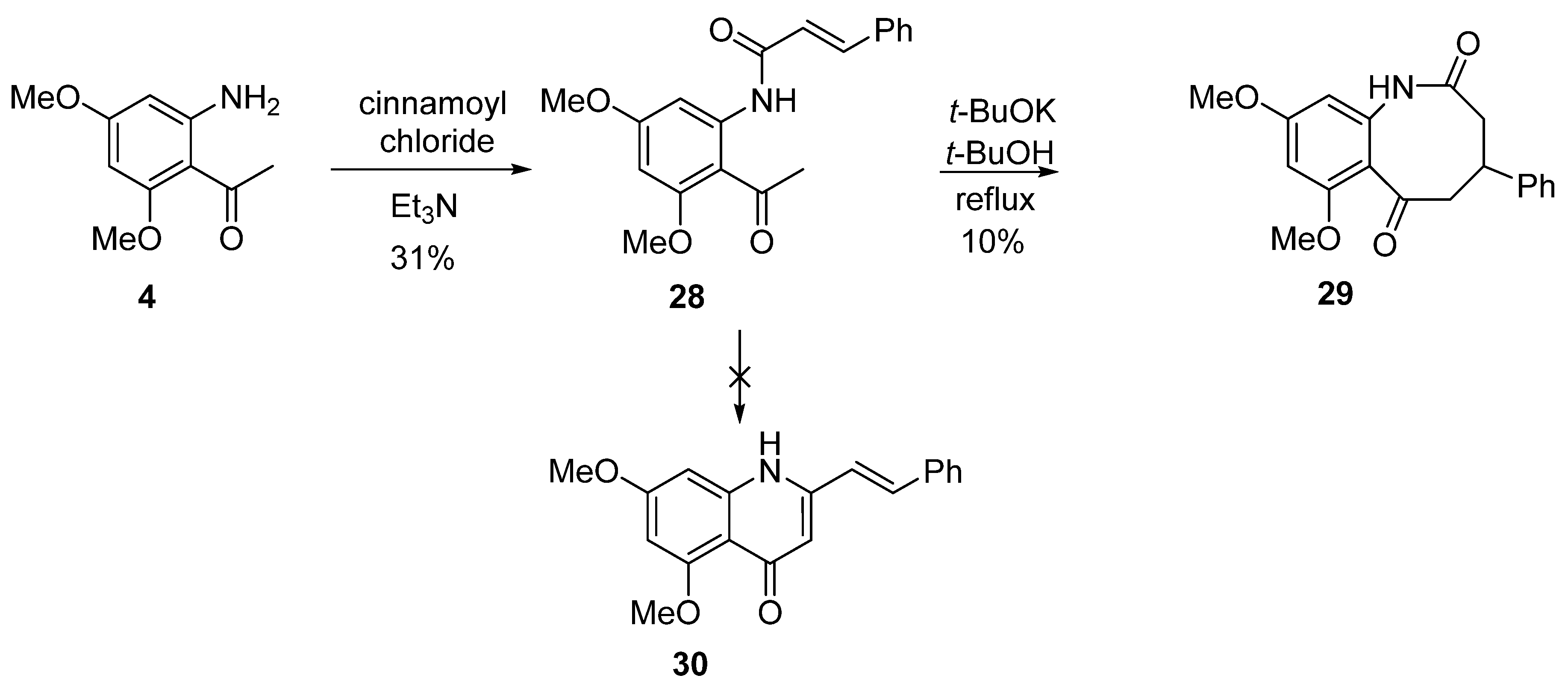
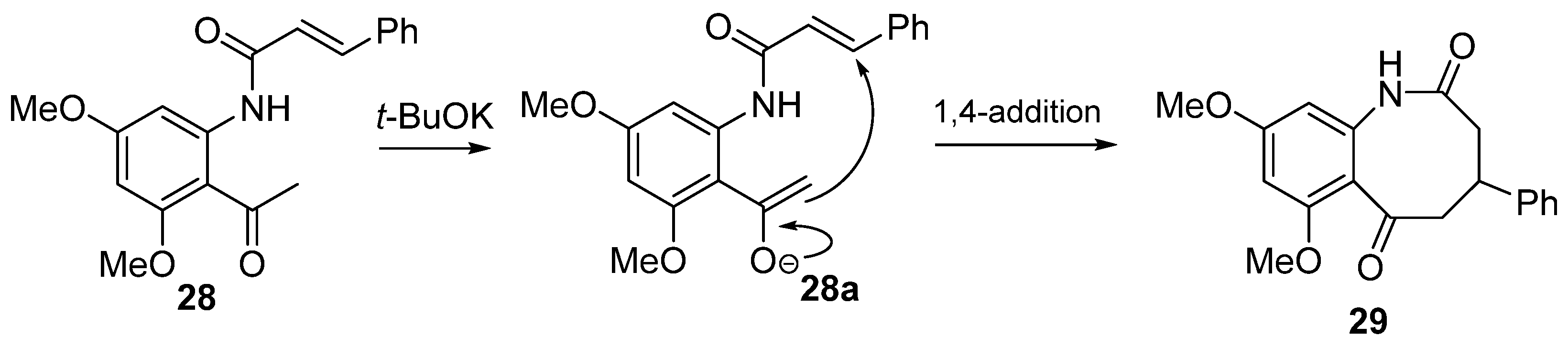
8. Potential Access to Eight-Membered N-Heterocyclic Ring Systems (Azocanes)
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Jones, L.H.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.L.; Osberger, T.J.; Mateu, N.; Sore, H.F.; Spring, D.R. Recent applications of diversity-oriented synthesis toward novel, 3-dimensional fragment collections. Front. Chem. 2018, 6, 460. [Google Scholar] [CrossRef]
- Pavlinov, I.; Gerlach, E.M.; Aldrich, L.N. Next generation diversity-oriented synthesis: A paradigm shift from chemical diversity to biological diversity. Org. Biomol. Chem. 2019, 17, 1608–1623. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394. [Google Scholar] [PubMed]
- Meanwell, M.; Fehr, G.; Ren, W.; Adluri, B.; Rose, V.; Lehmann, J.; Silverman, S.M.; Rawshanpour, R.; Bergeron-Blerk, M.; Foy, H.; et al. Diversity-oriented synthesis of glycomimetics. Commun. Chem. 2021, 4, 96. [Google Scholar]
- Murlykina, M.V.; Morozova, A.D.; Zviagin, I.M.; Sakhno, Y.I.; Desenko, S.M.; Chebanov, V.A. Aminoazole-Based Diversity-Oriented Synthesis of Heterocycles. Front. Chem. 2018, 6, 527. [Google Scholar] [PubMed]
- Alderton, G. Drug repurposing. Science 2020, 368, 840–842. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Shang, S.; Tan, D.S. Advancing chemistry and biology through diversity-oriented synthesis of natural product-like libraries. Curr. Opin. Chem. Biol. 2005, 9, 248–258. [Google Scholar] [CrossRef]
- Prabhu, G.; Agarwal, S.; Sharma, V.; Madurkar, S.M.; Munshi, P.; Singh, S.; Sen, S. A natural product based DOS library of hybrid systems. Eur. J. Med. Chem. 2015, 95, 41–48. [Google Scholar] [CrossRef]
- Chauhan, J.; Luthra, T.; Gundla, R.; Ferraro, A.; Holzgrabe, U.; Sen, S. A diversity oriented synthesis of natural product inspired molecular libraries. Org. Biomol. Chem. 2017, 5, 9108–9120. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Deb, P.K.; Priya, S.; Medina, K.D.; Devi, R.; Walode, S.G.; Rudrapal, M. Dietary Flavonoids: Cardioprotective Potential with Antioxidant Effects and Their Pharmacokinetic, Toxicological and Therapeutic Concerns. Molecules 2021, 26, 4021. [Google Scholar] [PubMed]
- Yammine, A.; Namsi, A.; Vervandier-Fasseur, D.; Mackrill, J.J.; Lizard, G.; Latruffe, N. Polyphenols of the Mediterranean Diet and Their Metabolites in the Prevention of Colorectal Cancer. Molecules 2021, 26, 3483. [Google Scholar] [PubMed]
- Pacifici, F.; Rovella, V.; Pastore, D.; Bellia, A.; Abete, P.; Donadel, G.; Santini, S.; Beck, H.; Ricordi, C.; Daniele, N.D.; et al. Polyphenols and Ischemic Stroke: Insight into One of the Best Strategies for Prevention and Treatment. Nutrients 2021, 13, 1967. [Google Scholar] [PubMed]
