3.2.5. General Procedure for the Synthesis of Compounds 3, 15, 23–28, 46–49, 50 and 51
To a solution of 15% (m/m) palladium on activated carbon in dry methanol (100 mL), the corresponding nitro compound (2.00 mmol) was added. The reduction of the nitro group was performed in an atmosphere of 50 psi hydrogen at the Parr-apparatus at room temperature for 24 h. After that, the reaction mixture was filtered and the solvent was evaporated in vacuo yielding the corresponding amino compound, which was either purified by column chromatography or used without further purification.
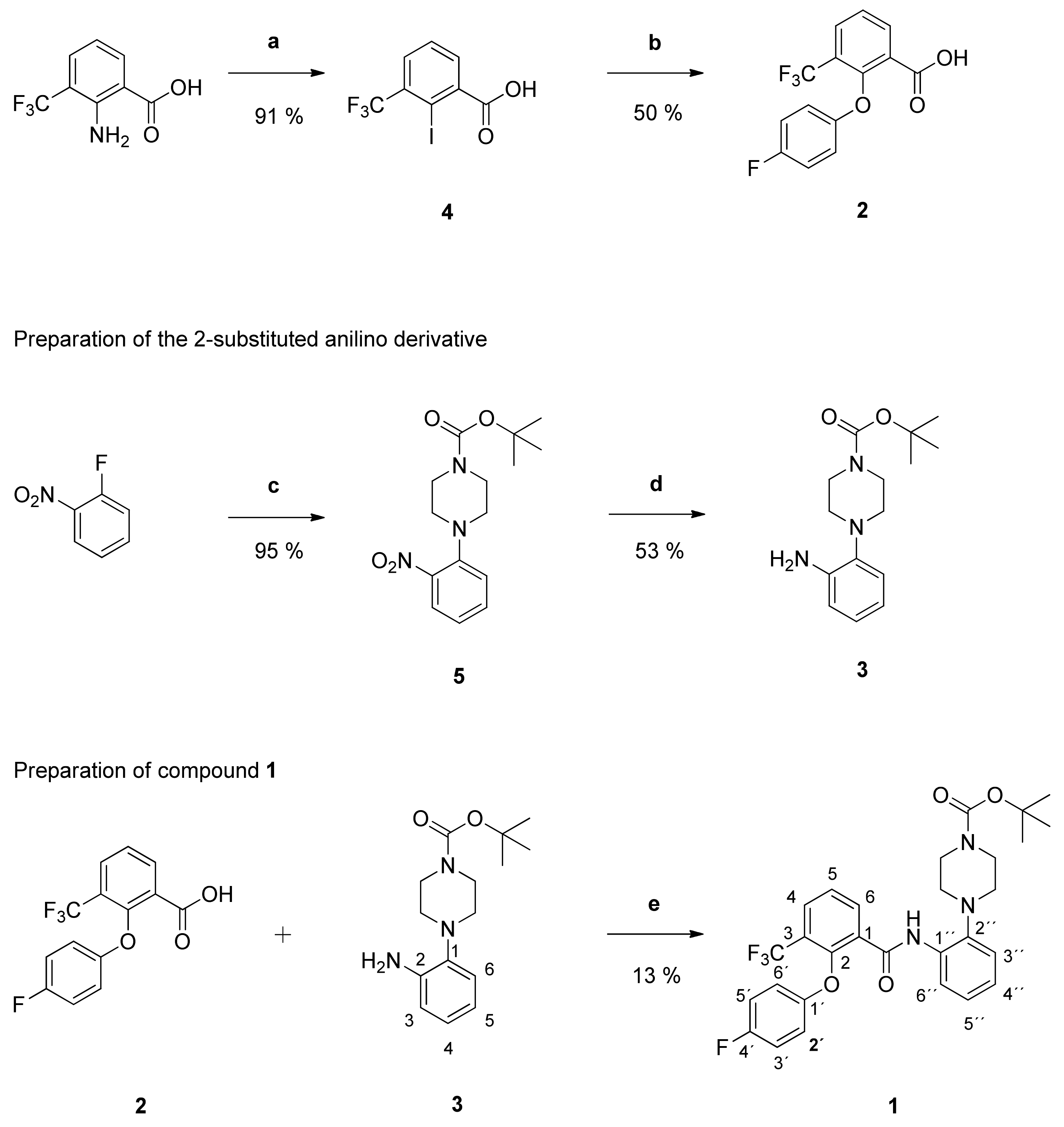
tert-Butyl-4-(2-aminophenyl)piperazine-1-carboxylate (
3): Reaction of compound
5 (3.67 g (11.93 mmol)) with PdC (560 mg) in dry methanol (100 mL) gave the raw anilino derivative. It was purified by column chromatography (silica gel, CH
2Cl
2/
MeOH 79:1) yielding compound
3 as pale brown solid (1.75 g (53%)). NMR data were in accordance with literature data [
23].
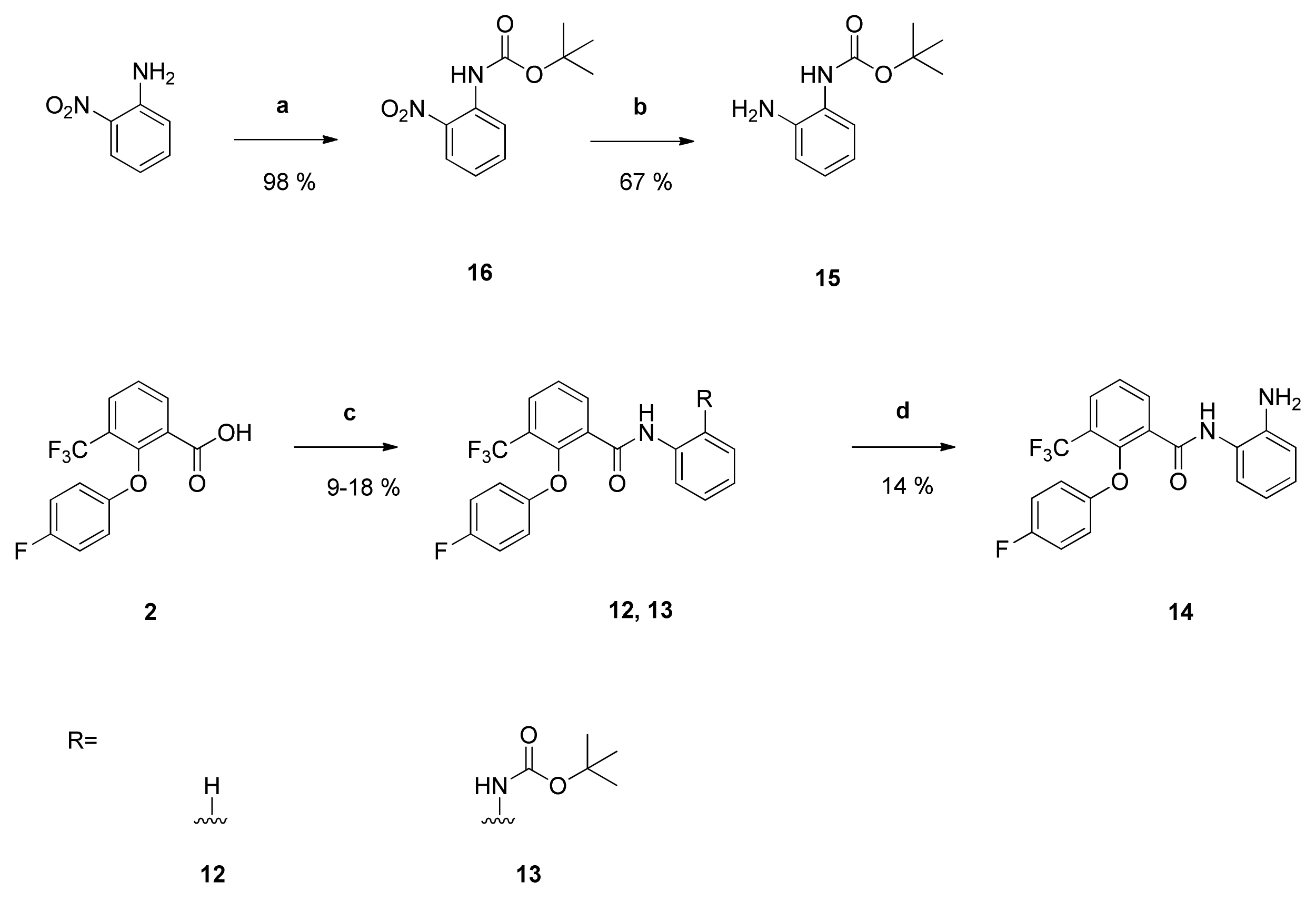
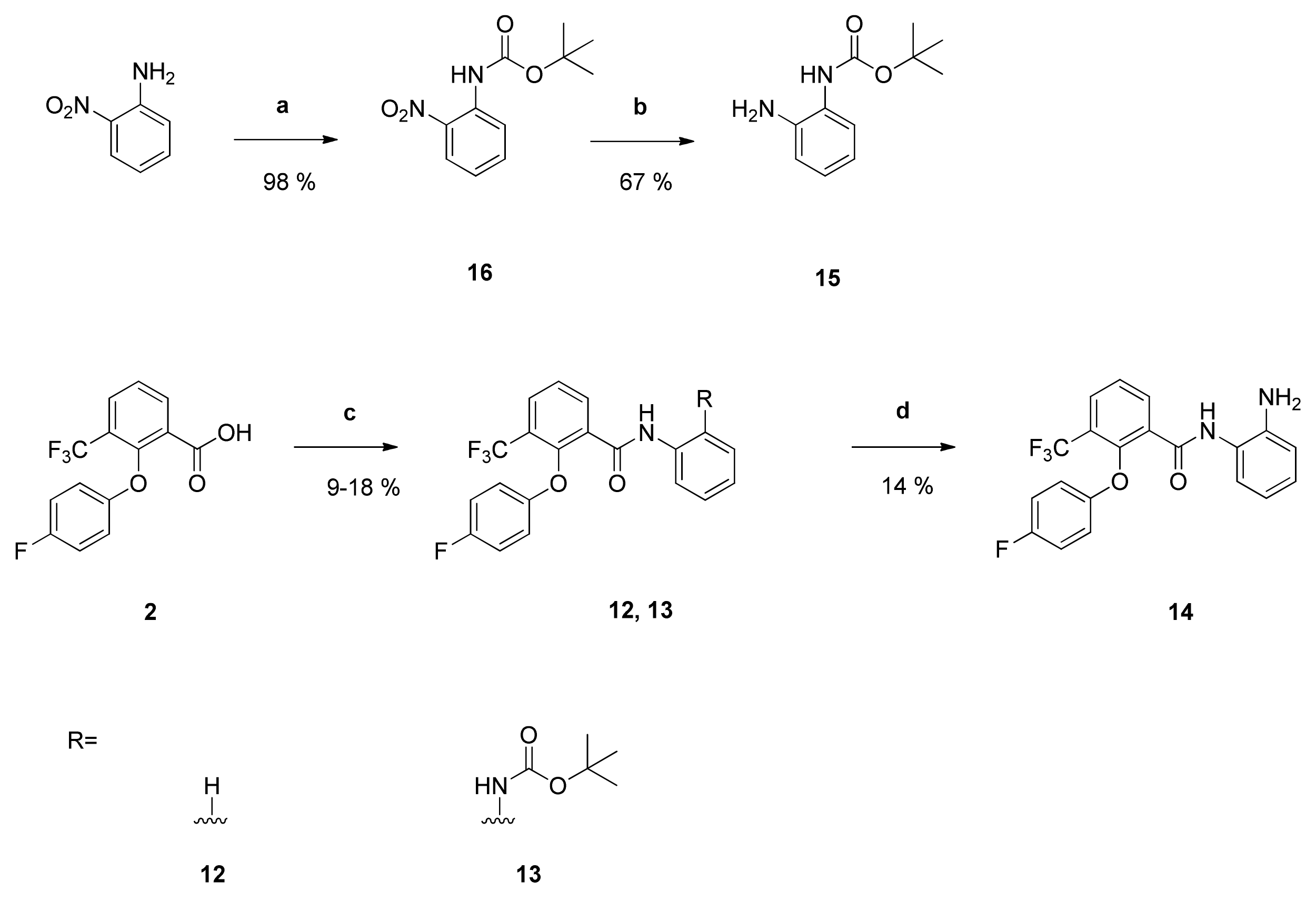
tert-Butyl-
N-(2-aminophenyl)carbamate (
15): Reaction of compound
16 (1.12 g (4.71 mmol)) with PdC (172 mg) in dry methanol (100 mL) yielded compound
15 as orange solid (657 mg (67%)), which was used without further purification. NMR data were in accordance with literature data [
40].
