
Repurposing of the ALK Inhibitor Crizotinib for Acute Leukemia and Multiple Myeloma Cells

 ,
,  ,
, 
Abstract

1. Introduction
2. Results
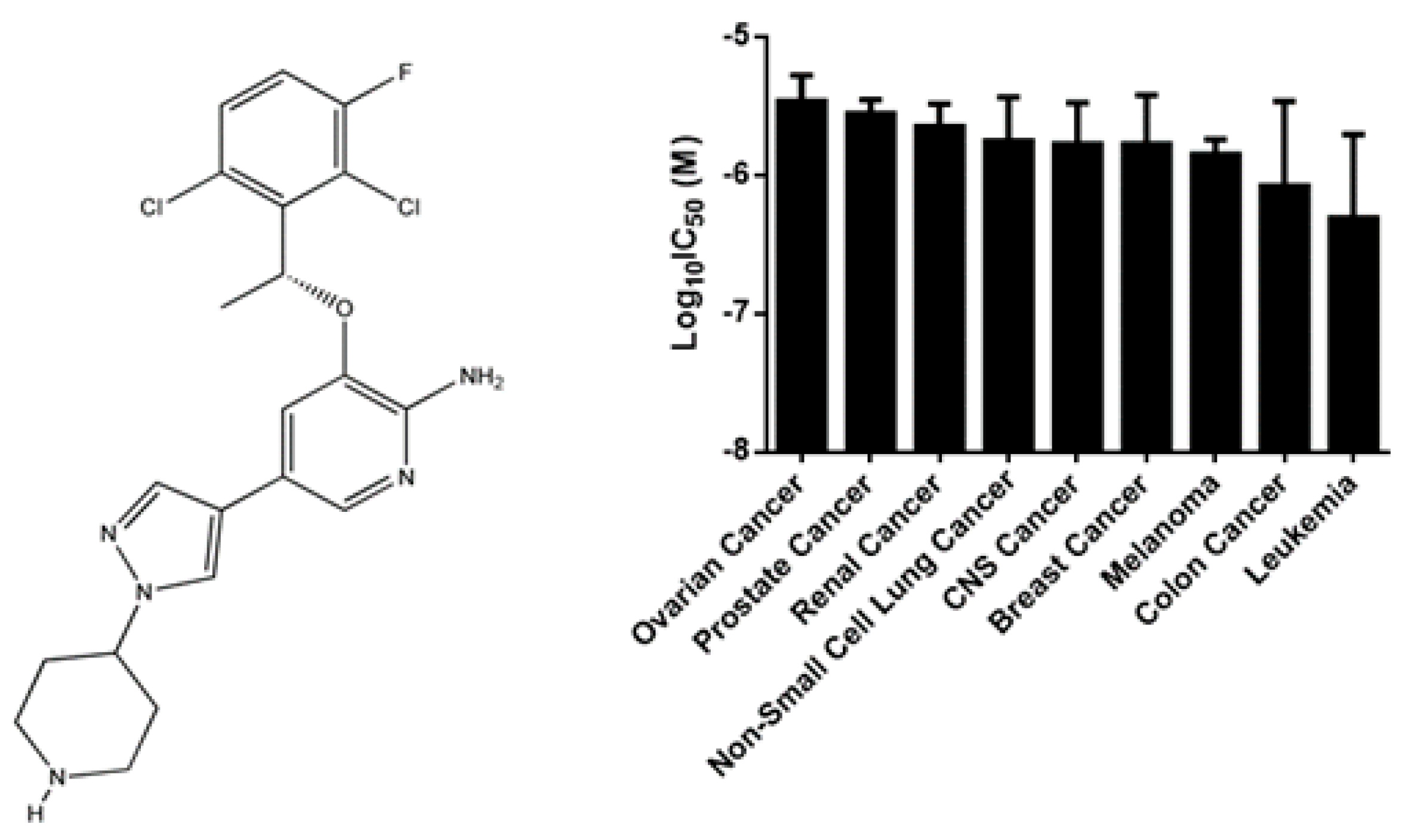
2.1. Cytotoxicity of Crizotinib
2.2. Cross-Resistance of Crizotinib to Established Anti-Cancer Drugs
2.3. COMPARE and Hierarchical Cluster Analyses of Transcriptome-Based Expression Profiling of the NCI Tumor Cell Line Panel
2.4. Transcription Factor Binding Motif Analysis
2.5. Transcriptome-Based Expression Profiling of Crizotinib-Treated NCI-H929 Cells
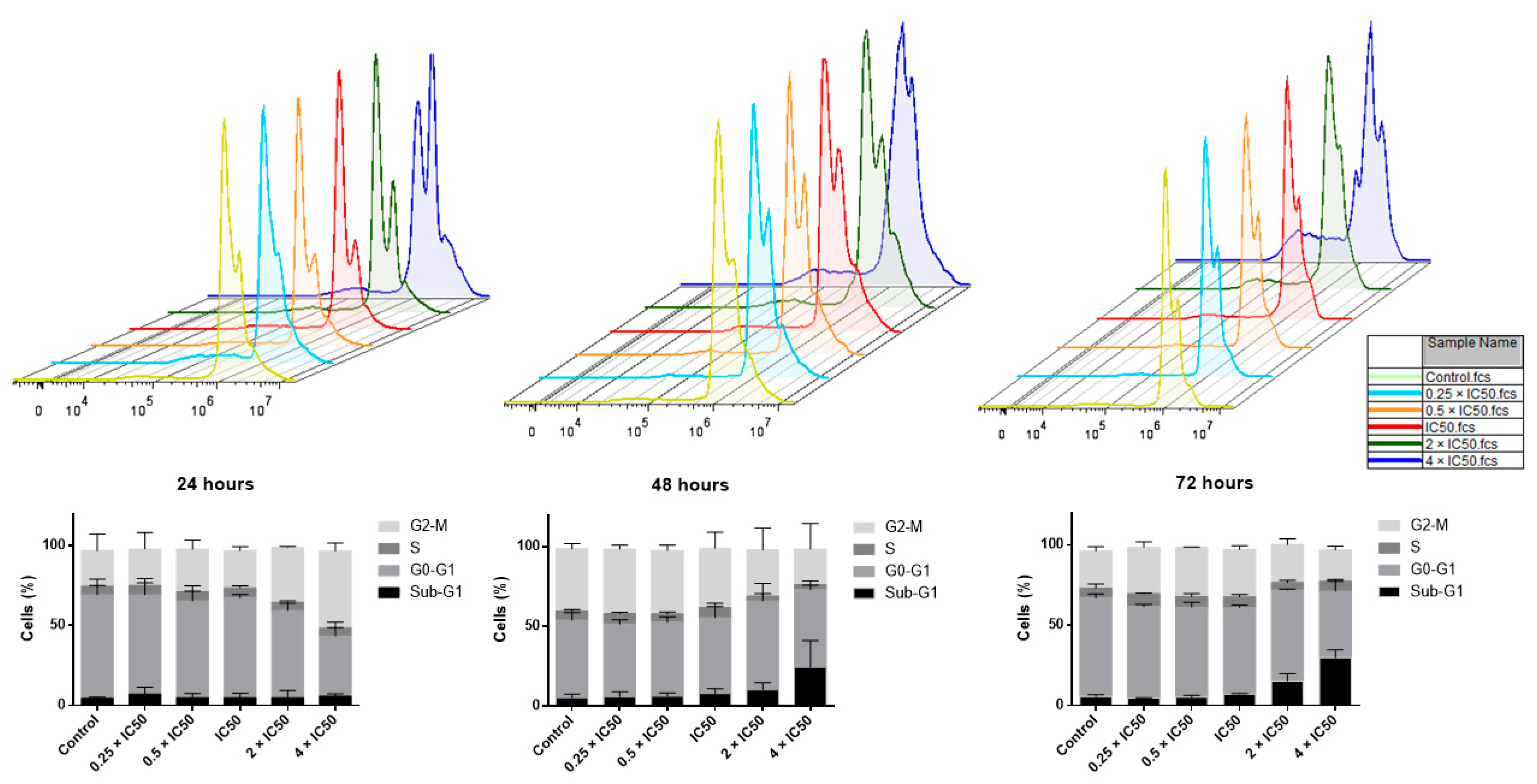
2.6. Cell Cycle Analysis
2.7. Immunofluorescence of Phospho-Histone-3 (Ser10)
2.8. Effect of Crizotinib on Microtubules
2.9. Effect of Crizotinib on Microtubule Polymerization
2.10. Molecular Docking of Crizotinib to Colchicine and Vinca Alkaloids Binding Sites
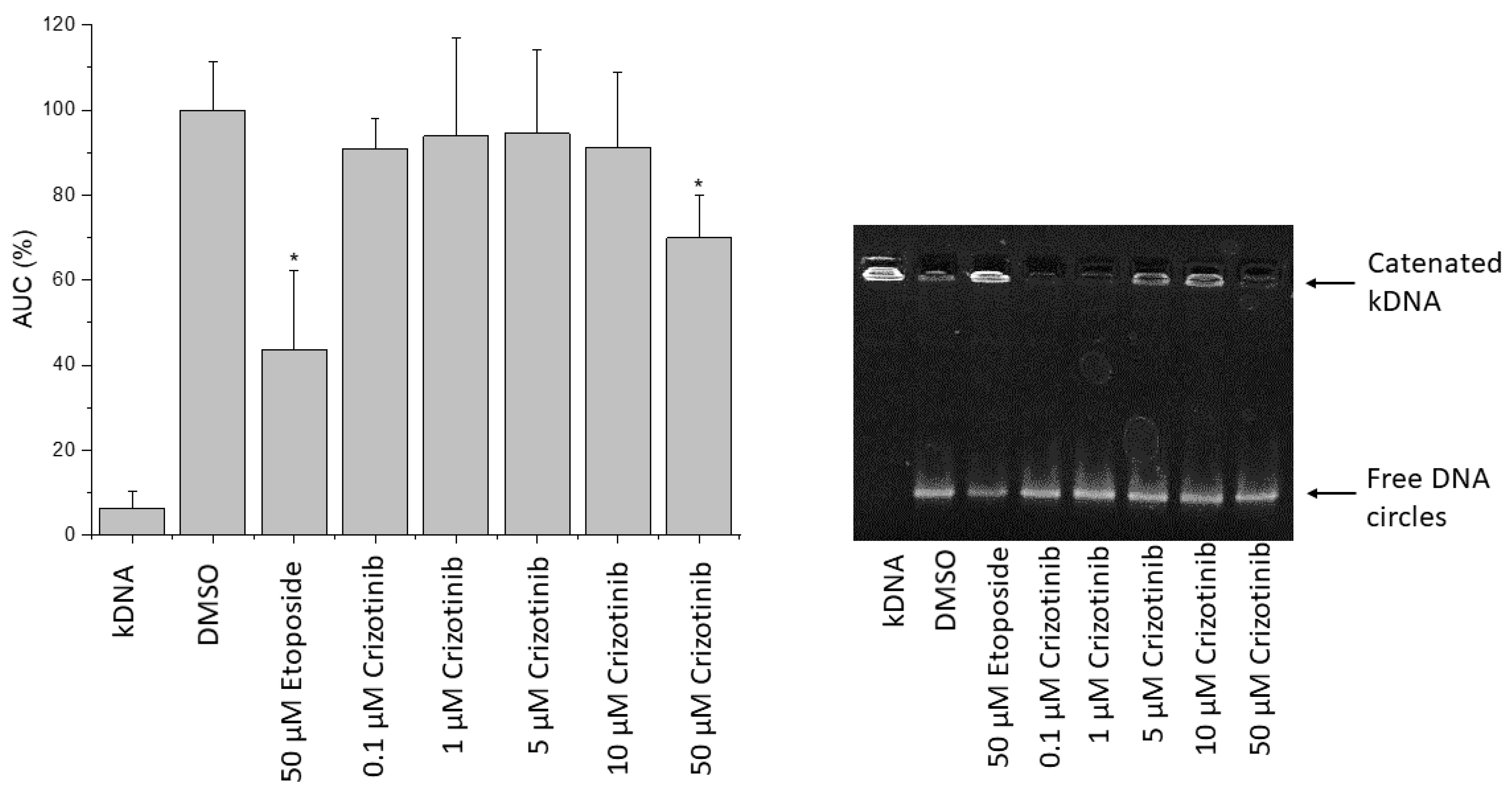
2.11. Influence of Crizotinib on Human Topoisomerase II Activity
3. Discussion
4. Materials and Methods
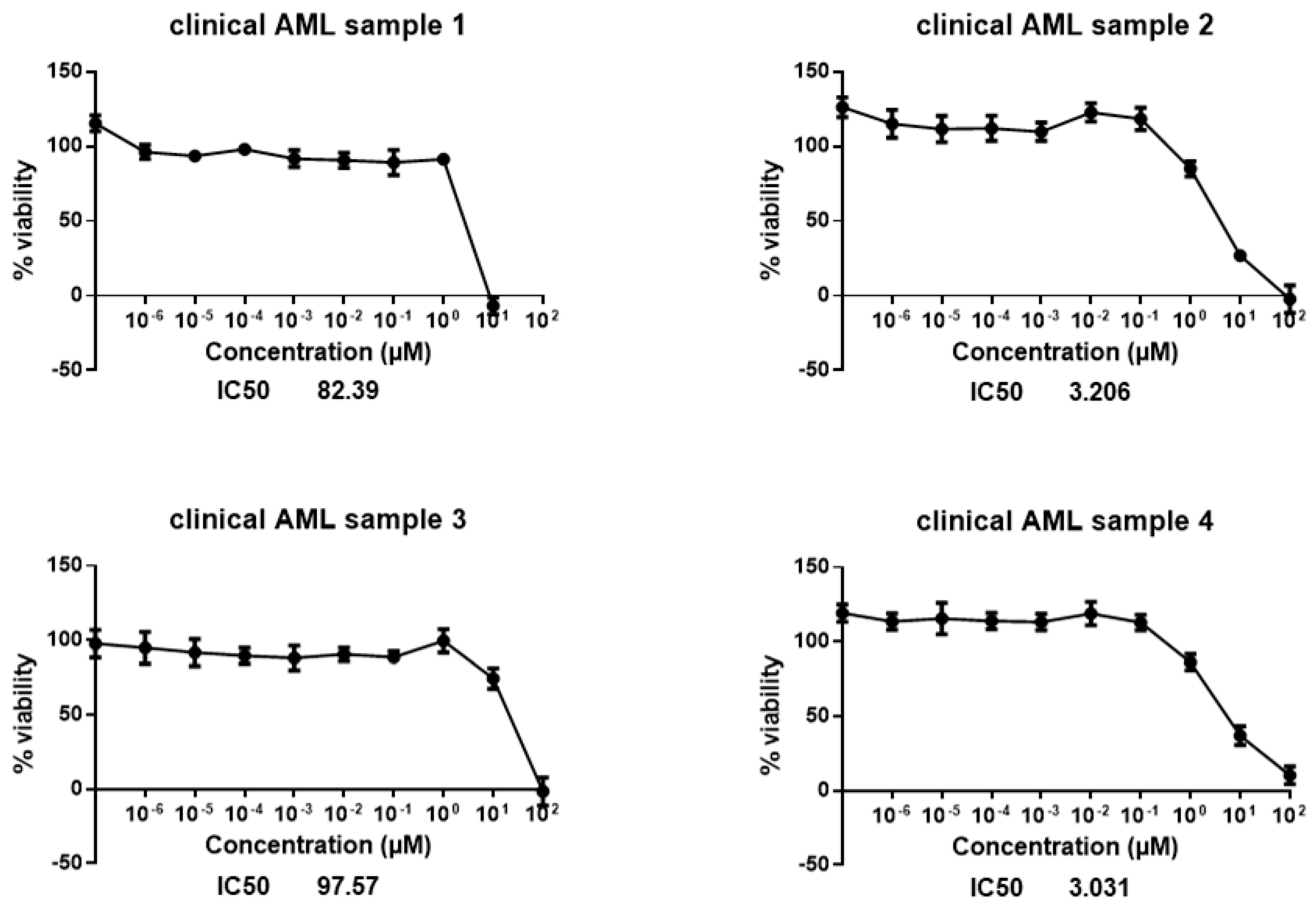
4.1. Cell Lines and Patient Samples
4.2. Cytotoxicity Assay
4.3. COMPARE and Hierarchical Cluster Analysis
4.4. Transcription Factor Promoter Binding Motif Analysis
4.5. Gene Expression Profiles
4.6. Cell Cycle Analysis
4.7. Fluorescence Microscopy of p-H3(Ser10)
4.8. Fluorescence Microscopy of the Microtubule Cytoskeleton
4.9. Tubulin Polymerization Assay
4.10. Molecular Docking of Tubulin
4.11. Decatenation Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D. Global burden of multiple myeloma: A systematic analysis for the global burden of disease study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.-J. Novel mechanism of drug resistance to proteasome inhibitors in multiple myeloma. World J. Clin. Oncol. 2019, 10, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef]
- Ria, R.; Reale, A.; Vacca, A. Novel agents and new therapeutic approaches for treatment of multiple myeloma. World J. Methodol. 2014, 4, 73. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Liu, X.-Y.; Liu, Y.-Z.; Liu, L.-N.; Lin, Q.-D.; Song, Y.-P.; Fang, B.-J. Analysis of the efficacy and safety of bortezomib for treating newly diagnosed multiple myeloma through different administration methods. Cancer Manag. Res. 2019, 11, 8295–8302. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Fuhrmann, S.; Strifler, S.; Schmidt-Hieber, M.; Einsele, H.; Knop, S. The diagnosis and treatment of multiple myeloma. Dtsch. Ärzteblatt Int. 2016, 113, 470. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Nishimura, Y.; Hara, H. Drug repositioning: Current advances and future perspectives. Front. Pharmacol. 2018, 9, 1068. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91. [Google Scholar] [PubMed]