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Synthesis and antitumor activity of topologically-related analogs of flavoneacetic acid. Anti-Cancer Drug Des. 1989, 4, 161–169. [Google Scholar]
- Brana, M.F.; Castellano, J.M.; Emling, F.; Schlick, E. Synthesis and in vitro cytotoxic evaluation of new compounds with hybrid structures of 8-flavoneacetic acid and quinolones. An. Quim. 1994, 90, 491–496. [Google Scholar]
- Devulapally, S.; Chandraiah, G.; Pramod, K.D. A Review on Pharmacological Properties of Coumarins. Mini Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar]
- Hadjeri, M.; Beney, C.; Boumendjel, A. Recent Advances in the Synthesis of Conveniently Substituted Flavones, Quinolones, Chalcones and Aurones: Potential Biologically Active Molecules. Curr. Org. Chem. 2003, 7, 679–689. [Google Scholar] [CrossRef]
- Long, L.M.; Troutman, H.D. Chloramphenicol (Chloromycetin). IV. A synthetic approach. J. Am. Chem. Soc. 1949, 71, 2469–2472. [Google Scholar] [CrossRef]
- Traxler, P.; Green, J.; Mett, H.; Séquin, U.; Furet, P. Use of a pharmacophore model for the design of EGFR tyrosine kinase inhibitors: Isoflavones and 3-phenyl-4(1H)-quinolones. J. Med. Chem. 1999, 42, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Hadjeri, M.; Mariotte, A.-M.; Boumendjel, A. Deacetylation of activated acetophenones with tin(IV) chloride. J. Chem. Res. 2002, 2002, 463–464. [Google Scholar] [CrossRef]
- Khazeei-Tabari, M.A.; Iranpanah, A.; Bahramsoltani, R.; Rahimi, R. Flavonoids as Promising Antiviral Agents against SARS-CoV-2 Infection: A Mechanistic Review. Molecules 2021, 26, 3900. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.; Akter, Y.; Siddika, A.; Nayeem, S.M.A.; Nahrin, A.; Afrose, S.S.; Ezaj, M.M.A.; Alam, M.S. Nature-derived hit, lead, and drug-like small molecules: Current status and future aspects against key target proteins of Coronaviruses. Mini Rev. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Boniface, P.K.; Elizabeth, F.I. Flavones as a Privileged Scaffold in Drug Discovery: Current Developments. Curr. Org. Synth. 2019, 16, 968–1001. [Google Scholar]
- Ibrahim, N.; Bonnet, P.; Brion, J.D.; Peyrat, J.F.; Bignon, J.; Levaique, H.; Josselin, B.; Robert, T.; Colas, P.; Bach, S.; et al. Identification of a new series of flavopiridol-like structures as kinase inhibitors with high cytotoxic potency. Eur. J. Med. Chem. 2020, 199, 112355. [Google Scholar] [CrossRef]
- Fabijańska, M.; Kasprzak, M.M.; Ochock, J. Ruthenium(II) and Platinum(II) Complexes with Biologically Active Aminoflavone Ligands Exhibit In Vitro Anticancer Activity. Int. J. Mol. Sci. 2021, 22, 7568. [Google Scholar] [CrossRef]
- Szappanos, Á.; Mándi, A.; Gulácsi, K.; Lisztes, E.; Tóth, B.I.; Bíró, T.; Kónya-Ábrahám, A.; Kiss-Szikszai, A.; Bényei, A.; Antus, S.; et al. Synthesis and HPLC-ECD Study of Cytostatic Condensed O,N-Heterocycles Obtained from 3-Aminoflavanones. Biomolecules 2020, 10, 1462. [Google Scholar]