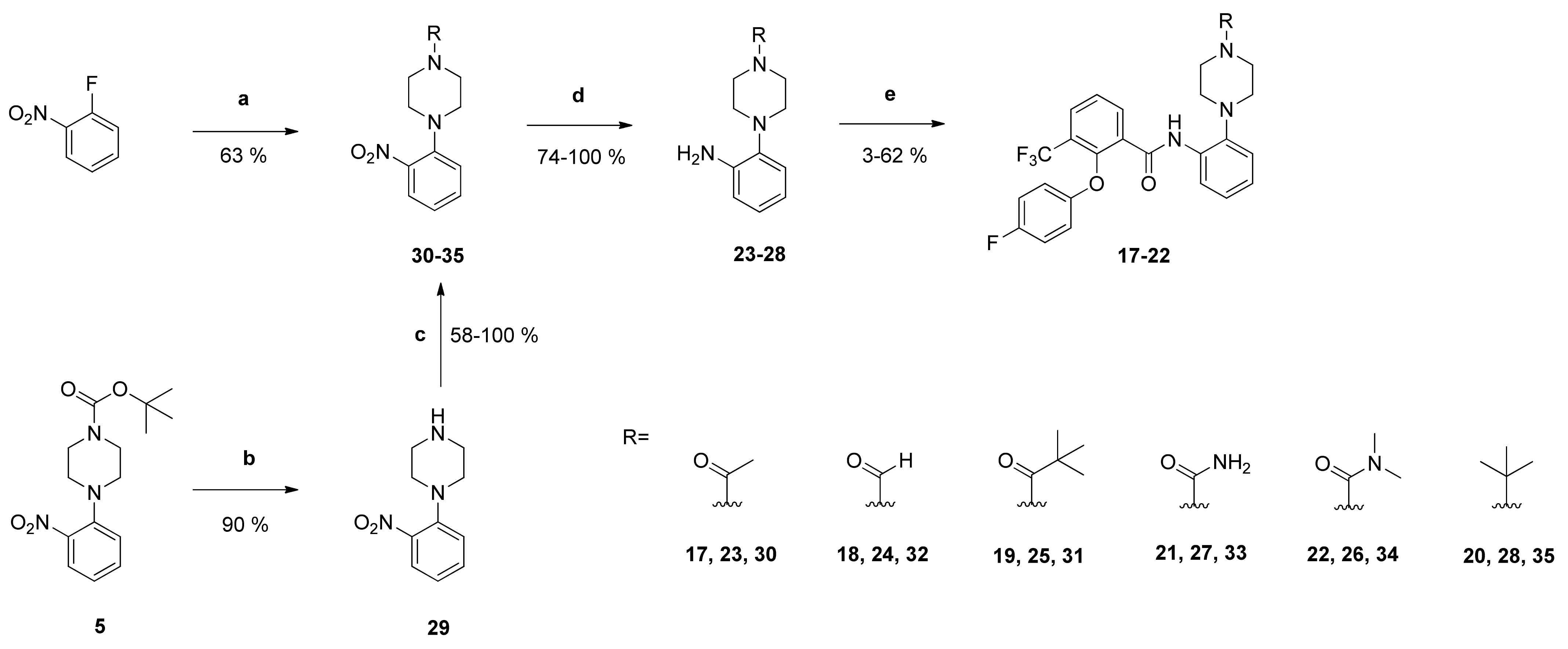
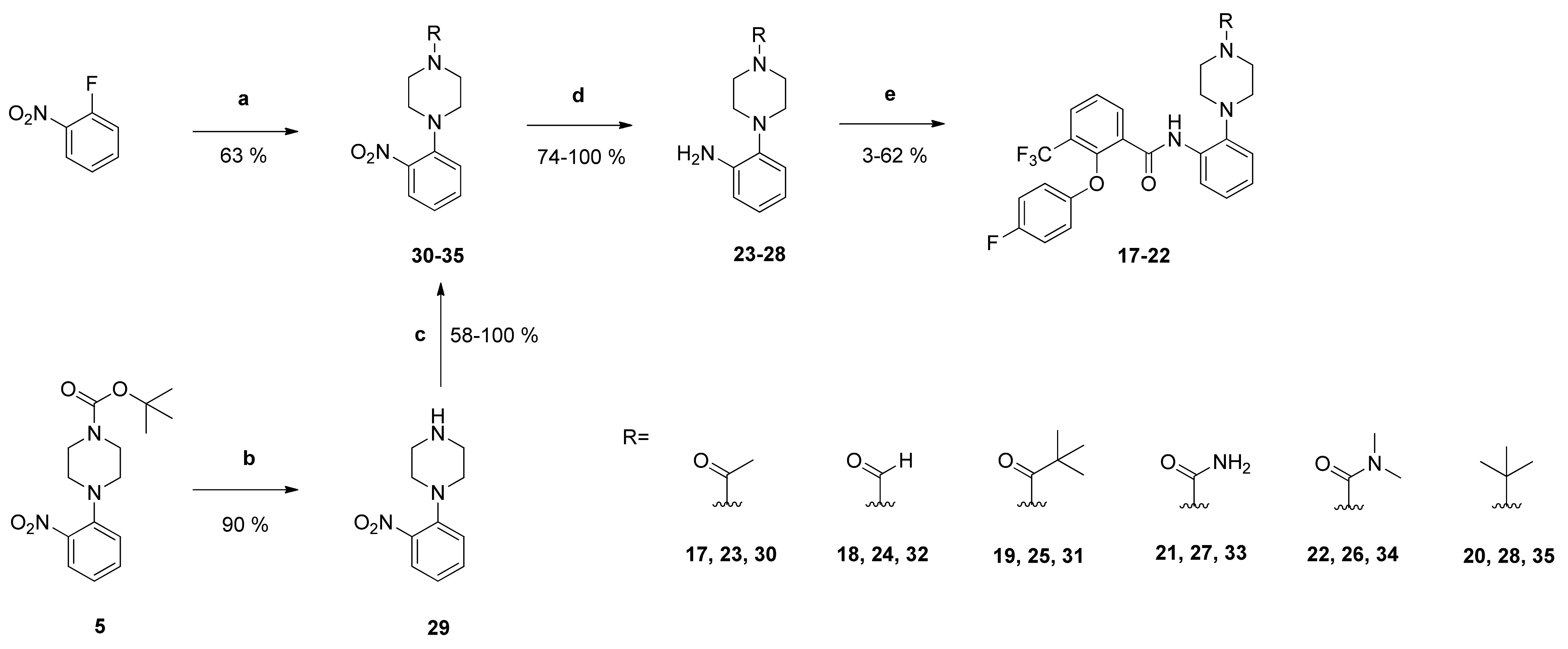
1-[4-(2-Aminophenyl)piperazin-1-yl]ethan-1-one (
23): Reaction of compound
30 (1.78 g (7.14 mmol)) with PdC (268 mg) in dry methanol (80 mL) yielded compound
23 as dark-green oil (1.57 g (100%)), which was used without further purification. NMR data were in accordance with literature data [
23].
4-(2-Aminophenyl)piperazine-1-carbaldehyde (24): Reaction of compound 32 (600 mg (2.55 mmol)) with PdC (123 mg) in dry methanol (90 mL) yielded compound 24 as pale brown solid (508 mg (97%)), which was used without further purification. IR = 3419, 3323, 2923, 2825, 1654, 1619, 1586, 1503, 1442, 1397, 1365, 1303, 1270, 1235, 1191, 1135, 1012, 918, 756; 1H NMR (CDCl3, 400 MHz) δ = 2.88–2.96 (m, 4H, N(CH2)2), 3.52 (t, J = 5.0 Hz, 2H, NCH2), 3.70 (br, 2H, NCH2), 4.00 (br s, 2H, NCH2), 6.73–6.77 (m, 2H, 3-H, 5-H), 6.94–7.00 (m, 2H, 4-H, 6-H), 8.10 (s, 1H, C=O); 13C NMR (CDCl3, 100 MHz) δ = 40.67 (NCH2), 46.32 (NCH2), 50.54 (NCH2), 51.70 (NCH2), 115.33 (C-3), 118.65 (C-5), 119.90 (C-6), 125.25 (C-4), 138.29 (C-1), 141.31 (C-2), 160.87 (C=O); HRMS (EI+) calcd for C11H16N3O [M+H]+: 206.1293; found: 206.1292.
1-[4-(2-Aminophenyl)piperazin-1-yl]-2,2-dimethylpropan-1-one (25): Reaction of compound 31 (555 mg (1.90 mmol)) with PdC (111 mg) in dry methanol (90 mL) yielded compound 25 as silver-grey solid (367 mg (74%)), which was used without further purification. IR = 3397, 3320, 2965, 2825, 1614, 1500, 1477, 1426, 1360, 1300, 1276, 1228, 1196, 1152, 1042, 1018, 934, 752; 1H NMR (CDCl3, 400 MHz) δ = 1.32 (s, 9H, (CH3)3), 2.88–2.91 (br, 4H, N(CH2)2), 3.78 (br, 4H, N(CH2)2), 3.99 (s, 2H, NH2), 6.72–6.76 (m, 2H, 3-H, 5-H), 6.93–6.97 (m, 2H, 4-H, 6-H); 13C NMR (CDCl3, 100 MHz) δ = 28.44 ((CH3)3), 38.67 (CMe3), 45.82 (N(CH2)2), 51.23 (N(CH2)2), 115.27 (C-3), 118.64 (C-5), 119.84 (C-6), 125.05 (C-4), 138.47 (C-1), 141.42 (C-2), 176.46 (C=O); HRMS (ESI +) calcd for C15H24N3O [M+H]+: 262.1919; found: 262.1919.
4-(2-Aminophenyl)-N,N-dimethylpiperazine-1-carboxamide (26): Reaction of compound 34 (407 mg (1.46 mmol)) with PdC (61 mg) in dry methanol (90 mL) yielded compound 28 as white solid (330 mg (91%)), which was used without further purification. IR = 3397, 3315, 2811, 1621, 1502, 1455, 1392, 1365, 1212, 1107, 1069, 1002, 928, 753; 1H NMR (CDCl3, 400 MHz) δ = 2.87 (s, 6H, N(CH3)2), 2.89–2.92 (m, 4H, N(CH2)2), 3.38 (br, 4H, N(CH2)2), 3.98 (br, 2H, NH2), 6.72–6.76 (m, 2H, 3-H, 5-H), 6.94 (td, J = 7.6, 1.2 Hz, 1H, 4-H), 6.98 (dd, J = 8.2, 1.3 Hz, 1H, 6-H); 13C NMR (CDCl3, 100 MHz) δ = 38.50 (N(CH3)2), 47.49 (N(CH2)2), 50.95 (N(CH2)2), 115.20 (C-3), 118.58 (C-5), 119.93 (C-6), 124.88 (C-4), 138.86 (C-1), 141.47 (C-2), 164.82 (C=O); HRMS (ESI +) calcd for C13H21N4O [M+H]+: 249.1715; found: 249.1714.
4-(2-Aminophenyl)piperazine-1-carboxamide (27): Reaction of compound 33 (404 mg (1.61 mmol)) with PdC (62 mg) in dry methanol (90 mL) yielded compound 27 as pale brown solid (333 mg (94%)), which was used without further purification. IR = 3424, 1645, 1592, 1503, 1440, 1283, 993, 754; 1H NMR (CDCl3, 400 MHz) δ = 2.69–2.73 (m, 4H, N(CH2)2), 3.44 (br, 4H, N(CH2)2), 4.77 (s, 2H, NH2), 6.00 (s, 2H, (C=O)NH2), 6.53 (td, J = 7.5, 1.5 Hz, 1H, 5-H), 6.67 (td, J = 7.9, 1.5 Hz, 1H, 3-H), 6.80 (td, J = 7.6, 1.3 Hz, 1H, 4-H),6.87 (dd, J = 7.8, 1.4 Hz, 1H, 6-H); 13C NMR (CDCl3, 100 MHz) δ = 44.12 (N(CH2)2), 50.63 (N(CH2)2), 114.55 (C-3), 116.72 (C-5), 119.32 (C-6), 124.31 (C-4), 138.13 (C-1), 142.51 (C-2), 158.33 (C=O); HRMS (EI+) calcd for C11H17N4 [M+H]+: 221.1402; found: 221.1402.
2-(4-tert-Butylpiperazin-1-yl)aniline (28): Reaction of compound 35 (619 mg (2.35 mmol)) with PdC (112 mg) in dry methanol (90 mL) yielded compound 26 as pale brown solid (472 mg (86%)), which was used without further purification. IR = 3395, 2974, 2829, 1610, 1503, 1457, 1363, 1279, 1220, 1132, 963, 760, 739; 1H NMR (CDCl3, 400 MHz) δ = 1.12 (s, 9H, (CH3)3), 2.73 (br, 4H, N(CH2)2), 2.95 (br, 4H, N(CH2)2), 3.97 (br, 2H, NH2), 6.71–6.76 (m, 2H, 3-H, 5-H), 6.92 (td, J = 7.6, 1.5 Hz, 1H, 4-H), 7.02 (dd, J = 8.3, 1.4 Hz, 1H, 6-H); 13C NMR (CDCl3, 100 MHz) δ = 25.92 ((CH3)3), 46.44 (N(CH2)2), 51.68 (N(CH2)2), 53.74 (CMe3), 115.00 (C-3), 118.57 (C-5), 119.91 (C-6), 124.45 (C-4), 139.34 (C-1), 141.53 (C-2); HRMS (ESI +) calcd for C14H24N3 [M+H]+: 234.1970; found: 234.1972.