- Barrows, S.M.; Wright, K.; Copley-Merriman, C.; Kaye, J.A.; Chioda, M.; Wiltshire, R.; Torgersen, K.M.; Masters, E.T. Systematic review of sequencing of ALK inhibitors in ALK-positive non-small-cell lung cancer. Lung Cancer Targets Ther. 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M.; Al-Shami, K.M.; Alqudah, M.A.; Mhaidat, N.M. Crizotinib inhibits growth, migration, and invasion of breast cancer cells in vitro and synergizes with chemotherapeutic agents. AACR 2018, 78, 2937. [Google Scholar] [CrossRef]
- Rochigneux, P.; Thomassin-Piana, J.; Laibe, S.; Brunelle, S.; Salem, N.; Escudier, B.; Vassal, G.; Gravis, G. Long-term efficacy of crizotinib in a metastatic papillary renal carcinoma with MET amplification: A case report and literature review. BMC Cancer 2018, 18, 1159. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef]
- Farina, F.; Stasia, A.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; Mussolin, L.; Deeren, D. High response rates to crizotinib in advanced, chemoresistant ALK+ lymphoma patients. Am. Soc. Hematol. 2013, 122, 368. [Google Scholar]
- Foyil, K.V.; Bartlett, N.L. Brentuximab vedotin and crizotinib in anaplastic large-cell lymphoma. Cancer J. 2012, 18, 450–456. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Tsiara, A.; Tsironis, G.; Lykka, M.; Liontos, M.; Bamias, A.; Dimopoulos, M.-A. Treating ALK-positive non-small cell lung cancer. Ann. Transl. Med. 2018, 6, 141. [Google Scholar] [CrossRef]
- Heigener, D.F.; Reck, M. Crizotinib. In Small Molecules in Oncology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 57–65. [Google Scholar]
- Boulos, J.C.; Idres, M.R.Y.; Efferth, T. Investigation of cancer drug resistance mechanisms by phosphoproteomics. Pharmacol. Res. 2020, 160, 105091. [Google Scholar] [CrossRef]
- Volm, M.; Efferth, T. Prediction of cancer drug resistance and implications for personalized medicine. Front. Oncol. 2015, 5, 282. [Google Scholar] [CrossRef]