- Shelke, R.N.; Pansare, D.N.; Sarkate, A.P.; Narula, I.K.; Lokwani, D.K.; Tiwari, S.V.; Azad, R.; Thopate, S.R. Synthesis and evaluation of novel sulfonamide analogues of 6/7-aminoflavones as anticancer agents via topoisomerase II inhibition. Bioorg. Med. Chem. Lett. 2020, 30, 127246. [Google Scholar] [CrossRef]
- Brinkman, A.M.; Chen, G.; Wang, Y.; Hedman, C.J.; Sherer, N.M.; Havighurst, T.C.; Gong, S.; Xu, W. Aminoflavone-loaded EGFR-targeted unimolecular micelle nanoparticles exhibit anti-cancer effects in triple negative breast cancer. Biomaterials 2016, 101, 20–31. [Google Scholar] [CrossRef]
- Thorat, N.M.; Sarkate, A.P.; Lokwani, D.K.; Tiwari, S.V.; Azad, R.; Thopate, S.R. N-Benzylation of 6-aminoflavone by reductive amination and efficient access to some novel anticancer agents via topoisomerase II inhibition. Mol. Divers. 2020, 25, 937–948. [Google Scholar] [CrossRef]
- Fabijańska, M.; Orzechowska, M.; Rybarczyk-Pirek, A.J.; Dominikowska, J.; Bieńkowska, A.; Małecki, M.; Ochocki, J. Simple trans-platinum complex bearing 3-aminoflavone ligand could be a useful drug: Structure-activity relationship of platinum complex in comparison with cisplatin. Int. J. Mol. Sci. 2020, 21, 2116. [Google Scholar]
- Campbell, P.S.; Mavingire, N.; Khan, S.; Rowland, L.K.; Wooten, J.V.; Opoku-Agyeman, A.; Guevara, A.; Soto, U.; Cavalli, F.; Loaiza-Pérez, A.I.; et al. AhR ligand aminoflavone suppresses alpha6-integrin-Src-Akt signaling to attenuate tamoxifen resistance in breast cancer cells. J. Cell. Physiol. 2018, 234, 108–121. [Google Scholar] [PubMed]
- Stompor, M.; Świtalska, M.; Bajek, A.; Wietrzyk, J. Influence of amide versus ester linkages on the anticancer properties of the new flavone–biotin conjugates. Z. Naturforsch. C 2019, 74, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Kuffel, M.J.; Schroeder, J.C.; Pobst, L.J.; Naylor, S.; Reid, J.M.; Kaufmann, S.H.; Ames, M.M. Activation of the antitumor agent aminoflavone (NSC 686288) is mediated by induction of tumor cell cytochrome P450 1A1/1A2. Mol. Pharmacol. 2002, 62, 143–153. [Google Scholar] [PubMed]
- McLean, L.; Soto, U.; Agama, K.; Francis, J.; Jimenez, R.; Pommier, Y.; Sowers, L.; Brantley, E. Aminoflavone induces oxidative DNA damage and reactive oxidative species-mediated apoptosis in breast cancer cells. Int. J. Cancer 2008, 122, 1665–1674. [Google Scholar] [CrossRef]
- Itkin, B.; Breen, A.; Turyanska, L.; Sandes, E.O.; Bradshaw, T.D.; Loaiza-Perez, A.I. New Treatments in Renal Cancer: The AhR Ligands. Int. J. Mol. Sci. 2020, 21, 3551. [Google Scholar] [CrossRef]
