
Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
2.1. Synthesis of MSPA and Its Derivatives
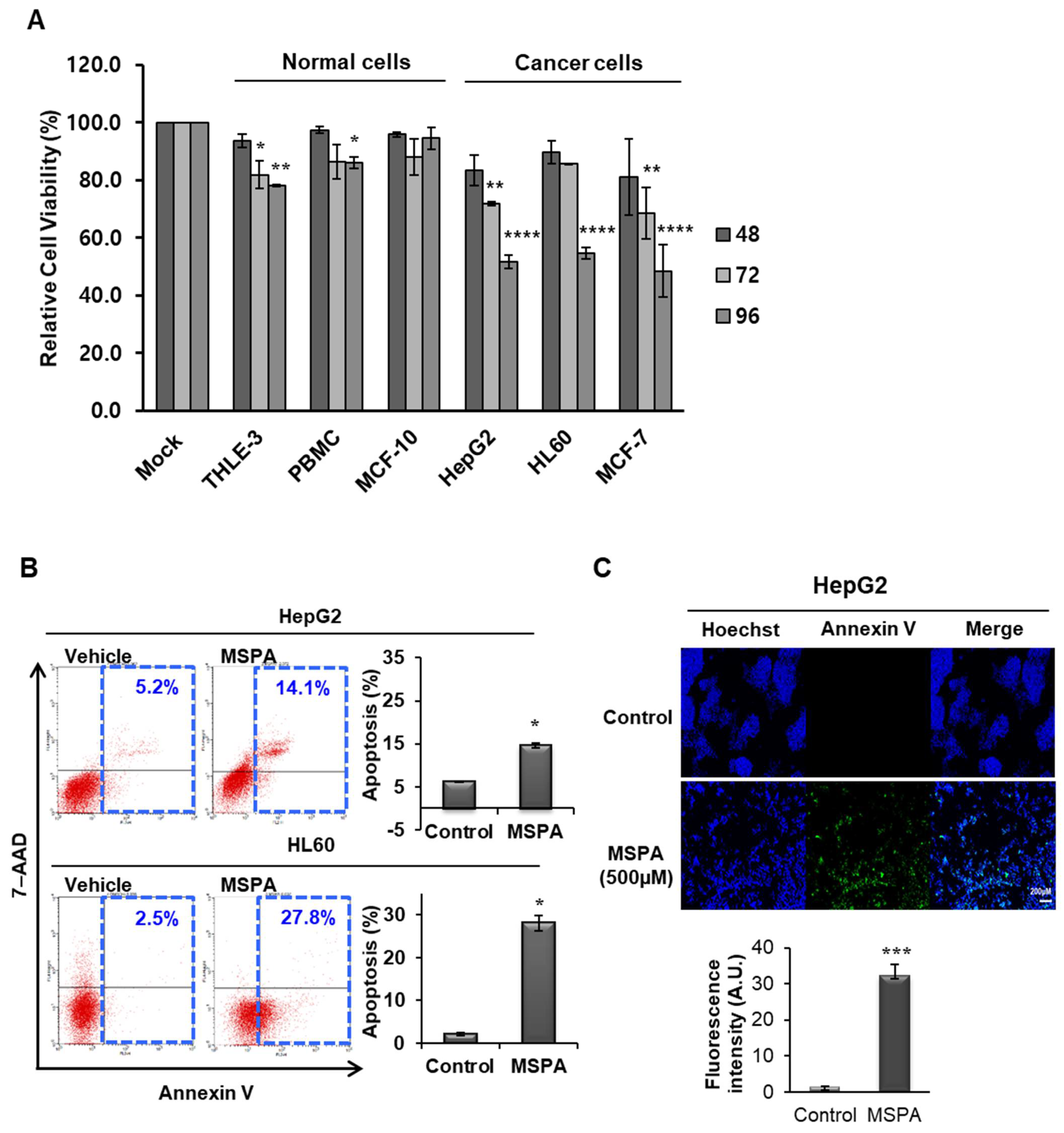