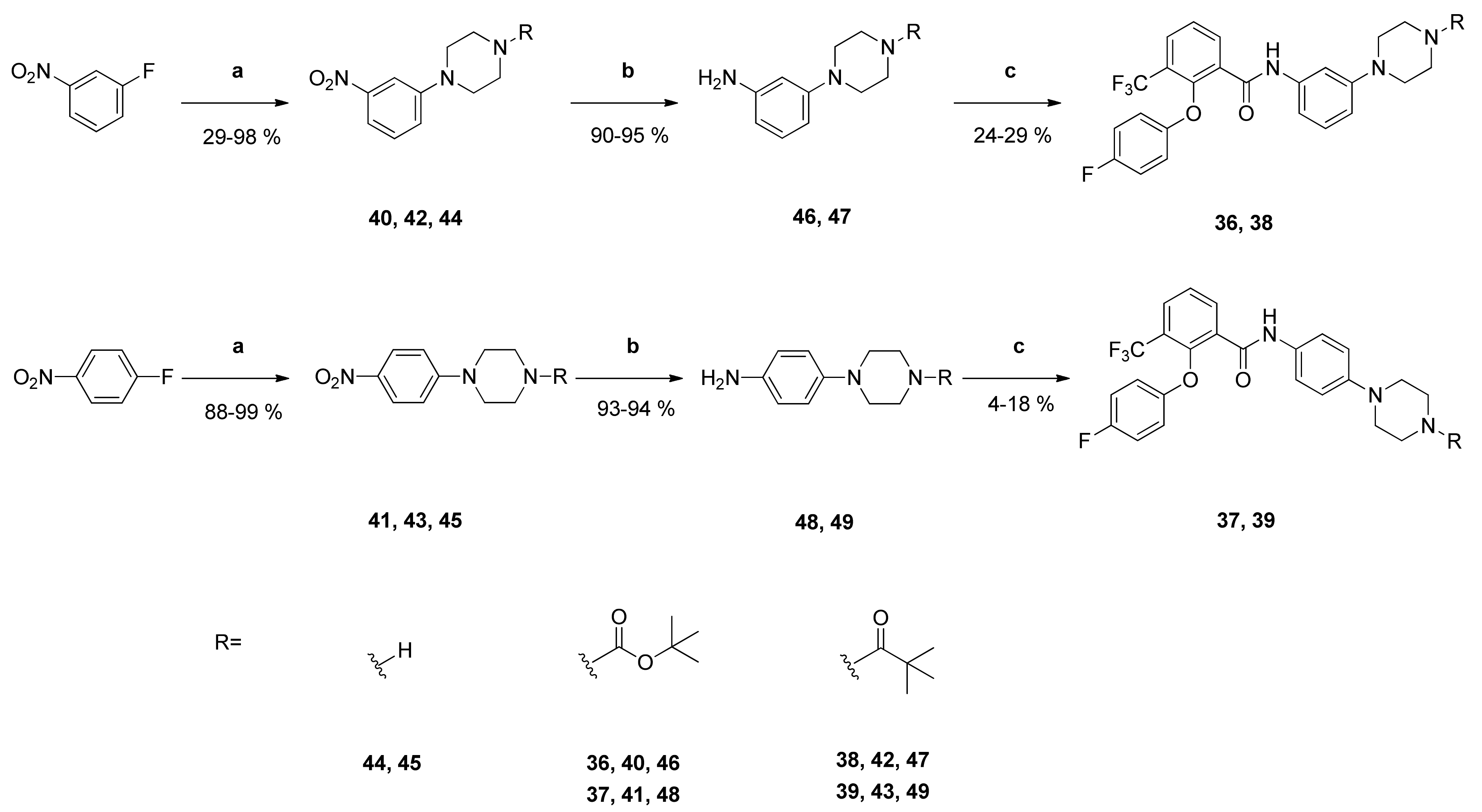
tert-Butyl-4-(3-aminophenyl)piperazine-1-carboxylate (
46): Reaction of compound
40 (809 mg (2.63 mmol)) with PdC (125 mg) in dry methanol (100 mL) yielded compound
46 as brown oil (657 mg (90%)), which was used without further purification. NMR data were in accordance with literature data [
41].
1-[4-(3-Aminophenyl)piperazin-1-yl]-2,2-dimethylpropan-1-one (47): Reaction of compound 42 (291 mg (1.00 mmol)) with PdC (60 mg) in dry methanol (80 mL) gave the raw anilino derivative. The residue was dissolved in ethyl acetate and extracted with 2N HCl. The aqueous phases were combined and basified with 2N NaOH to a pH of 14. The aqueous phase was extracted with ethyl acetate. The organic phase was washed with 8% aq NaHCO3, dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo yielding compound 47 as pale brown solid (248 mg (95%)). IR = 3471, 3338, 2972, 1614, 1503, 1426, 1364, 1283, 1210, 1193, 974, 841, 761, 689; 1H NMR (CDCl3, 400 MHz) δ = 1.31 (s, 9H, (CH3)3), 3.12–3.15 (m, 4H, N(CH2)2), 3.63 (br, 2H, NH2), 3.77–3.80 (m, 4H, N(CH2)2), 6.24–6.26 (m, 2H, 2-H, 2-H, 4-H), 6.35 (dd, J = 8.2, 2.0 Hz, 1H, 6-H), 7.06 (t, J = 8.2 Hz, 1H, 5-H); 13C NMR (CDCl3, 100 MHz) δ = 28.40 ((CH3)3), 38.63 (CMe3), 45.00 (N(CH2)2), 49.47 (N(CH2)2), 103.14 (C-2), 107.00 (C-6), 107.57 (C-4), 129.99 (C-5), 147.35 (C-3), 152.18 (C-1), 176.33 (C=O); HRMS (ESI +) calcd for C15H24N3O [M+H]+: 262.1919; found: 262.1920.
tert-Butyl-4-(4-aminophenyl)piperazine-1-carboxylate (
48): Reaction of compound
41 (1.98 g (6.45 mmol)) with PdC (299 mg) in dry methanol (100 mL) yielded compound
48 as dark-red oil (1.66 g (93%)), which was used without further purification. NMR data were in accordance with literature data [
41].
1-[4-(4-Aminophenyl)piperazin-1-yl]-2,2-dimethylpropan-1-one (49): Reaction of compound 43 (410 mg (1.41 mmol)) with PdC (69 mg) in dry methanol (100 mL) yielded compound 49 as dark-red oil (346 mg (94%)). IR = 3435, 2966, 1610, 1515, 1423, 1364, 1269, 1229, 1190, 1017, 831; 1H NMR (CDCl3, 400 MHz) δ = 1.31 (s, 9H, (CH3)3), 2.99–3.02 (m, 4H, N(CH2)2), 3.46 (br, 2H, NH2), 3.77–3.80 (m, 4H, N(CH2)2), 6.66 (d, J = 8.7 Hz, 2H, 3-H, 5-H), 6.80 (d, J = 8.7 Hz, 2H, 2-H, 6-H); 13C NMR (CDCl3, 100 MHz) δ = 28.43 ((CH3)3), 38.64 (CMe3), 45.23 ((NCH2)2), 51.34 ((NCH2)2), 116.14 (C-3, C-5), 118.96 (C-2, C-6), 140.68 (C-4), 144.04 (C-1), 176.30 (C=O); HRMS (ESI +) calcd for C15H24N3O [M+H]+: 262.1919; found: 262.1913.
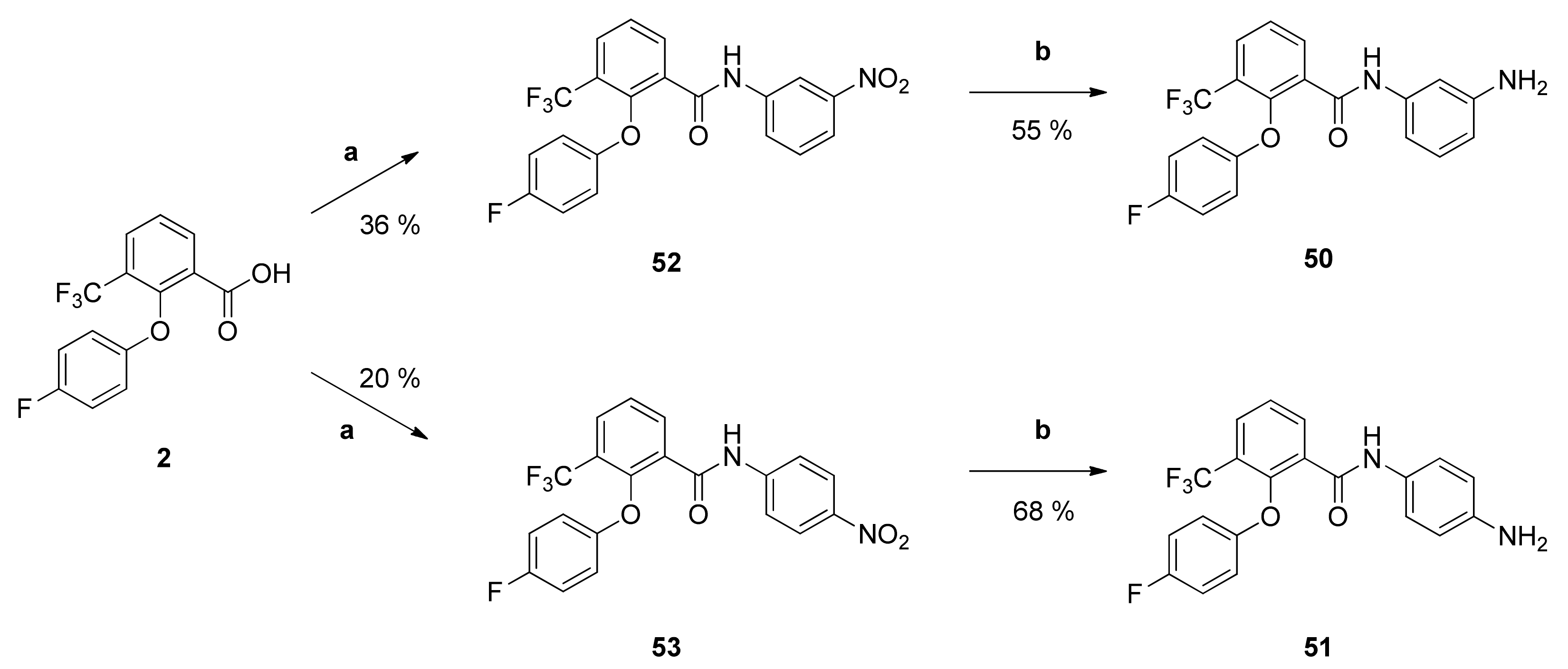
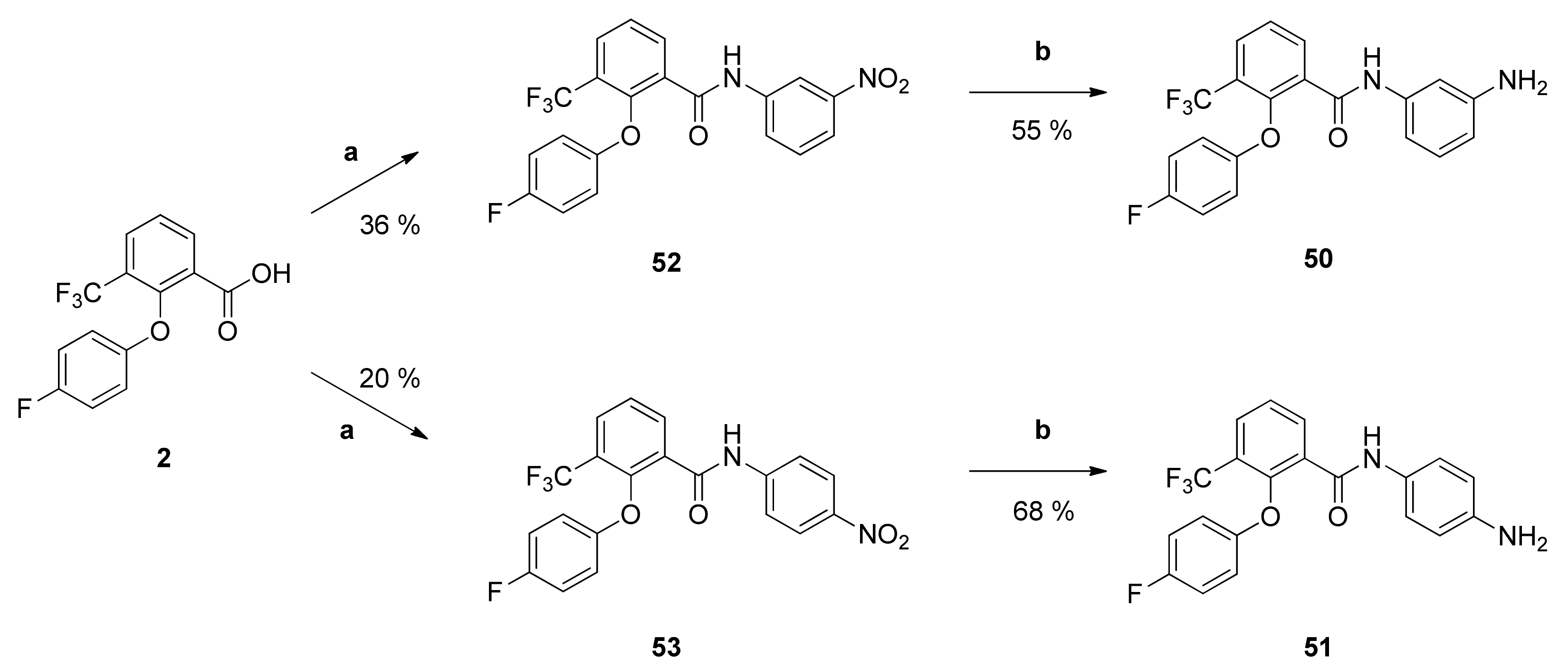
N-(3-Aminophenyl)-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (50): Reaction of compound 52 (90 mg (0.21 mmol)) with PdC (15 mg) in dry methanol (80 mL) yielded compound 50 as pale yellow solid (45 mg (55%)). IR = 3253, 1656, 1597, 1547, 1500, 1450, 1325, 1222, 1160, 776, 686; 1H NMR (CDCl3, 400 MHz) δ = 6.43 (dd, J = 8.1, 2.2 Hz, 1H, 4″-H), 6.54 (dd, J = 8.0, 1.9 Hz, 1H, 6″-H), 6.74–6.78 (m, 2H, 2′-H, 6′-H), 6.90–6.95 (m, 2H, 3′-H, 5′-H), 7.02–7.06 (m, 2H, 2″-H, 5″-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.27 (dd, J = 7.8, 1.7 Hz, 1H, 6-H), 8.39 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 106.82 (C-2″), 110.02 (C-6″), 111.69 (C-4″), 116.20 (d, J = 8.3 Hz, C-2′, C-6′), 116.53 (d, J = 23.8 Hz, C-3′, C-5′), 122.67 (q, J = 273 Hz, CF3), 125.22 (q, J = 31.7 Hz, C-3), 126.28 (C-5), 129.71 (C-5″), 130.59 (q, J = 4.8 Hz, C-4), 130.97 (C-1), 135.89 (C-6), 138.18 (C-1″), 147.19 (C-3″), 149.43 (q, J = 1.8 Hz, C-2), 154.01 (d, J = 2.5 Hz, C-1′), 158.50 (d, J = 242 Hz, C-4′), 161.52 (C=O); HRMS (ESI +) calcd for C20H15F4N2O2 [M+H]+: 391.1070; found: 391.1061.