- Syrykh, C.; Ysebaert, L.; Péricart, S.; Evrard, S.M.; Meggetto, F.; Kanoun, S.; Brousset, P.; Laurent, C. ALK-positive histiocytosis associated with chronic lymphocytic leukaemia/small lymphocytic lymphoma: A multitarget response under ibrutinib. Virchows Arch. 2020, 478, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, W.; Zhang, J.-P.; Song, Z.; Li, Y.; Xiao, W.; Liu, Y.; Zeng, M.-S.; Petrus, M.N.; Thomas, C.J. A novel model of alternative NF-κB pathway activation in anaplastic large cell lymphoma. Leukemia 2020, 35, 1976–1989. [Google Scholar] [CrossRef]
- Hao, X.; Xiao, H.; Ju, J.; Hewitt, S.M.; Morse, H.C. Decreased Expression of Retinoid X Receptors During Human and Azoxymethane-induced Colorectal Carcinogenesis in the Rat. Anticancer Res. 2016, 36, 2659–2664. [Google Scholar] [PubMed]
- Halftermeyer, J.; Le, M.B. RXR, a key member of the oncogenic complex in acute promyelocytic leukemia. Med. Sci. M/S 2011, 27, 973–978. [Google Scholar]
- Huang, G.; Chen, Q.; Shen, D. The Function of Retinoid X Receptor α in Cancer Cells. Biol. Syst. Open Access 2016, 5, 2. [Google Scholar] [CrossRef]
- Crowe, D.L.; Chandraratna, R.A.S. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-α is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res. 2004, 6, R546. [Google Scholar] [CrossRef]
- Yen, W.-C.; Lamph, W.W. The selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced breast carcinoma. Mol. Cancer Ther. 2005, 4, 824–834. [Google Scholar] [CrossRef]
- Kiss, M.; Czimmerer, Z.; Nagy, G.; Bieniasz-Krzywiec, P.; Ehling, M.; Pap, A.; Poliska, S.; Boto, P.; Tzerpos, P.; Horvath, A. Retinoid X receptor suppresses a metastasis-promoting transcriptional program in myeloid cells via a ligand-insensitive mechanism. Proc. Natl. Acad. Sci. USA 2017, 114, 10725–10730. [Google Scholar] [CrossRef]
- Zheng, R.; Blobel, G.A. GATA transcription factors and cancer. Genes Cancer 2010, 1, 1178–1188. [Google Scholar] [CrossRef]
- Evans, T.; Reitman, M.; Felsenfeld, G. An erythrocyte-specific DNA-binding factor recognizes a regulatory sequence common to all chicken globin genes. Proc. Natl. Acad. Sci. USA 1988, 85, 5976–5980. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, A.; Shimizu, R. GATA1 Activity Governed by Configurations of cis-Acting elements. Front. Oncol. 2017, 6, 269. [Google Scholar] [CrossRef]