- Callero, M.A.; Rodriguez, C.E.; Sólimo, A.; Offé, E.B.; Loaiza-Perez, A.I. The Immune System As a New Possible Cell Target for AFP 464 in a Spontaneous Mammary Cancer Mouse Model. J. Cell. Biochem. 2017, 118, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, S.; Yang, J.; Gao, W.; Cui, J.; Zhuang, T. A Novel approach to the synthesis of 6-amino-7-hydroxy-flavone. Molecules 2004, 9, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Patoilo, D.T.; Silva, A.M.S.; Cavaleiro, J.A.S. Regioselective 3-nitration of flavones: A new synthesis of 3-nitro- and 3-aminoflavones. Synlett 2010, 9, 1381–1385. [Google Scholar]
- Deka, N.; Hadjeri, M.; Lawson, M.; Beney, C.; Mariotte, A.-M.; Boumendjel, A. Acetylated dimethoxyaniline as a key intermediate for the synthesis of aminoflavones and quinolones. Heterocycles 2002, 57, 123–128. [Google Scholar]
- Kuo, S.-C.; Lee, H.Z.; Juang, J.P.; Lin, Y.T.; Wu, T.S.; Cheng, J.J.; Lednicer, D.; Paull, K.D.; Lin, C.M.; Hamel, E.; et al. Synthesis and cytotoxicity of 1,6,7,8- and 4′-substituted 2-phenyl-4-quinolones and related compounds: Identification as antimitotic agents interacting with tubulin. J. Med. Chem. 1993, 36, 1146–1156. [Google Scholar]
- Li, L.; Wang, H.K.; Kuo, S.-C.; Wu, T.S.; Mauger, A.; Lin, C.M.; Hamel, E.; Lee, K.-H. Synthesis and biological evaluation of 3′,6,7-substituted 2-phenyl-4-quinolones as antimitotic antitumor agents. J. Med. Chem. 1994, 37, 3400–3407. [Google Scholar]
- Li, L.; Wuang, H.K.; Kuo, S.-C.; Lednicer, D.; Lin, C.M.; Hamel, E.; Lee, K.-H. 2′,3′,4′,5′,5,6,7-Substituted 2-phenyl-4-quinolones and related compounds: Their synthesis and inhibition of tubulin polymerization. J. Med. Chem. 1994, 37, 1126–1135. [Google Scholar] [CrossRef]
- Lee, H.Z.; Lin, W.C.; Yeh, F.T.; Wu, C.H. 2-Phenyl-4-quinolone prevents serotonin-induced increases in endothelial permeability to albumin. Eur. J. Pharmacol. 1998, 354, 205–213. [Google Scholar] [CrossRef]
- Nilsson, J.; Nielsen, E.Ø.; Liljefors, T.; Nielsen, M.; Sterner, O. Azaflavones compared to flavones as ligands to the benzodiazepine binding site of brain GABA(A) receptors. Bioorg. Med. Chem. Lett. 2008, 18, 5713–5716. [Google Scholar] [CrossRef]
- Dhiman, R.; Sharma, S.; Singh, G.; Nepali, K.; Singh Bedi, P.M. Design and synthesis of aza-flavones as a new class of xanthine oxidase inhibitors. Arch. Pharm. 2013, 346, 7–16. [Google Scholar]
- Chong, R.J.; Siddiqui, M.A.; Snieckus, V. Synthetic connections to the aromatic directed metalation reaction. A modified von Niementowski quinoline synthesis from anthranilamides. Tetrahedron Lett. 1982, 27, 5323–5326. [Google Scholar]
- Chen, B.C.; Huang, X.; Wang, J. A versatile synthesis of 2-alkyl and 2-aryl-4-quinolones. Synthesis 1987, 1987, 482–483. [Google Scholar] [CrossRef]