N-(4-Aminophenyl)-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (51): Reaction of compound 53 (58 mg (0.14 mmol)) with PdC (10 mg) in dry methanol (80 mL) gave the raw anilino derivative. It was purified by column chromatography (silica gel, CH/EtAc 1:1) yielding compound 51 as pale yellow solid (37 mg (68%)). IR = 3362, 1654, 1517, 1500, 1449, 1315, 1217, 1167, 1135, 1097, 828, 779, 685; 1H NMR (CDCl3, 400 MHz) δ = 3.61 (br s, 2H, NH2), 6.59 (d, J = 8.6 Hz, 2H, 3″-H, 5″-H), 6.75–6.79 (m, 2H, 2′-H, 6′-H), 6.91–6.96 (m, 2H, 3′-H, 5′-H), 7.09 (d, J = 8.6 Hz, 2H, 2″-H, 6″-H), 7.52 (t, J = 7.8 Hz, 1H, 5-H), 7.88 (dd, J = 7.8, 1.7 Hz, 1H, 4-H), 8.26–8.29 (m, 1H, 6-H, NH); 13C NMR (CDCl3, 100 MHz) δ = 115.27 (C-3″, C-5″), 116.19 (d, J = 8.2 Hz, C-2′, C-6′), 116.51 (d, J = 23.7 Hz, C-3′, C-5′), 122.35 (C-2″, C-6″), 122.71 (q, J = 273 Hz, CF3), 125.13 (q, J = 31.8 Hz, C-3), 126.22 (C-5), 128.31 (C-1″), 130.38 (q, J = 4.9 Hz, C-4), 131.02 (C-1), 135.88 (C-6), 143.91 (C-4″), 149.39 (q, J = 1.8 Hz, C-2), 154.03 (d, J = 2.5 Hz, C-1′), 158.47 (d, J = 242 Hz, C-4′), 161.41 (C=O); HRMS (ESI +) calcd for C20H15F4N2O2 [M+H]+: 391.1070; found: 391.1062.
3.2.6. General Procedure for the Synthesis of Compounds 1, 6–8, 12, 13, 17–22, 36–39 and 52–56
Carboxylic acid (1.00 mmol) and anilino derivative (1.00 mmol) were dissolved in dry CH2Cl2 and cooled to 0 °C in an ice-bath. 2-Chloro-N-methylpyridinium iodide and diisopropylethylamine were added whereupon the ice-bath was removed. The reaction mixture was stirred at room temperature for 24–48 h. Reaction progress was monitored by TLC. Afterwards, 20% aq NH4Cl (50 mL) was added. The aqueous and organic phases were separated, and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were washed with 8% aq NaHCO3 and brine, dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo giving the raw carboxamide that was purified by recrystallization or column chromatography.
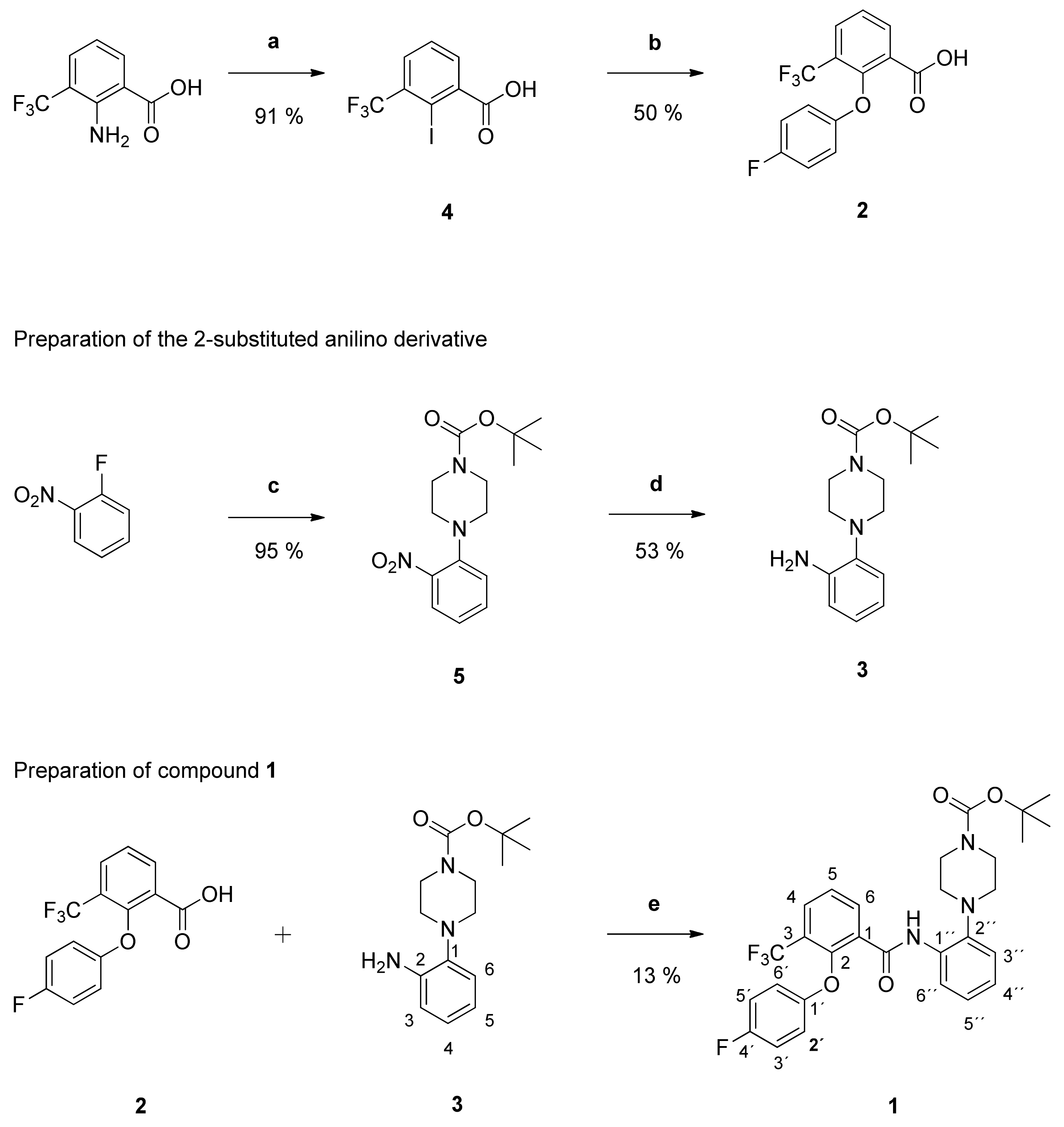
tert-Butyl-4-{2-[2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (1): Reaction of the carboxylic acid 2 (210 mg (0.70 mmol)) with the amine 3 (194 mg (0.70 mmol)), 2-chloro-N-methylpyridinium iodide (316 mg (1.24 mmol)) and DIPEA (452 mg (3.50 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/MeOH 99:1) yielded compound 1 as pale-yellow solid (51 mg (13%)). IR = 3440, 1690, 1539, 1523, 1500, 1450, 1366, 1320, 1216, 1166, 1135, 837, 777, 689; 1H NMR (CDCl3, 400 MHz) δ = 1.50 (s, 9H, (CH3)3), 2.81 (t, J = 4.8 Hz, 4H, N(CH2)2), 3.62 (br s, 4H, N(CH2)2), 6.68–6.72 (m, 2H, 2′-H, 6′-H), 6.84–6.89 (m, 2H, 3′-H, 5′-H), 7.04–7.14 (m, 3H, 3″-H, 4″-H, 5″-H), 7.53 (t, J = 7.6 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.6 Hz, 1H, 4-H), 8.21 (dd, J = 7.6, 1.6 Hz, 1H, 6-H), 8.31 (dd, J = 8.3, 1.6 Hz, 1H, 6″-H), 9.69 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.39 ((CH3)3), 44.12 (N(CH2)2), 52.26 (N(CH2)2), 80.13 (CMe3), 116.28 (d, J = 23.6 Hz, C-3′, C-5′), 116.44 (d, J = 7.6 Hz, C-2′, C- 6′), 119.68 (C-6″), 120.53 (C-3″), 122.67 (q, J = 274 Hz, CF3), 124.41 (C-4″), 125.31 (q, J = 31.8 Hz, C-3), 125.80 (C-5″), 126.18 (C-5), 130.42 (q, J = 4.9 Hz, C-4), 131.94 (C-1), 133.11 (C-1″), 135.28 (C-6), 141.07 (C-2″), 149.77 (q, J = 1.8 Hz, C-2), 154.14 (d, J = 2.6 Hz, C-1′), 154.65 (C=O), 158.35 (d, J = 242 Hz, C-4′), 161.74 ((C=O)NH); HRMS (EI+) calcd for C29H29F4N3O4 [M+]: 559.2094; found: 559.2094.
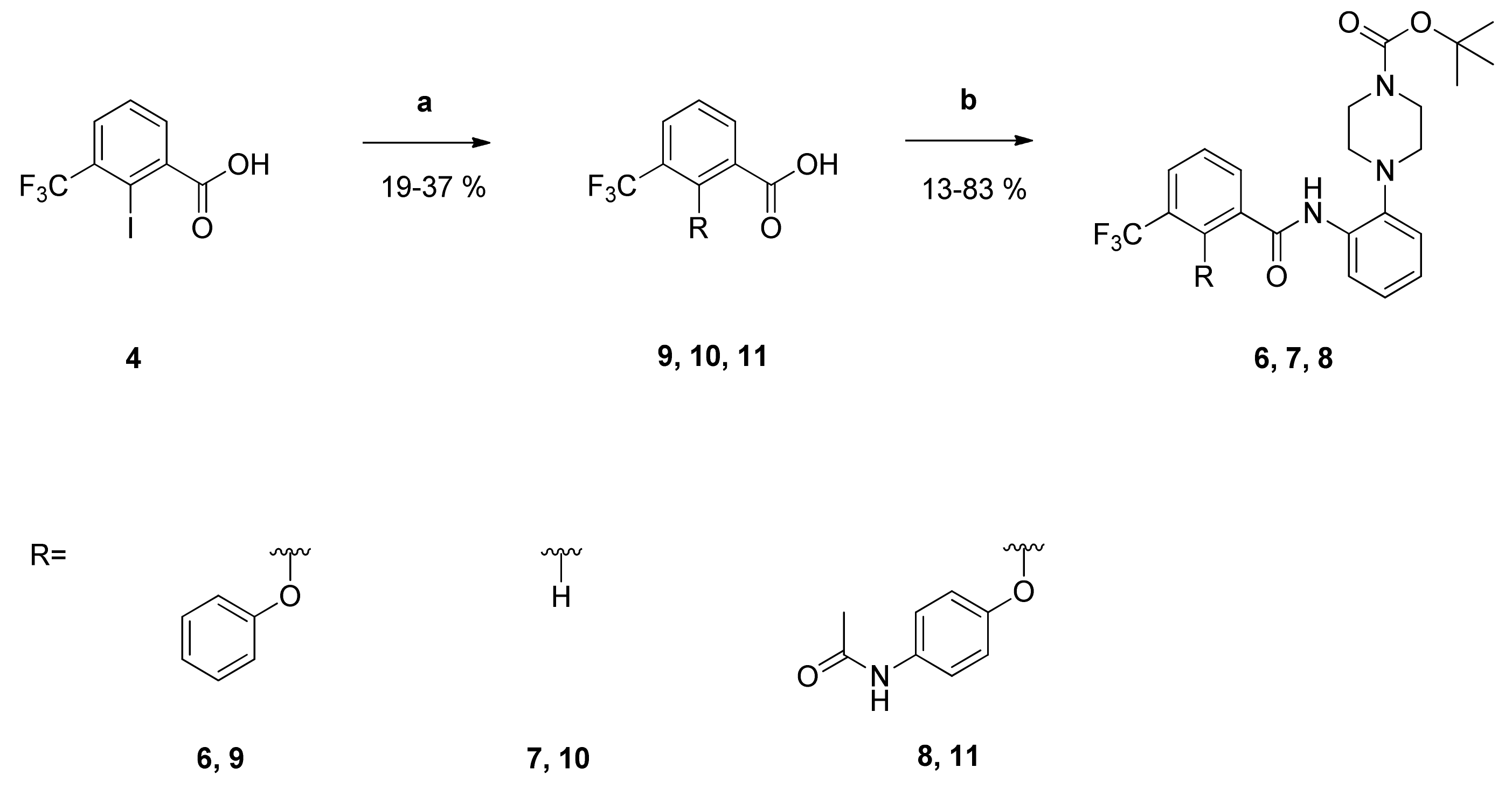
tert-Butyl-4-{2-[2-phenoxy-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (6): Reaction of the carboxylic acid 9 (414 mg (1.47 mmol)) with the amine 3 (411 mg (1.48 mmol)), 2-chloro-N-methylpyridinium iodide (657 mg (2.57 mmol)) and DIPEA (949 mg (7.34 mmol)) in dry CH2Cl2 (32 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/EtOH 79:1) yielded compound 6 as white solid (414 mg (52%)). IR = 3330, 2976, 1687, 1592, 1522, 1449, 1356, 1321, 1229, 1136, 911, 871, 752, 690; 1H NMR (CDCl3, 400 MHz) δ = 1.50 (s, 9H, (CH3)3), 2.81 (t, J = 4.9 Hz, 4H, N(CH2)2), 3.64 (br t, J = 5.0 Hz, 4H, N(CH2)2), 6.74 (br d, J = 7.9 Hz, 2H, 2′-H, 6′-H), 6.95 (br t, J = 7.4 Hz, 1H, 4′-H), 7.02–7.12 (m, 3H, 3″-H, 4″-H, 5″-H), 7.14–7.20 (m, 2H, 3′-H, 5′-H), 7.53 (br t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 8.0, 1.7 Hz, 1H, 4-H), 8.24 (dd, J = 7.8, 1.7 Hz, 1H, 6-H), 8.29 (dd, J = 8.1, 1.8 Hz, 1H, 6″-H), 9.76 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.42 ((CH3)3), 44.07 (N(CH2)2), 52.26 (N(CH2)2), 80.07 (CMe3), 115.22 (C-2′, C-6′), 119.77 (C-6″), 120.48 (C-3″), 122.72 (q, J = 273 Hz, CF3), 123.13 (C-4′), 124.27 (C-4″), 125.41 (q, J = 31.7 Hz, C-3), 125.69 (C-5″), 126.02 (C-5), 129.72 (C-3′, C-5′), 130.41 (q, J = 4.9 Hz, C-4), 131.98 (C-1), 133.23 (C-1″), 135.29 (C-6), 141.16 (C-2″), 149.68 (q, J = 2.0 Hz, C-2), 154.66 (COO), 158.16 (C-1′), 161.80 (C=O); HRMS (EI+) calcd for C29H30F3N3O4 [M+]: 551.2110; found: 542.2278 [M+H]+.