- Caldwell, J.T.; Edwards, H.; Dombkowski, A.A.; Buck, S.A.; Matherly, L.H.; Ge, Y.; Taub, J.W. Overexpression of GATA1 confers resistance to chemotherapy in acute megakaryocytic Leukemia. PLoS ONE 2013, 8, e68601. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Zhang, Y.; Liu, J.; Guan, C.; Gu, X.; Yang, Z.; Ye, Q.; Ding, L.; Liu, R. GATA1 Promotes Gemcitabine Resistance in Pancreatic Cancer through Antiapoptotic Pathway. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef]
- Prigent, C.; Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 2003, 116, 3677–3685. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The mechanism of tubulin assembly into microtubules: Insights from structural studies. Iscience 2020, 23, 101511. [Google Scholar] [CrossRef]
- Jordan, M.A.; Kamath, K. How do microtubule-targeted drugs work? An overview. Curr. Cancer Drug Targets 2007, 7, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Borys, F.; Joachimiak, E.; Krawczyk, H.; Fabczak, H. Intrinsic and extrinsic factors affecting Microtubule dynamics in normal and cancer cells. Molecules 2020, 25, 3705. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kaverina, I.; Straube, A. Regulation of Cell Migration by Dynamic Microtubules; Elsevier: Amsterdam, The Netherlands, 2011; pp. 968–974. [Google Scholar]
- Usui, T. Actin-and microtubule-targeting bioprobes: Their binding sites and inhibitory mechanisms. Biosci. Biotechnol. Biochem. 2007, 71, 300–308. [Google Scholar] [CrossRef][Green Version]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Červinka, M.; Cerman, J.; Rudolf, E. Apoptosis in Hep2 cells treated with etoposide and colchicine. Cancer Detect Prev. 2004, 28, 214–226. [Google Scholar] [CrossRef]
- Skok, Z.i.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 2019, 63, 884–904. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1117. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Chi, G.F.; Bonsou, I.N.; Abdelfatah, S.; Tamfu, A.N.; Yeboah, E.M.O.; Kuete, V.; Efferth, T. N-acetylglycoside of oleanolic acid (aridanin) displays promising cytotoxicity towards human and animal cancer cells, inducing apoptotic, ferroptotic and necroptotic cell death. Phytomedicine 2020, 76, 153261. [Google Scholar] [CrossRef]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Boulos, J.C.; Elhaboub, G.; Rigano, D.; Saab, A.; Loizzo, M.R.; Hassan, L.E.A.; Sugimoto, Y.; Piacente, S.; Tundis, R. Cytotoxicity of cucurbitacin E from Citrullus colocynthis against multidrug-resistant cancer cells. Phytomedicine 2019, 62, 152945. [Google Scholar] [CrossRef] [PubMed]