- Torii, S.; Okumoto, H.; He-Xu, L.; Sadakane, M.; Shostakovsky, M.V.; Ponomaryov, A.B.; Kalinin, V. Syntheses of chromones and quinolones via palladium catalysed carbonylation of O-iodophenols and anilines in the presence of acetylenes. Tetrahedron 1993, 49, 6773–6784. [Google Scholar]
- Hadjeri, M.; Peiller, E.L.; Beney, C.; Deka, N.; Lawson, M.A.; Dumontet, C.; Boumendjel, A. Antimitotic activity of 5-hydroxy-7-methoxy-2-phenyl-4-quinolones. J. Med. Chem. 2004, 47, 4964–4970. [Google Scholar] [CrossRef]
- Hadjeri, M.; Mariotte, A.-M.; Boumendjel, A. Alkylation of 2-Phenyl-4-quinolones: Synthetic and Structural Studies. Chem. Pharm. Bull. 2001, 49, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Rauhamäki, S.; Postila, P.A.; Niinivehmas, S.; Kortet, S.; Schildt, E.; Pasanen, M.; Manivannan, E.; Ahinko, M.; Koskimies, P.; Nyberg, N.; et al. Structure-Activity Relationship Analysis of 3-Phenylcoumarin-Based Monoamine Oxidase B Inhibitors. Front. Chem. 2018, 2, 41. [Google Scholar]
- Pisano, M.B.; Kumar, A.; Medda, R.; Gatto, G.; Pal, R.; Fais, A.; Era, B.; Cosentino, S.; Uriarte, E.; Santana, L.; et al. Antibacterial Activity and Molecular Docking Studies of a Selected Series of Hydroxy-3-arylcoumarins. Molecules 2019, 24, 2815. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, D.; Sancho, R.; Bedoya, L.M.; López-Pérez, J.L.; del Olmo, E.; Muñoz, E.; Alcamí, J.; Gupta, M.P. Feliciano, A.S. 3-Phenylcoumarins as Inhibitors of HIV-1 Replication. Molecules 2012, 17, 9245–9257. [Google Scholar]
- Hong, W.P.; Shin, I.; Lim, H.N. Recent advances in one-pot modular synthesis of 2-Quinolones. Molecules 2020, 25, 5450. [Google Scholar] [CrossRef]
- Doléans-Jordheim, A.; Veron, J.-B.; Fendrich, O.; Bergeron, E.; Montagut-Romans, A.; Wong, Y.-S.; Furdui, B.; Freney, J.; Dumontet, C.; Boumendjel, A. 3-Aryl-4-methyl-2-quinolones targeting multiresistant Staphylococcus aureus bacteria. ChemMedChem 2013, 8, 652–657. [Google Scholar]
- Joseph, B.; Darro, F.; Béhard, A.; Lesur, B.; Collignon, F.; Decaestecker, C.; Frydman, A.; Guillaumet, G.; Kiss, R. 3-Aryl-2-quinolone derivatives: synthesis and characterization of in vitro and in vivo antitumor effects with emphasis on a new therapeutical target connected with cell Migration. J. Med. Chem. 2002, 45, 2543–2555. [Google Scholar] [CrossRef]
- Manley, P.J.; Bilodeau, M.T. A new synthesis of naphthyridinones and quinolinones: Palladium-catalyzedamidation ofo-carbonyl-substituted aryl halides. Org. Lett. 2004, 6, 2433–2435. [Google Scholar] [CrossRef]
- Sugasawa, T.; Adachi, M.; Sasakura, K.; Kitagawa, A. Aminohaloborane in Organic Synthesis. 2. Simple Synthesis of Indoles and l-Acyl-3-indolinones Using Specific Ortho-Chloroacetylation of Anilines. J. Org. Chem. 1979, 44, 578–586. [Google Scholar] [CrossRef]