tert-Butyl-4-{2-[3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (7): Reaction of the carboxylic acid 10 (323 mg (1.70 mmol)) with the amine 3 (287 mg (1.04 mmol)), 2-chloro-N-methylpyridinium iodide (463 mg (1.81 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (44 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/MeOH 79:1) yielded compound 7 as white solid (99 mg (13%)). IR = 3324, 2854, 1687, 1591, 1521, 1456, 1395, 1368, 1247, 1166, 1125, 1072, 911, 773, 695; 1H NMR (CDCl3, 400 MHz) δ = 1.50 (s, 3H, (CH3)3), 2.88 (t, J = 5.0 Hz, 4H, N(CH2)2), 3.61 (br, 4H, N(CH2)2), 7.14 (ddd, J = 8.0, 7.3, 1.5 Hz, 1H, 4″-H), 7.23 (ddd, J = 9.6, 7.3, 1.1 Hz, 1H, 3″-H), 7.25–7.28 (m, 1H, 5″-H), 7.68 (br t, J = 7.7 Hz, 1H, 5-H), 7.83 (br d, J = 7.8 Hz, 1H, 4-H), 8.10 (br d, J = 7.8 Hz, 1H, 6-H), 8.20 (br s, 1H, 2-H), 8.56 (dd, J = 8.1, 1.4 Hz, 1H, 6″-H), 9.58 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.39 ((CH3)3), 44.53 (N(CH2)2), 52.35 (N(CH2)2), 80.22 (CMe3), 119.45 (C-6″), 123.64 (q, J = 273 Hz, CF3), 120.92 (C-3″), 123.98 (q, J = 3.8 Hz, C-2), 124.33 (C-4″), 126.27 (C-5″), 128.38 (q, J = 3.6 Hz, C-4), 129.61 (C-5), 129.95 (C-6), 131.49 (q, J = 32.7 Hz, C-3), 133.33 (C-1″), 135.89 (C-1), 141.09 (C-2″), 154.61 (COO), 163.10 (C=O); HRMS (ESI +) calcd for C23H27F3N3O3 [M+H]+: 450.2005; found: 450.2008.
tert-Butyl-4-{2-[2-(4-acetamidophenoxy)-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (8): Reaction of the carboxylic acid 11 (708 mg (2.09 mmol)) with the amine 3 (579 mg (2.09 mmol)), 2-chloro-N-methylpyridinium iodide (933 mg (3.65 mmol)) and DIPEA (1.35 g (10.44 mmol)) in dry CH2Cl2 (100 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/MeOH 29:1) yielded compound 8 as white solid (1.04 g (83%)). IR = 3333, 1673, 1603, 1505, 1449, 1367, 1324, 1233, 1163, 760; 1H NMR (DMSO-d6, 400 MHz) δ = 1.43 (s, 9H, (CH3)3), 1.96 (s, 3H, CH3), 2.76 (t, J = 4.9 Hz, 4H, N(CH2)2), 3.50 (t, J = 4.9 Hz, 4H, N(CH2)2), 6.70 (d, J = 9.0 Hz, 2H, 2′-H, 6′-H), 7.01 (td, J = 7.7, 1.5 Hz, 1H, 5″-H), 7.06 (td, J = 7.6, 1.6 Hz, 1H, 4″-H), 7.19 (br d, J = 7.8 Hz, 1H, 3″-H), 7.41 (d, J = 9.0 Hz, 2H, 3′-H, 5′-H), 7.64 (t, J = 7.8 Hz, 1H, 5-H), 7.68 (br d, J = 8.0 Hz, 1H, 6″-H), 8.00–8.06 (m, 2H, 4-H, 6-H), 9.62 (s, 1H, NH), 9.81 (s, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.74 (CH3), 28.03 ((CH3)3), 43.65 (N(CH2)2), 51.39 (N(CH2)2), 79.00 (CMe3), 115.46 (C-2′, C-6′), 120.31 (C-3′, C-5′), 120.67 (C-3″), 120.95 (C-6″), 123.02 (q, J = 273 Hz, CF3), 123.39 (q, J = 30.9 Hz, C-3), 124.38 (C-5″), 124.78 (C-4″), 126.17 (C-5), 129.61 (q, J = 4.2 Hz, C-4), 132.08 (C-1), 132.22 (C-1″), 134.41 (C-4′), 134.64 (C-6), 142.77 (C-2″), 149.42 (q, J = 1.7 Hz, C-2), 153.48 (C-1′), 153.89 (COO), 162.07 (ArC=O), 167.85 (CH3C=O); HRMS (ESI +) calcd for C31H34F3N4O5 [M+H]+: 599.2481; found: 599.2487.
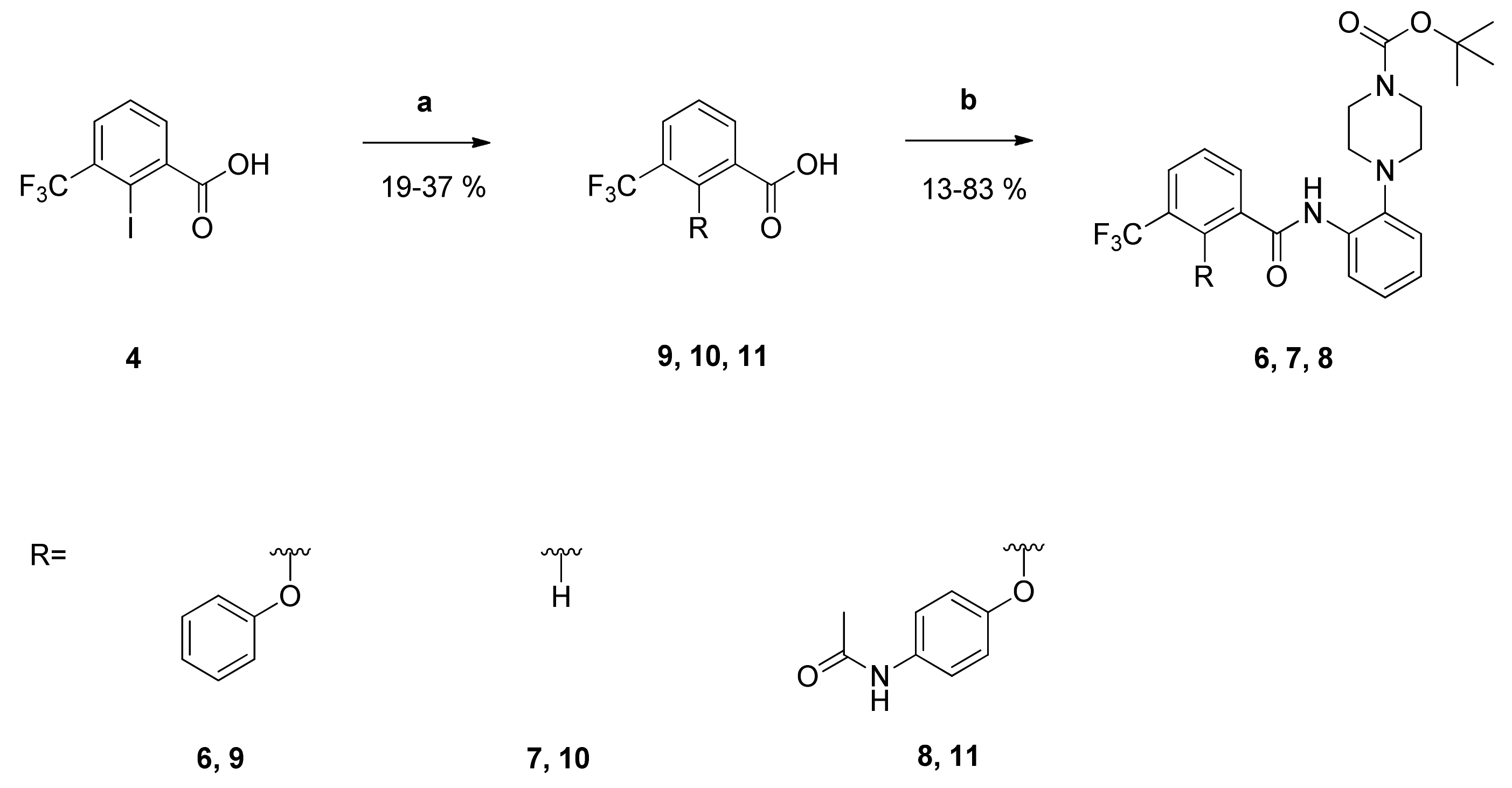
2-(4-Fluorophenoxy)-N-phenyl-3-(trifluoromethyl)benzamide (12): Reaction of the carboxylic acid 2 (303 mg (1.01 mmol)) with aniline (93 mg (1.00 mmol)), 2-chloro-N-methylpyridinium iodide (464 mg (1.82 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 3:1) yielded compound 12 as white solid (68 mg (18%)). IR = 3317, 1657, 1579, 1529, 1502, 1444, 1339, 1313, 1249, 1217, 1166, 1132, 821, 779, 755, 687; 1H NMR (CDCl3, 400 MHz) δ = 6.75–6.79 (m, 2H, 2′-H, 6′-H), 6.91–6.95 (m, 2H, 3′-H, 5′-H), 7.11 (t, J = 7.4 Hz, 1H, 4″-H), 7.29 (t, J = 7.9 Hz, 2H, 3″-H, 5″-H), 7.38 (d, J = 7.6 Hz, 2H, 2″-H, 6″-H), 7.54 (t, J = 7.8 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.30 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.49 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 116.15 (d, J = 8.4 Hz, C-2′, C-6′), 116.57 (d, J = 23.8 Hz, C-3′, C- 5′), 120.22 (C-2″, C-6″), 122.66 (q, J = 273 Hz, CF3), 125.01 (C-4″), 125.24 (q, J = 32.2 Hz, C-3), 126.33 (C-5), 129.04 (C-3″, C-5″), 130.70 (q, J = 4.6 Hz, C-4), 135.95 (C-6), 137.12 (C-1″), 149.46 (q, J = 1.6 Hz, C-2), 154.00 (d, J = 2.1 Hz, C-1′), 158.51 (d, J = 242 Hz, C-4′), 161.61 (C=O); HRMS (ESI +) calcd for C20H13F4NO2 [M+]: 375.0882; found: 375.0894.