- Özenver, N.; Boulos, J.C.; Efferth, T. Activity of Cordycepin From Cordyceps sinensis Against Drug-Resistant Tumor Cells as Determined by Gene Expression and Drug Sensitivity Profiling. Nat. Prod. Commun. 2021, 16, 1934578X21993350. [Google Scholar]
- Kadioglu, O.; Fu, Y.; Wiench, B.; Zu, Y.; Efferth, T. Synthetic cajanin stilbene acid derivatives inhibit c-MYC in breast cancer cells. Arch. Toxicol. 2016, 90, 575–588. [Google Scholar] [CrossRef]
- Seo, E.-J.; Klauck, S.M.; Efferth, T.; Panossian, A. Adaptogens in chemobrain (Part I): Plant extracts attenuate cancer chemotherapy-induced cognitive impairment–Transcriptome-wide microarray profiles of neuroglia cells. Phytomedicine 2019, 55, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Dawood, M.; Böckers, M.; Klauck, S.M.; Fottner, C.; Weber, M.M.; Efferth, T. Multiple modes of cell death in neuroendocrine tumors induced by artesunate. Phytomedicine 2020, 79, 153332. [Google Scholar] [CrossRef]
- Telford, W.G.; King, L.E.; Fraker, P.J. Comparative evaluation of several DNA binding dyes in the detection of apoptosis-associated chromatin degradation by flow cytometry. Cytometry 1992, 13, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Adem, F.A.; Mbaveng, A.T.; Kuete, V.; Heydenreich, M.; Ndakala, A.; Irungu, B.; Yenesew, A.; Efferth, T. Cytotoxicity of isoflavones and biflavonoids from Ormocarpum kirkii towards multi-factorial drug resistant cancer. Phytomedicine 2019, 58, 152853. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, M.-E.F.; Abdelfatah, S.; Hamed, A.R.; Mohamed, T.A.; Elshamy, A.A.; Saleh, I.A.; Reda, E.H.; Abdel-Azim, N.S.; Shams, K.A.; Sakr, M. Cytotoxicity of 40 Egyptian plant extracts targeting mechanisms of drug-resistant cancer cells. Phytomedicine 2019, 59, 152771. [Google Scholar] [CrossRef]
- Özenver, N.; Dawood, M.; Fleischer, E.; Klinger, A.; Efferth, T. Chemometric and transcriptomic profiling, microtubule disruption and cell death induction by secalonic acid in tumor cells. Molecules 2020, 25, 3224. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Cell Line | IC50 (µM) |
|---|---|---|
| Multiple myeloma | NCI-H929 | 0.53 ± 0.04 |
| MolP8 | 0.77 ± 0.03 | |
| KMS12BM | 0.80 ± 0.18 | |
| AMO1 | 1.21 ± 2.02 | |
| KMS11 | 1.40 ± 0.40 | |
| OPM2 | 1.46 ± 0.21 | |
| L363 | 1.71 ± 0.40 | |
| RPMI8226 | 1.91 ± 0.18 | |
| JJN3 | 3.07 ± 0.39 | |