- Blanc, M.; Martin, M.; Okombi, S.; Boumendjel, A. 2-Amino-α-chloroacetophenone as a valuable tool for the synthesis of conveniently substituted α,β-epoxychalcone derivatives. Chem. Pharm. Bull. 2005, 53, 435–436. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points forDrug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar]
- Bérubé, C.; Barbeau, X.; Cardinal, S.; Boudreault, P.L.; Bouchard, C.; Delcey, N.; Lagüe, P.; Voyer, N. Interfacial supramolecular biomimetic epoxidation catalyzed by cyclic dipeptides. Supramol. Chem. 2016, 29, 1–20. [Google Scholar]
- Hussein, K.A.; Shihab, N.L.; Saeed, B.A. Synthesis, Identification and Study of the Anti-microbial activity of Novel Chalcone and Epoxy chalcone compounds. Egypt. J. Chem. 2021, 64, 2297–2304. [Google Scholar]
- Saba, F.; Zainab, N. One Pot and Two Pot Synthetic Strategies and Biological Applications of Epoxy-Chalcones. Chem. Afr. 2020, 3, 291–302. [Google Scholar]
- Ono, E.; Fukuchi-Mizutani, M.; Nakamura, N.; Fukui, Y.; Yonekura-Sakakibara, K.; Yamaguchi, M.; Nakayama, T.; Tanaka, T.; Kusumi, T.; Tanaka, Y. Yellow flowers generated by expression of the aurone biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 11075–11080. [Google Scholar] [CrossRef]
- Sui, G.; Li, T.; Zhang, B.; Wang, R.; Hao, H.; Zhou, W. Recent advances on synthesis and biological activities of aurones. Bioorg. Med. Chem. 2021, 29, 115895. [Google Scholar]
- Tóth, S.; Szepesi, Á.; Tran-Nguyen, V.K.; Sarkadi, B.; Német, K.; Falson, P.; Di Pietro, A.; Szakács, G.; Boumendjel, A. Synthesis and anticancer cytotoxicity of azaaurones overcoming multidrug resistance. Molecules 2020, 25, 764. [Google Scholar] [CrossRef]
- Campaniço, A.; Carrasco, M.P.; Njoroge, M.; Seldon, R.; Chibale, K.; Perdigão, J.; Portugal, I.; Warner, D.F.; Moreira, R.; Lopes, F. Azaaurones as potent antimycobacterial agents active against MDR- and XDR-TB. ChemMedChem 2019, 14, 1537–1546. [Google Scholar] [CrossRef]
- Souard, F.; Okombi, S.; Beney, C.; Chevally, S.; Valentin, A.; Boumendjel, A. Azaaurones derived from the naturally occurring aurones as potential antimalarial drugs. Bioorg. Med. Chem. 2010, 18, 5724–5731. [Google Scholar]
- Carrasco, M.P.; Machado, M.; Gonçalves, L.; Sharma, M.; Gut, J.; Lukens, A.K.; Wirth, D.F.; André, V.; Duarte, M.T.; Guedes, R.C.; et al. Probing the azaaurone scaffold against the hepatic and rrythrocytic stages of malaria parasites. ChemMedChem 2016, 11, 2194–2204. [Google Scholar] [CrossRef]
- Pirovano, V.; Brambilla, E.; Riva, M.; Leoni, S.; Rizzato, S.; Garanzini, D.; Abbiati, G.; Rossi, E. Stereoselective synthesis of 2-spirocyclopropyl-indolin-3-ones through cyclopropanation of aza-aurones with tosylhydrazones. Org. Biomol. Chem. 2021, 19, 3925–3931. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Schedler, M.; Daniliuc, C.G.; Glorius, F. N-Heterocyclic carbene catalyzed formal [3+2] annulation reaction of enals: An efficient enantioselective access to spiro-heterocycles. Angew. Chem. Int. Ed. Engl. 2014, 53, 10232–10236. [Google Scholar] [CrossRef] [PubMed]