tert-Butyl-N-{2-[2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamido]phenyl}carbamate (13): Reaction of the carboxylic acid 2 (311 mg (1.04 mmol)) with the amine 15 (217 mg (1.04 mmol)), 2-chloro-N-methylpyridinium iodide (490 mg (1.92 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 3:1) yielded compound 13 as dark-red solid (46 mg (9%)). IR = 3276, 1730, 1639, 1604, 1503, 1455, 1314, 1223, 1164, 842, 754; 1H NMR (CDCl3, 400 MHz) δ = 1.50 (s, 9H, (CH3)3), 6.50 (br s, 1H, NH), 6.80–6.84 (m, 2H, 2′-H, 6′-H), 6.92–6.97 (m, 2H, 3′-H, 5′-H), 7.10 (td, J = 7.4, 1.6 Hz, 1H, 5″-H), 7.15 (td, J = 7.6, 1.7 Hz, 1H, 4″-H), 7.24–7.27 (m, 1H, 6″-H), 7.34 (dd, J = 7.8, 1.6 Hz, 1H, 3″-H), 7.51 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.8, 1.7 Hz, 1H, 4-H), 8.23 (dd, J = 7.8, 1.7 Hz, 1H, 6-H), 9.02 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.19 ((CH3)3), 81.41 (CMe3), 116.39 (d, J = 23.8 Hz, C-3′, C-5′), 116.47 (d, J = 8.1 Hz, C-2′, C-6′), 122.66 (q, J = 273 Hz, CF3), 124.54 (C-3″), 124.97 (C-6″), 125.27 (q, J = 31.8 Hz, C-3), 125.77 (C-5″), 126.02 (C-5), 126.49 (C-4″), 129.83 (C-1″), 130.27 (C-2″), 130.60 (q, J = 4.9 Hz, C-4), 130.89 (C-1), 135.66 (C-6), 149.83 (q, J = 1.9 Hz, C-2), 153.95 (N(C=O)O), 154.20 (d, J = 2.4 Hz, C-1′), 158.40 (d, J = 242 Hz, C-4′), 162.67 (C=O); HRMS (ESI -) calcd for C25H20F4N2O4 [M-H]−: 489.1437; found: 489.1442
N-[2-(4-Acetylpiperazin-1-yl)phenyl]-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (17): Reaction of the carboxylic acid 2 (623 mg (2.08 mmol)) with the amine 23 (462 mg (2.11 mmol)), 2-chloro-N-methylpyridinium iodide (928 mg (3.63 mmol)) and DIPEA (1.29 g (10.00 mmol)) in dry CH2Cl2 (90 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/MeOH 39:1) yielded compound 17 as colorless oil (31 mg (3%)). IR = 3419, 1653, 1591, 1520, 1501, 1448, 1324, 1219, 1137, 999, 834, 780; 1H NMR (CDCl3, 400 MHz) δ = 2.16 (s, 3H, CH3), 2.82–2.88 (m, 4H, 2 NCH2), 3.63–3.67 (m, 2H, NCH2), 3.82 (br, 2H, NCH2), 6.68–6.72 (m, 2H, 2′-H, 6′-H), 6.85–6.89 (m, 2H, 3′-H, 5′-H), 7.05–7.19 (m, 3H, 3″-H, 4″-H, 5″-H), 7.54 (t, J = 7.9 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.23 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.32 (dd, J = 8.0, 1.4 Hz, 1H, 6″-H), 9.70 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 21.36 (CH3), 41.94 (NCH2), 46.83 (NCH2), 52.18 (NCH2), 52.55 (NCH2), 116.37 (d, J = 23.7 Hz, C-3′, C-5′), 116.41 (d, J = 8.3 Hz, C-2′, C-6′), 119.81 (C-6″), 120.57 (C-3″), 122.66 (q, J = 273 Hz, CF3), 124.48 (C-4″), 125.33 (q, J = 31.8 Hz, C-3), 126.09 (C-5″), 126.28 (C-5), 130.50 (q, J = 4.8 Hz, C-4), 131.98 (C-1), 131.98 (C-1″), 135.35 (C-6), 140.59 (C-2″), 149.72 (q, J = 1.8 Hz, C-2), 154.10 (d, J = 2.3 Hz, C-1′), 158.40 (d, J = 242 Hz, C-4′), 161.73 ((C=O)NH), 169.07 (MeC=O); HRMS (ESI +) calcd for C26H24F4N3O3 [M+H]+: 502.1754; found: 502.1774.
2-(4-Fluorophenoxy)-N-[2-(4-formylpiperazin-1-yl)phenyl]-3-(trifluoromethyl)benzamide (18): Reaction of the carboxylic acid 2 (312 mg (1.04 mmol)) with the amine 24 (222 mg (1.08 mmol)), 2-chloro-N-methylpyridinium iodide (451 mg (1.77 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (40 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, EtAc/CH 3:1) yielded compound 18 as white solid (314 mg (62%)). IR = 3441, 1668, 1521, 1500, 1447, 1322, 1218, 1010, 780; 1H NMR (CDCl3, 400 MHz) δ = 2.83–2.86 (m, 2H, NCH2), 2.87–2.90 (m, 2H, NCH2), 3.56–3.59 (m, 2H, NCH2), 3.76 (br, 2H, NCH2), 6.68–6.72 (m, 2H, 2′-H, 6′-H), 6.86–6.90 (m, 2H, 3′-H, 5′-H), 7.05–7.19 (m, 3H, 3″-H, 4″-H, 5″-H), 7.55 (t, J = 7.9 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.12 (s, 1H, HC=O), 8.24 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.33 (dd, J = 8.1, 1.3 Hz, 1H, 6″-H), 9.67 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 40.43 (NCH2), 46.07 (NCH2), 51.87 (NCH2), 52.98 (NCH2), 116.39 (d, J = 23.6 Hz, C-3′, C-5′), 116.41 (d, J = 8.1 Hz, C-2′, C-6′), 119.88 (C-6″), 120.60 (C-3″), 122.64 (q, J = 273 Hz, CF3), 124.52 (C-4″), 125.32 (q, J = 31.7 Hz, C-3), 126.21 (C-5″), 126.32 (C-5), 130.53 (q, J = 4.8 Hz, C-4), 131.99 (C-1), 133.05 (C-1″), 135.34 (C-6), 140.47 (C-2″), 149.69 (q, J = 1.8 Hz, C-2), 154.07 (d, J = 2.5 Hz, C-1′), 158.41 (d, J = 242 Hz, C-4′), 160.81 (H(C=O)NR2), 161.75 ((C=O)NH); HRMS (ESI +) calcd for C25H22F4N3O3 [M+H]+: 488.1597; found: 488.1587.
N-{2-[4-(2,2-Dimethylpropanoyl)piperazin-1-yl]phenyl}-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (19): Reaction of the carboxylic acid 2 (299 mg (0.99 mmol)) with the amine 25 (221 mg (0.85 mmol)), 2-chloro-N-methylpyridinium iodide (454 mg (1.78 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (40 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 3:1) yielded compound 19 as white solid (194 mg (42%)). IR = 3441, 1631, 1520, 1501, 1449, 1325, 1219, 1139, 1016, 781; 1H NMR (CDCl3, 400 MHz) δ = 1.33 (s, 9H, (CH3)3), 2.83–2.86 (m, 4H, N(CH2)2), 3.85 (br, 4H, N(CH2)2), 6.68–6.74 (m, 2H, 2′-H, 6′-H), 6.84–6.90 (m, 2H, 3′-H, 5′-H), 7.07 (td, J = 7.5, 1.6 Hz, 1H, 4″-H), 7.11–7.16 (m, 2H, 3″-H, 5″-H), 7.54 (td, J = 7.9, 0.9 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.24 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.32 (dd, J = 8.3, 1.6 Hz, 1H, 6″-H), 9.74 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.40 ((CH3)3), 38.71 (CMe3), 45.61 (N(CH2)2), 52.50 (N(CH2)2), 116.35 (d, J = 23.7 Hz, C-3′, C-5′), 116.45 (d, J = 8.3 Hz, C-2′, C-6′), 119.77 (C-6″), 120.51 (C-3″), 122.67 (q, J = 273 Hz, CF3), 124.45 (C-4″), 125.34 (q, J = 31.7 Hz, C-3), 125.95 (C-5″), 126.25 (C-5), 130.47 (q, J = 4.9 Hz, C-4), 131.91 (C-1), 133.14 (C-1″), 135.37 (C-6), 140.63 (C-2″), 149.78 (q, J = 1.9 Hz, C-2), 154.13 (d, J = 2.5 Hz, C-1′), 158.38 (d, J = 242 Hz, C-4′), 161.67 ((C=O)NH), 176.52 (C=O); HRMS (ESI +) calcd for C29H30F4N3O3 [M+H]+: 544.2223; found: 544.2214.
2-(4-Fluorophenoxy)-N-[2-(4-tert-butylpiperazin-1-yl)phenyl]-3-(trifluoromethyl)benzamide (20): Reaction of the carboxylic acid 2 (304 mg (1.01 mmol)) with the amine 26 (220 mg (0.94 mmol)), 2-chloro-N-methylpyridinium iodide (453 mg (1.77 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (40 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, EtAc) was followed by recrystallization from CH yielding compound 20 as white solid (99 mg (20%)). m.P. 142–145 °C; IR = 3447, 2974, 1672, 1588, 1501, 1447, 1323, 1214, 1165, 1129, 782; 1H NMR (CDCl3, 400 MHz) δ = 1.14 (s, 9H, (CH3)3), 2.82 (br, 4H, N(CH2)2), 2.90–2.93 (m, 4H, N(CH2)2), 6.69–6.73 (m, 2H, 2′-H, 6′-H), 6.83–6.87 (m, 2H, 3′-H, 5′-H), 7.03–7.12 (m, 2H, 4″-H, 5″-H), 7.18 (dd, J = 7.6, 1.8 Hz, 3″-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.8, 1.7 Hz, 1H, 4-H), 8.26 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.33 (dd, J = 7.8, 1.7 Hz, 1H, 6″-H), 9.96 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 25.80 ((CH3)3), 46.32 (N(CH2)2), 53.03 (N(CH2)2), 53.84 (CMe3), 116.25 (d, J = 23.7 Hz, C-3′, C-5′), 116.33 (d, J = 8.0 Hz, C-2′, C-6′), 119.24 (C-6″), 120.56 (C-3″), 122.76 (q, J = 273 Hz, CF3), 124.25 (C-4″), 125.34 (q, J = 31.7 Hz, C-3), 125.50 (C-5″), 126.11 (C-5), 130.28 (q, J = 4.9 Hz, C-4), 132.00 (C-1), 133.36 (C-1″), 135.57 (C-6), 141.39 (C-2″), 149.78 (q, J = 1.9 Hz, C-2), 154.30 (d, J = 2.5 Hz, C-1′), 158.30 (d, J = 242 Hz, C-4′), 161.48 ((C=O)NH); HRMS (EI+) calcd for C28H30F4N3O2 [M+H]+: 516.2274; found: 516.2266.
N-[2-(4-Carbamoylpiperazin-1-yl)phenyl]-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (21): Reaction of the carboxylic acid 2 (297 mg (0.99 mmol)) with the amine 27 (220 mg (1.00 mmol)), 2-chloro-N-methylpyridinium iodide (453 mg (1.77 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (40 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, EtAc) yielded compound 21 as white solid (239 mg (48%)). IR = 3355, 1656, 1592, 1500, 1448, 1325, 1219, 1139, 988, 831, 780; 1H NMR (CDCl3, 400 MHz) δ = 2.85–2.88 (m, 4H, N(CH2)2), 3.59–3.61 (m, 4H, N(CH2)2), 4.60 (s, 2H, NH2), 6.68–6.72 (m, 2H, 2′-H, 6′-H), 6.84–6.89 (m, 2H, 3′-H, 5′-H), 7.05–7.16 (m, 3H, 3″-H, 4″-H, 5″-H), 7.54 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.6 Hz, 1H, 4-H), 8.22 (dd, J = 7.8, 1.6 Hz, 1H, 6-H), 8.30 (dd, J = 7.9, 2.0 Hz, 1H, 6″-H), 9.67 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 44.63 (N(CH2)2), 52.09 (N(CH2)2), 116.34 (d, J = 23.8 Hz, C-3′, C-5′), 116.45 (d, J = 8.3 Hz, C-2′, C-6′), 119.80 (C-6″), 120.55 (C-3″), 122.67 (q, J = 273 Hz, CF3), 124.49 (C-4″), 125.34 (q, J = 31.7 Hz, C-3), 126.01 (C-5″), 126.24 (C-5), 130.46 (q, J = 4.9 Hz, C-4), 131.99 (C-1), 133.12 (C-1″), 135.29 (C-6), 140.74 (C-2″), 149.75 (q, J = 2.2 Hz, C-2), 154.12 (d, J = 2.5 Hz, C-1′), 157.80 ((C=O)NH2), 158.39 (d, J = 242 Hz, C-4′), 161.75 ((C=O)NH); HRMS (ESI +) calcd for C25H23F4N4O3 [M+H]+: 503.1706; found: 503.1700.