| Leukemia | CCRF-CEM | 0.43 ± 0.07 |
| NB4 | 0.63 ± 0.01 | |
| HL60 | 0.74 ± 0.02 | |
| MolT4 | 1.18 ± 0.46 | |
| CEM/ADR5000 | 29.15 ± 2.59 | |
| Normal leukocytes | PBMCs | 4.02 ± 0.49 |
| COMPARE Coefficient | Symbol | Name | Function |
|---|---|---|---|
| 0.5363 | TIMP4 | TIMP metallopeptidase inhibitor 4 | Inhibitor of matrix metalloproteinases |
| 0.561 | CRADD | CASP2 and RIPK1 domain containing adaptor with death domain | Apoptosis |
| 0.559 | NTRK1 | Neurotrophic tyrosine kinase, receptor, type 1 | Cell differentiation |
| 0.549 | KIF21B | Kinesin family member 21B | Regulation of microtubule dynamics |
| 0.541 | CD53 | CD53 cell surface molecule | Cell development, activation, growth and motility |
| 0.541 | UBE2N | Ubiquitin-conjugating enzyme E2N (UBC13 homologue, yeast) | DNA post-replication repair |
| 0.541 | NDUFA5 | NADH dehydrogenase (ubiquinone) 1α subcomplex, 5, 13 kDa | Mitochondrial respiratory chain |
| 0.536 | SH2D2A | SH2 domain containing 2A | Control of T-cell activation |
| 0.534 | NUP98 | Nucleoporin, 98 kDa | Transport of macromolecules between nucleus and cytoplasm |
| 0.533 | OSTF1 | Osteoclast stimulating factor 1 | Induction of osteoclast formation and bone resorption |
| 0.521 | PTPRC | Protein tyrosine phosphatase, receptor type, C | T-cell activation |
| 0.516 | MTHFD2 | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, methenyl tetrahydrofolate cyclohydrolase | Nuclear-encoded mitochondrial bifunctional enzyme |
| 0.509 | SLA | SRC-like-adaptor | Negative regulation T-cell receptor (TCR) signaling |
| 0.509 | TNFRSF8 | Tumor necrosis factor receptor superfamily, member 8 | Cellular growth and transformation of activated lymphoblasts |
| 0.509 | KIR3DL1 | Killer cell immunoglobulin-like receptor, three domains, long cytoplasmic tail | Inhibition of NK cells activity |
| 0.505 | TMPRSS11D | Transmembrane protease, serine 11D | Host defense system on the mucous membrane |
| 0.504 | CASP2 | Caspase 2, apoptosis-related cysteine peptidase | Apoptosis |
| 0.504 | KIR2DL3 | Killer cell immunoglobulin-like receptor, two domains, long cytoplasmic tail, 3 | Inhibition of NK cells activity |
| 0.503 | SPINK4 | Serine peptidase inhibitor, Kazal type 4 | Serine peptidase inhibitor |
| 0.501 | SLC35D1 | Solute carrier family 35 (UDP-glucuronic acid/UDP-N-acetyl galactosamine dual transporter), member D1 | Transport of UDP-glucuronic acid (UDP-GlcA) and UDP-N-acetyl galactosamine (UDP-GalNAc) from the cytoplasm into the endoplasmic reticulum |