- Roeder, E. Medicinal plants in Europe containing pyrrolizidine alkaloids. Pharmazie 1995, 50, 83–98. [Google Scholar] [PubMed]
- Schramm, S.; Köhler, N.; Rozhon, W. Pyrrolizidine alkaloids: Biosynthesis, biological activities and occurrence in crop plants. Molecules 2019, 24, 498. [Google Scholar] [CrossRef] [PubMed]
- Clayton, M.J.; Davis, T.Z.; Knoppel, E.L.; Stegelmeier, B.L. Hepatotoxic Plants that Poison Livestock. Vet. Clin. North Am. Food Anim. Pract. 2020, 36, 715–723. [Google Scholar] [PubMed]
- Sun, H.; Nikolovska-Coleska, Z.; Lu, J.; Meagher, J.L.; Yang, C.-Y.; Qiu, S.; Tomita, Y.; Ueda, Y.; Jiang, S.; Krajewski, K.; et al. Design, Synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J. Am. Chem. Soc. 2007, 129, 15279–15294. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; He, H.; Bertekap, R.; Westphal, R.; Lelas, S.; Newton, A.; Wallace, T.; Taber, M.; Davis, C.; Macor, J.E.; et al. Discovery of disubstituted piperidines and homopiperidines as potent dual NK1 receptor antagonists-serotonin. Reuptake transporter inhibitors for the treatment of depression. Bioorg. Med. Chem. 2013, 21, 2217–2228. [Google Scholar] [PubMed]
- Shaw, M.H.; Croft, R.A.; Whittingham, W.G.; Bower, J.F. Modular access to substituted azocanes via a rhodium-catalyzed cycloaddition–fragmentation strategy. J. Am. Chem. Soc. 2015, 137, 8054–8057. [Google Scholar] [CrossRef]
- Chauhan, P.S.; Sacher, J.R.; Weinreb, S.M. Synthesis of the tetracyclic skeleton of the Lycopodium alkaloid lycopladine H via a pivotal double hydroformylation/intramolecular reductive amination sequence. Org. Lett. 2015, 17, 806–808. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eymery, M.; Tran-Nguyen, V.-K.; Boumendjel, A. Diversity-Oriented Synthesis: Amino Acetophenones as Building Blocks for the Synthesis of Natural Product Analogs. Pharmaceuticals 2021, 14, 1127. https://doi.org/10.3390/ph14111127
Eymery M, Tran-Nguyen V-K, Boumendjel A. Diversity-Oriented Synthesis: Amino Acetophenones as Building Blocks for the Synthesis of Natural Product Analogs. Pharmaceuticals. 2021; 14(11):1127. https://doi.org/10.3390/ph14111127
Chicago/Turabian StyleEymery, Mathias, Viet-Khoa Tran-Nguyen, and Ahcène Boumendjel. 2021. "Diversity-Oriented Synthesis: Amino Acetophenones as Building Blocks for the Synthesis of Natural Product Analogs" Pharmaceuticals 14, no. 11: 1127. https://doi.org/10.3390/ph14111127
APA StyleEymery, M., Tran-Nguyen, V.-K., & Boumendjel, A. (2021). Diversity-Oriented Synthesis: Amino Acetophenones as Building Blocks for the Synthesis of Natural Product Analogs. Pharmaceuticals, 14(11), 1127. https://doi.org/10.3390/ph14111127