N-{2-[4-(N,N-Dimethylcarbamoyl)piperazin-1-yl]phenyl}-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (22): Reaction of the carboxylic acid 2 (339 mg (1.13 mmol)) with the amine 28 (279 mg (1.12 mmol)), 2-chloro-N-methylpyridinium iodide (509 mg (1.99 mmol)) and DIPEA (724 mg (5.60 mmol)) in dry CH2Cl2 (50 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, EtAc/CH 2:1) yielded compound 22 as yellow oil (244 mg (41%)). IR = 3333, 2848, 1649, 1591, 1500, 1448, 1394, 1328, 1218, 1138, 828, 781; 1H NMR (CDCl3, 400 MHz) δ = 2.84–2.87 (m, 4H, N(CH2)2), 2.87 (s, 6H, N(CH3)2), 3.42–3.46 (m, 4H, N(CH2)2), 4.60 (s, 2H, NH2), 6.69–6.73 (m, 2H, 2′-H, 6′-H), 6.83–6.88 (m, 2H, 3′-H, 5′-H), 7.04–7.18 (m, 3H, 3″-H, 4″-H, 5″-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.20 (dd, J = 7.8, 1.7 Hz, 1H, 6-H), 8.28 (dd, J = 7.9, 1.6 Hz, 1H, 6″-H), 9.66 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 38.52 (N(CH3)2), 47.30 (N(CH2)2), 52.23 (N(CH2)2), 116.28 (d, J = 23.7 Hz, C-3′, C-5′), 116.53 (d, J = 8.2 Hz, C-2′, C-6′), 119.70 (C-6″), 120.65 (C-3″), 122.70 (q, J = 273 Hz, CF3), 124.44 (C-4″), 125.38 (q, J = 31.7 Hz, C-3), 125.80 (C-5″), 126.16 (C-5), 130.39 (q, J = 4.8 Hz, C-4), 132.01 (C-1), 133.16 (C-1″), 135.18 (C-6), 141.07 (C-2″), 149.87 (q, J = 1.9 Hz, C-2), 154.20 (d, J = 2.5 Hz, C-1′), 158.36 (d, J = 242 Hz, C-4′), 161.81 ((C=O)NH), 164.52 ((C=O)NR2); HRMS (ESI +) calcd for C27H27F4N4O3 [M+H]+: 531.2019; found: 531.2026
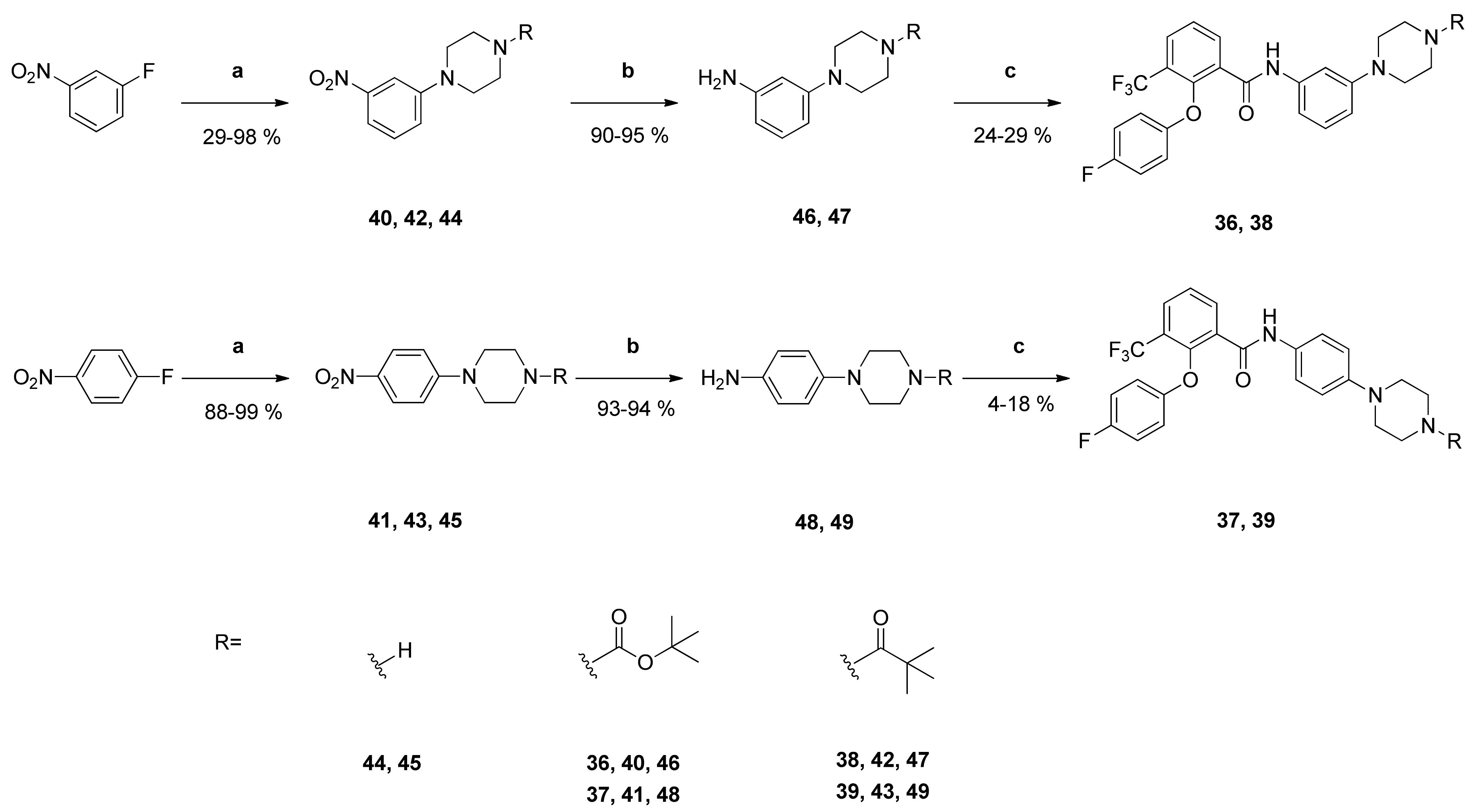
tert-Butyl-4-{3-[2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (36): Reaction of the carboxylic acid 2 (329 mg (1.10 mmol)) with the amine 46 (302 mg (1.09 mmol)), 2-chloro-N-methylpyridinium iodide (486 mg (1.90 mmol)) and DIPEA (698 mg (5.40 mmol)) in dry CH2Cl2 (35 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 2:1) yielded compound 36 as pale-yellow solid (177 mg (29%)). IR = 3422, 1691, 1609, 1501, 1450, 1332, 1248, 1221, 1166, 998, 777, 688; 1H NMR (CDCl3, 400 MHz) δ = 1.48 (s, 9H, (CH3)3), 3.09–3.12 (m, 4H, N(CH2)2), 3.54–3.57 (m, 4H, N(CH2)2), 6.65–6.70 (m, 2H, 4″-H, 6″-H), 6.75–6.79 (m, 2H, 2′-H, 6′-H), 6.91–6.96 (m, 2H, 3′-H, 5′-H), 7.16 (t, J = 8.1 Hz, 1H, 5″-H), 7.18 (d, J = 2.5 Hz, 1H, 2″-H), 7.54 (t, J = 7.8 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.29 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.44 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.42 ((CH3)3), 43.46 ((NCH2)2), 49.04 ((NCH2)2), 79.92 (CMe3), 108.47 (C-2″), 111.75 (C-6″), 112.95 (C-4″), 116.17 (d, J = 7.3 Hz, C-2′, C-6′), 116.57 (d, J = 23.7 Hz, C-3′, C-5′), 122.66 (q, J = 274 Hz, CF3), 125.25 (q, J = 31.8 Hz, C-3), 126.35 (C-5), 129.54 (C-5″), 130.69 (q, J = 5.0 Hz, C-4), 130.87 (C-1), 135.90 (C-6), 138.09 (C-1″), 149.40 (q, J = 2.0 Hz, C-2), 151.86 (C-3″), 154.01 (d, J = 2.2 Hz, C-1′), 154.69 (COO), 158.50 (d, J = 242 Hz, C-4′), 161.59 (C=O); HRMS (ESI +) calcd for C29H30F4N3O4 [M+H]+: 560.2172; found: 560.2163.
tert-Butyl-4-{4-[2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (37): Reaction of the carboxylic acid 2 (305 mg (1.02 mmol)) with the amine 48 (280 mg (1.01 mmol)), 2-chloro-N-methylpyridinium iodide (452 mg (1.77 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 3:1) yielded compound 37 as pale-yellow solid (23 mg (4%)). IR = 3422, 1662, 1502, 1450, 1315, 1219, 1163, 824, 782; 1H NMR (CDCl3, 400 MHz) δ = 1.48 (s, 9H, (CH3)3), 3.06–3.19 (m, 4H, N(CH2)2), 3.54–3.57 (m, 4H, N(CH2)2), 6.75–6.79 (m, 2H, 2′-H, 6′-H), 6.84 (d, J = 8.8 Hz, 2H, 3″-H, 5″-H), 6.91–6.96 (m, 2H, 3′-H, 5′-H), 7,24 (d, J = 8.8 Hz, 2H, 2″-H, 6″-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.88 (dd, J = 7.4, 1.5 Hz, 1H, 4-H), 8.28 (dd, J = 7.9, 1.8 Hz, 1H, 6-H), 8.37 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.41 ((CH3)3), 43.51 ((NCH2)2), 49.55 ((NCH2)2), 79.92 (CMe3), 116.16 (d, J = 8.3 Hz, C-2′, C-6′), 116.53 (d, J = 23.7 Hz, C-3′, C-5′), 116.98 (C-3″, C-5″), 121.73 (C-2″, C-6″), 122.68 (q, J = 273 Hz, CF3), 125.17 (q, J = 31.8 Hz, C-3), 126.27 (C-5), 129.84 (C-1″), 130.50 (q, J = 4.9 Hz, C-4), 130.90 (C-1), 135.89 (C-6), 148.70 (C-4″), 149.39 (q, J = 2.8 Hz, C-2), 154.01 (d, J = 2.3 Hz, C-1′), 154.67 (COO), 158.48 (d, J = 242 Hz, C-4′), 161.42 (C=O); HRMS (ESI +) calcd for C29H30F4N3O4 [M+H]+: 560.2172; found: 560.2162.
N-{3-[4-(2,2-Dimethylpropanoyl)piperazin-1-yl]phenyl}-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (38): Reaction of the carboxylic acid 2 (221 mg (0.74 mmol)) with the amine 47 (183 mg (0.70 mmol)), 2-chloro-N-methylpyridinium iodide (337 mg (1.49 mmol)) and DIPEA (452 mg (3.50 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CHCl3/EtAc 2:1) yielded compound 38 as white solid (91 mg (24%)). IR = 2978, 1608, 1543, 1501, 1449, 1332, 1220, 1188, 997, 837, 777, 688; 1H NMR (CDCl3, 400 MHz) δ = 1.31 (s, 9H, (CH3)3), 3.12–3.15 (m, 4H, N(CH2)2), 3.76–3.79 (m, 4H, N(CH2)2), 6.65–6.68 (m, 2H, 4″-H, 6″-H), 6.75–6.79 (m, 2H, 2′-H, 6′-H), 6.91–6.95 (m, 2H, 3′-H, 5′-H), 7.17 (t, J = 7.6 Hz, 1H, 5″-H), 7.21 (t, J = 2.1 Hz, 1H, 2″-H), 7.54 (t, J = 7.8 Hz, 1H, 5-H), 7.90 (dd, J = 7.9, 1.6 Hz, 1H, 4-H), 8.28 (dd, J = 7.8, 1.6 Hz, 1H, 6-H), 8.45 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.40 ((CH3)3), 38.65 (CMe3), 44.88 (N(CH2)2), 49.17 (N(CH2)2), 108.26 (C-2″), 111.81 (C-6″), 112.75 (C-4″), 116.18 (d, J = 8.2 Hz, C-2′, C-6′), 116.56 (d, J = 23.7 Hz, C-3′, C-5′), 122.66 (q, J = 274 Hz, CF3), 125.26 (q, J = 31.6 Hz, C-3), 126.35 (C-5), 129.55 (C-5″), 130.69 (q, J = 4.9 Hz, C-4), 130.85 (C-1), 135.85 (C-6), 138.122 (C-1″), 149.41 (q, J = 1.8 Hz, C-2), 151.60 (C-3″), 154.02 (d, J = 2.5 Hz, C-1′), 158.49 (d, J = 242 Hz, C-4′), 161.65 ((C=O)NH), 176.38 (C=O); HRMS (ESI +) calcd for C29H30F4N3O3 [M+H]+: 544.2223; found: 544.2217.
N-{4-[4-(2,2-Dimethylpropanoyl)piperazin-1-yl]phenyl}-2-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide (39): Reaction of the carboxylic acid 2 (307 mg (1.03 mmol)) with the amine 49 (256 mg (0.98 mmol)), 2-chloro-N-methylpyridinium iodide (440 mg (1.72 mmol)) and DIPEA (633 mg (4.90 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/EtOH 59:1) yielded compound 39 as white solid (96 mg (18%)). IR = 3275, 1660, 1609, 1514, 1503, 1449, 1316, 1223, 1186, 1156, 1098, 1016, 824, 781; 1H NMR (CDCl3, 400 MHz) δ = 1.31 (s, 9H, (CH3)3), 3.09–3.12 (m, 4H, N(CH2)2), 3.77–3.80 (m, 4H, N(CH2)2), 6.75–6.78 (m, 2H, 2′-H, 6′-H), 6.84 (d, J = 8.9 Hz, 2H, 3″-H, 5″-H), 6.91–6.96 (m, 2H, 3′-H, 5′-H), 7.25 (d, J = 8.9 Hz, 2H, 2″-H, 6″-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.8, 1.6 Hz, 1H, 4-H), 8.29 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.38 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.41 ((CH3)3), 38.66 (CMe3), 44.93 ((NCH2)2), 49.70 ((NCH2)2), 116.16 (d, J = 8.2 Hz, C-2′, C-6′), 116.53 (d, J = 23.8 Hz, C-3′, C-5′), 116.78 (C-3″, C-5″), 121.72 (C-2″, C-6″), 122.68 (q, J = 273 Hz, CF3), 125.18 (q, J = 31.8 Hz, C-3), 126.76 (C-5), 129.97 (C-1″), 130.51 (q, J = 4.9 Hz, C-4), 130.88 (C-1), 135.89 (C-6), 148.40 (C-4″), 149.40 (q, J = 1.9 Hz, C-2), 154.01 (d, J = 2.5 Hz, C-1′), 158.48 (d, J = 242 Hz, C-4′), 161.43 (C=O), 176.37 ((C=O)NR2); HRMS (ESI +) calcd for C29H30F4N3O3 [M+H]+: 544.2223; found: 544.2214.