| −0.49 | AMOTL2 | Angiomotin like 2 | Angiogenesis inhibitor |
| −0.447 | BCKDK | Branched chain keto acid dehydrogenase kinase | Regulator of valine, leucine, and isoleucine catabolic pathways |
| −0.438 | IP6K1 | Inositol hexakisphosphate kinase 1 | Member of the inositol phosphokinase family |
| −0.432 | DHCR24 | 24-Dehydrocholesterol reductase | Protection from oxidative stress |
| −0.432 | ANKLE2 | Ankyrin repeat and LEM domain containing 2 | Mitotic regulator |
| −0.428 | GPR161 | G protein-coupled receptor 161 | Potential drug target for triple-negative breast cancer |
| −0.428 | PLS3 | Plastin 3 | Bone development |
| −0.426 | C2orf55 | Chromosome 2 open reading frame 55 | Unknown |
| −0.425 | RAI14 | Retinoic acid induced 14 | Actin regulation |
| −0.423 | HYAL2 | Hyaluronoglucosaminidase 2 | Cell proliferation, migration and differentiation |
| −0.42 | NUAK1 | NUAK family, SNF1-like kinase, 1 | Cell adhesion, cell ploidy and senescence, cell proliferation and tumor progression |
| −0.418 | APP | Amyloid β (A4) precursor protein | Involved in cell mobility and transcription regulation |
| −0.415 | STK25 | Serine/threonine kinase 25 | Protein transport, cell adhesion, and cell migration |
| −0.415 | TMEM115 | Transmembrane protein 115 | Retrograde protein transport from Golgi to endoplasmic reticulum |
| −0.415 | VEGFB | Vascular endothelial growth factor B | Blood vessels formation |
| −0.414 | LAPTM4B | Lysosomal protein transmembrane 4β | Inhibition of EGFR degradation |
| −0.41 | CHRNB4 | Cholinergic receptor, nicotinic, β4 | Opening of an ion-conducting channel across the plasma membrane |
| −0.41 | BRD3 | Bromodomain containing 3 | Transcription |
| −0.408 | HSF2BP | Heat shock transcription factor 2 binding protein | Inhibition BNC1 transcriptional activity during spermatogenesis |
| −0.408 | SORBS3 | Sorbin and SH3 domain containing 3 | Cytoskeletal organization, cell adhesion, migration, signaling, gene expression |
| Category | Partition (log10IC50) | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Cluster 7 |
|---|---|---|---|---|---|---|---|---|
| Sensitive | ≤−5.771 M | 3 | 4 | 3 | 0 | 5 | 5 | 10 |
| Resistant | >−5.771 M | 5 | 4 | 4 | 9 | 1 | 3 | 3 |
| Tubulin Binding Sites | Compounds | Lowest Binding Energy (Kcal/mol) | pKi (nM) | Amino Acids Involved in Ligand Interaction |
|---|---|---|---|---|
| Colchicine binding site | Colchicine | −7.25 | 4.83 × 103 | LYS254, THR145, ASP69, TYR224, ALA180, LEU248 |
| Crizotinib | −9.62 | 88.6 | GLU183, GLY142, SER140, ASP69, GLU71, LYS254, ASN101, LYS352, ALA12, LEU248, | |
| Vinca alkaloid binding site | Vinorelbine | −6.84 | 9.85 × 103 | GLU386, ARG390, ALA208, ALA304, LYS176, PRO175, CYS305, HIS309 |
| Crizotinib | −9.24 | 177.73 | ASN228, GLN15, VAL171, SER174, PRO173, GLU183, TYR224, CYS12, VAL177 |
| Sample ID | Primary Diagnosis | Cytogenetics | Molecular Genetics | Leukocytosis at Diagnosis | Treatment Received |
|---|---|---|---|---|---|
| Sample 1 | AML FAB M4 | 46 XY | FLT3-ITDhigh | 78.000/ul | Untreated |
| Sample 2 | Highly proliferative AML FAB M4Eo | 46 XY, CBFB-MYH11 = Inv (16) | FLT3-TKD Mutation | 140.000/ul | Two applications of hydroxyurea |
| Sample 3 | Common B-ALL (B-acute lymphoplastic leukemia) | 46 XX | m-Bcr-Abl (type e1a2) | 8.000/ul | Untreated |
| Sample 4 | Very proliferative AML FAB4 | 46 XY, t (3;12) (q26;p13) [20] | No detectable mutations | 269.000/ul | Two applications of hydroxyurea |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulos, J.C.; Saeed, M.E.M.; Chatterjee, M.; Bülbül, Y.; Crudo, F.; Marko, D.; Munder, M.; Klauck, S.M.; Efferth, T. Repurposing of the ALK Inhibitor Crizotinib for Acute Leukemia and Multiple Myeloma Cells. Pharmaceuticals 2021, 14, 1126. https://doi.org/10.3390/ph14111126
Boulos JC, Saeed MEM, Chatterjee M, Bülbül Y, Crudo F, Marko D, Munder M, Klauck SM, Efferth T. Repurposing of the ALK Inhibitor Crizotinib for Acute Leukemia and Multiple Myeloma Cells. Pharmaceuticals. 2021; 14(11):1126. https://doi.org/10.3390/ph14111126
Chicago/Turabian StyleBoulos, Joelle C., Mohamed E. M. Saeed, Manik Chatterjee, Yagmur Bülbül, Francesco Crudo, Doris Marko, Markus Munder, Sabine M. Klauck, and Thomas Efferth. 2021. "Repurposing of the ALK Inhibitor Crizotinib for Acute Leukemia and Multiple Myeloma Cells" Pharmaceuticals 14, no. 11: 1126. https://doi.org/10.3390/ph14111126
APA StyleBoulos, J. C., Saeed, M. E. M., Chatterjee, M., Bülbül, Y., Crudo, F., Marko, D., Munder, M., Klauck, S. M., & Efferth, T. (2021). Repurposing of the ALK Inhibitor Crizotinib for Acute Leukemia and Multiple Myeloma Cells. Pharmaceuticals, 14(11), 1126. https://doi.org/10.3390/ph14111126