2-(4-Fluorophenoxy)-N-(3-nitrophenyl)-3-(trifluoromethyl)benzamide (52): Reaction of the carboxylic acid 2 (309 mg (1.03 mmol)) with 3-nitroaniline (145 mg (1.05 mmol)), 2-chloro-N-methylpyridinium iodide (454 mg (1.78 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2) yielded compound 52 as pale-yellow solid (156 mg (36%)). IR = 3368, 1695, 1601, 1548, 1503, 1450, 1351, 1287, 1266, 1222, 1182, 1167, 1127, 1098, 825, 782, 738; 1H NMR (CDCl3, 400 MHz) δ = 6.77–6.80 (m, 2H, 2′-H, 6′-H), 6.93–6.98 (m, 2H, 3′-H, 5′-H), 7.47 (t, J = 8.2 Hz, 1H, 5″-H), 7.58 (t, J = 7.8 Hz, 1H, 5-H), 7.76 (dd, J = 8.1, 2.1 Hz, 1H, 6″-H), 7.94–7.98 (m, 2H, 4-H, 4″-H), 8.31–8.34 (m, 2H, 2″-H, 6-H), 8.76 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 114.89 (C-2″), 115.99 (d, J = 8.3 Hz, C-2′, C-6′), 116.80 (d, J = 23.8 Hz, C-3′, C-5′), 119.57 (C-4″), 122.53 (q, J = 273 Hz, CF3), 125.47 (q, J = 32.0 Hz, C-3), 125.62 (C-6″), 126.60 (C-5), 129.88 (C-1), 129.91 (C-5″), 131.35 (q, J = 5.0 Hz, C-4), 136.01 (C-6), 138.23 (C-1″), 148.56 (C-3″), 149.49 (q, J = 1.9 Hz, C-2), 153.89 (d, J = 2.7 Hz, C-1′), 158.62 (d, J = 243 Hz, C-4′), 161.94 (C=O); HRMS (EI+) calcd for C20H11F4N2O4 [M-H]−: 419.0655; found: 419.0660.
2-(4-Fluorophenoxy)-N-(4-nitrophenyl)-3-(trifluoromethyl)benzamide (53): Reaction of the carboxylic acid 2 (301 mg (1.00 mmol)) with 4-nitroaniline (141 mg (1.03 mmol)), 2-chloro-N-methylpyridinium iodide (467 mg (1.83 mmol)) and DIPEA (646 mg (5.00 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH2Cl2/AcOH 100:1) yielded compound 53 as white solid (84 mg (20%)). IR = 3299, 1660, 1597, 1502, 1449, 1406, 1345, 1304, 1256, 1219, 1167, 1134, 834, 778, 751, 696; 1H NMR (CDCl3, 400 MHz) δ = 6.74–6.78 (m, 2H, 2′-H, 6′-H), 6.92–6.96 (m, 2H, 3′-H, 5′-H), 7.59 (t, J = 7.8 Hz, 1H, 5-H), 7.62 (d, J = 9.1 Hz, 2H, 2″-H, 6″-H), 7.96 (dd, J = 7.7, 1.6 Hz, 1H, 4-H), 8.19 (d, J = 9.1 Hz, 2H, 3″-H, 5″-H), 8.32 (dd, J = 7.7, 1.6 Hz, 1H, 6-H), 8.85 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 115.98 (d, J = 8.3 Hz, C-2′, C-6′), 116.80 (d, J = 24.0 Hz, C-3′, C-5′), 119.39 (C-2″, C-6″), 122.50 (q, J = 274 Hz, CF3), 125.10 (C-3″, C-5″), 125.51 (q, J = 31.8 Hz, C-3), 126.64 (C-5), 129.83 (C-1), 131.49 (q, J = 4.8 Hz, C-4), 136.04 (C-6), 142.88 (C-1″), 144.02 (C-4″), 149.53 (q, J = 1.9 Hz, C-2), 153.85 (d, J = 2.5 Hz, C-1′), 158.62 (d, J = 243 Hz, C-4′), 161.91 (C=O); HRMS (ESI -) calcd for C20H11F4N2O4 [M-H]−: 419.0655; found: 419.0660.
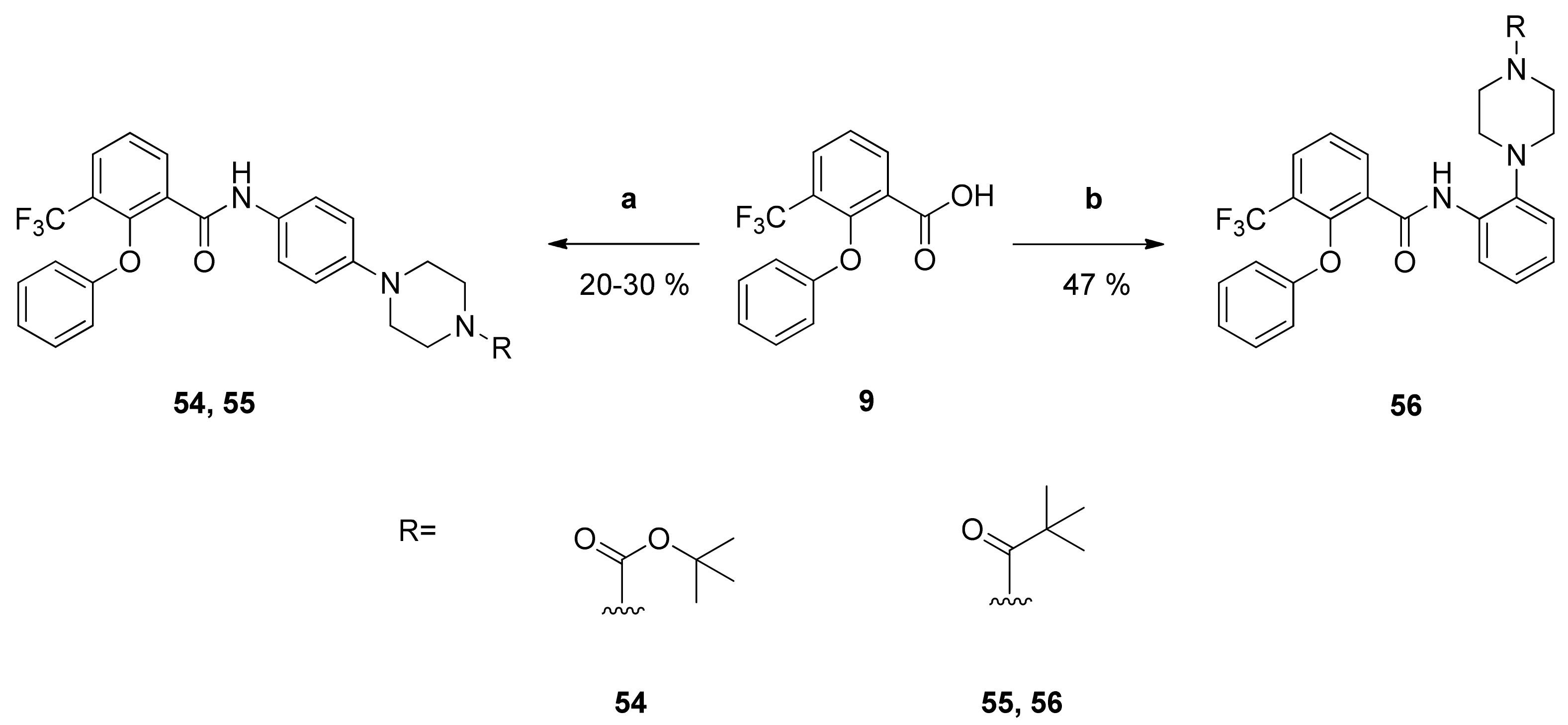
tert-Butyl-4-{4-[2-phenoxy-3-(trifluoromethyl)benzamido]phenyl}piperazine-1-carboxylate (54): Reaction of the carboxylic acid 9 (264 mg (0.94 mmol)) with the amine 48 (274 mg (0.99 mmol)), 2-chloro-N-methylpyridinium iodide (438 mg (1.71 mmol)) and DIPEA (607 mg (4.70 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 2:1) yielded compound 54 as pale-yellow solid (153 mg (30%)). IR = 3309, 1689, 1518, 1449, 1316, 1234, 1164, 750; 1H NMR (CDCl3, 400 MHz) δ = 1.48 (s, 9H, (CH3)3), 3.04–3.08 (m, 4H, N(CH2)2), 3.53–3.57 (m, 4H, N(CH2)2), 6.80–6.83 (m, 4H, 2′-H, 3″-H, 5″-H, 6′-H), 7.03 (t, J = 7.4 Hz, 1H, 4′-H), 7.21 (d, J = 8.8 Hz, 2H, 2″-H, 6″-H), 7.25 (t, J = 8.0 Hz, 2H, 3′-H, 5′-H), 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.33 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.48 (br s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.41 ((CH3)3), 43.50 (N(CH2)2), 49.58 (N(CH2)2), 79.90 (CMe3), 114.92 (C-2′, C-6′), 116.95 (C-3″, C-5″), 121.87 (C-2″, C-6″), 122.74 (q, J = 274 Hz, CF3), 123.32 (C-4′), 125.28 (q, J = 31.6 Hz, C-3), 125.13 (C-5), 126.33 (C-5), 129.97 (C-1″), 130.00 (C-3′, C- 5′), 130.52 (q, J = 5.0 Hz, C-4), 130.80 (C-1), 135.94 (C-6), 148.62 (C-4″), 154.67 (N(C=O)O), 158.03 (C-1″), 161.44 (C=O); HRMS (ESI +) calcd for C29H31F3N3O4 [M+H]+: 542.2267; found: 542.2255.
N-{4-[4-(2,2-Dimethylpropanoyl)piperazin-1-yl]phenyl}-2-phenoxy-3-(trifluoromethyl)benzamide (55): Reaction of the carboxylic acid 9 (260 mg (0.92 mmol)) with the amine 49 (252 mg (0.96 mmol)), 2-chloro-N-methylpyridinium iodide (445 mg (1.74 mmol)) and DIPEA (595 mg (4.60 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 1:1) yielded compound 55 as pale-yellow solid (97 mg (20%)). IR = 3423, 1625, 1516, 1448, 1316, 1234, 1162, 751, 688; 1H NMR (CDCl3, 400 MHz) δ = 1.30 (s, 9H, (CH3)3), 3.08–3.11 (m, 4H, N(CH2)2), 3.76–3.79 (m, 4H, N(CH2)2), 6.80–6.83 (m, 4H, 2′-H, 3″-H, 5″-H, 6′-H), 7.03 (t, J = 7.4 Hz, 1H, 4′-H), 7.20–7.28 (m, 4H, 2″-H, 3′-H, 5′-H, 6″-H) 7.53 (t, J = 7.8 Hz, 1H, 5-H), 7.89 (dd, J = 7.9, 1.7 Hz, 1H, 4-H), 8.33 (dd, J = 7.9, 1.7 Hz, 1H, 6-H), 8.49 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.41 ((CH3)3), 38.66 (CMe3), 44.94 (N(CH2)2), 49.74 (N(CH2)2), 114.92 (C-2′, C-6′), 116.75 (C-3″, C-5″), 121.87 (C-2″, C-6″), 122.73 (q, J = 274 Hz, CF3), 123.32 (C-4′), 125.29 (q, J = 31.7 Hz, C-3), 126.14 (C-5), 130.00 (C-3′, C-5′), 130.09 (C-1″), 130.53 (q, J = 4.9 Hz, C-4), 130.77 (C-1), 135.94 (C-6), 148.32 (C-4″), 149.30 (q, J = 1.8 Hz, C-2), 158.03 (C-1″), 161.46 (N(C=O)O), 176.36 (C=O); HRMS (ESI +) calcd for C29H31F3N3O3 [M+H]+: 526.2318; found: 526.2310.
N-{2-[4-(2,2-Dimethylpropanoyl)piperazin-1-yl]phenyl}-2-phenoxy-3-(trifluoromethyl)benzamide (56): Reaction of the carboxylic acid 9 (163 mg (0.58 mmol)) with the amine 25 (148 mg (0.57 mmol)), 2-chloro-N-methylpyridinium iodide (268 mg (1.05 mmol)) and DIPEA (368 mg (2.85 mmol)) in dry CH2Cl2 (30 mL) gave the raw carboxamide. Purification by column chromatography (silica gel, CH/EtAc 3:1) yielded compound 56 as white solid (141 mg (47%)). IR = 3442, 1685, 1636, 1588, 1521, 1448, 1326, 1160, 1122, 799, 752, 690; 1H NMR (CDCl3, 400 MHz) δ = 1.33 (s, 9H, (CH3)3), 2.84–2.87 (m, 4H, N(CH2)2), 3.87 (br s, 4H, N(CH2)2), 6.75 (d, J = 8.2 Hz, 2H, 2′-H, 6′-H), 6.97 (t, J = 7.4 Hz, 1H, 4′-H), 7.05 (t, J = 7.9 Hz, 1H; 4″-H), 7.09–7.14 (m, 2H, 3″-H, 5″-H), 7.19 (t, J = 7.9 Hz, 2H, 3′-H, 5′-H), 7.54 (t, J = 7.8 Hz, 1H, 5-H), 7.90 (dd, J = 7.8, 1.7 Hz, 1H, 4-H), 8.27 (br d, J = 7.8 Hz, 1H, 6-H), 8.31 (br d, J = 7.9 Hz, 1H, 6″-H), 9.82 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 28.42 ((CH3)3), 38.72 (CMe3), 45.62 (N(CH2)2), 52.49 (N(CH2)2), 115.22 (C-2′, C-6′), 119.87 (C-6″), 120.46 (C-3″), 122.72 (q, J = 274 Hz, CF3), 123.23 (C-4′), 124.32 (C-4″), 125.43 (q, J = 31.6 Hz, C-3), 125.84 (C-5″), 126.10 (C-5), 129.78 (C-3′, C-5′), 130.48 (q, J = 4.9 Hz, C-4), 131.93 (C-1), 133.26 (C-1″), 135.40 (C-6), 140.73 (C-2″), 149.68 (q, J = 1.9 Hz, C-2), 154.14 (C-1′), 161.73 (C=O), 176.50 ((C=O)NH); HRMS (ESI +) calcd for C29H31F3N3O3 [M+H]+: 525.2318; found: 526.2309.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}