2.2. Selective Cytotoxic Activity of MSPA on Cancer Cells
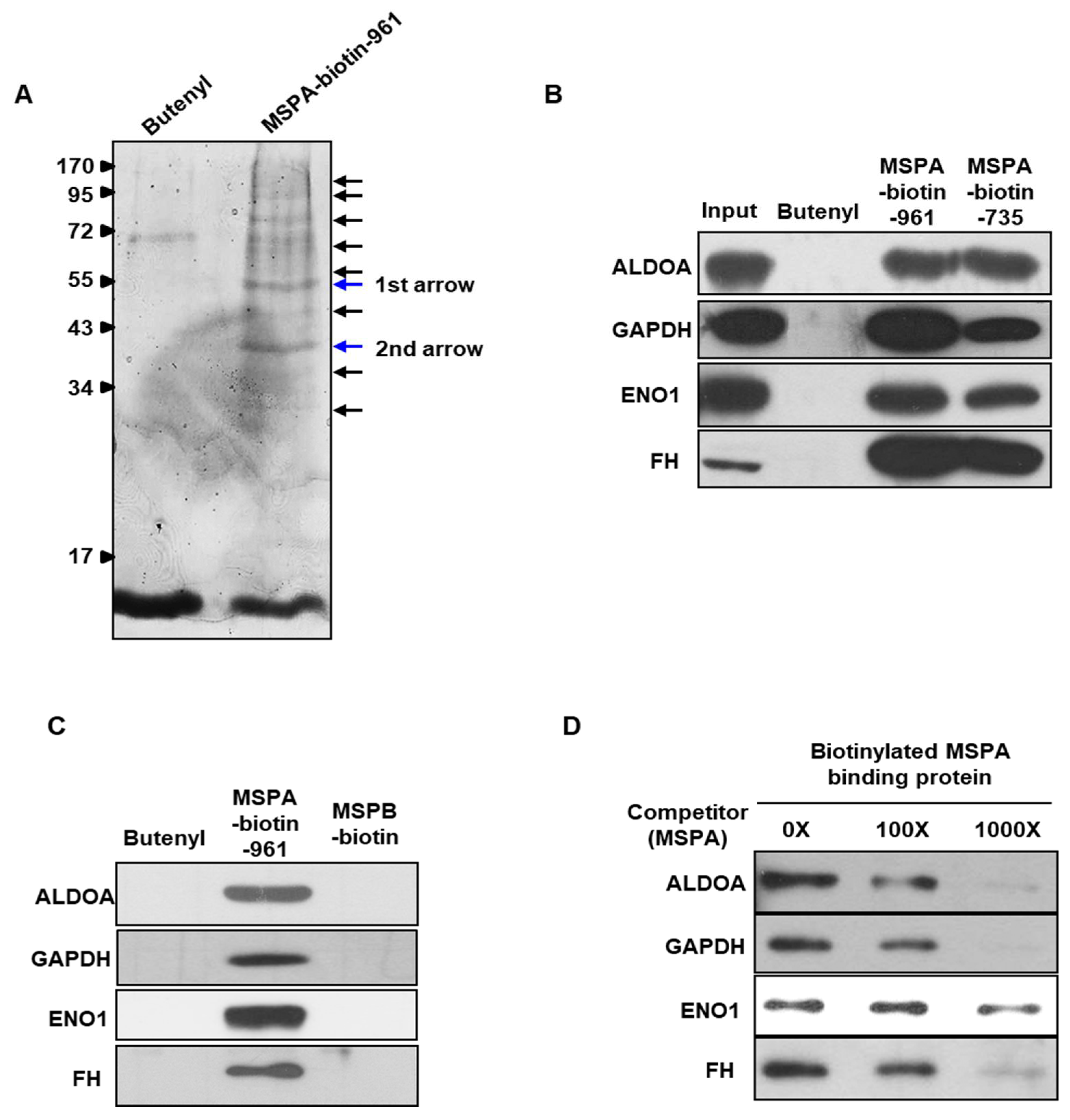
2.3. Identification of MSPA Target Molecules
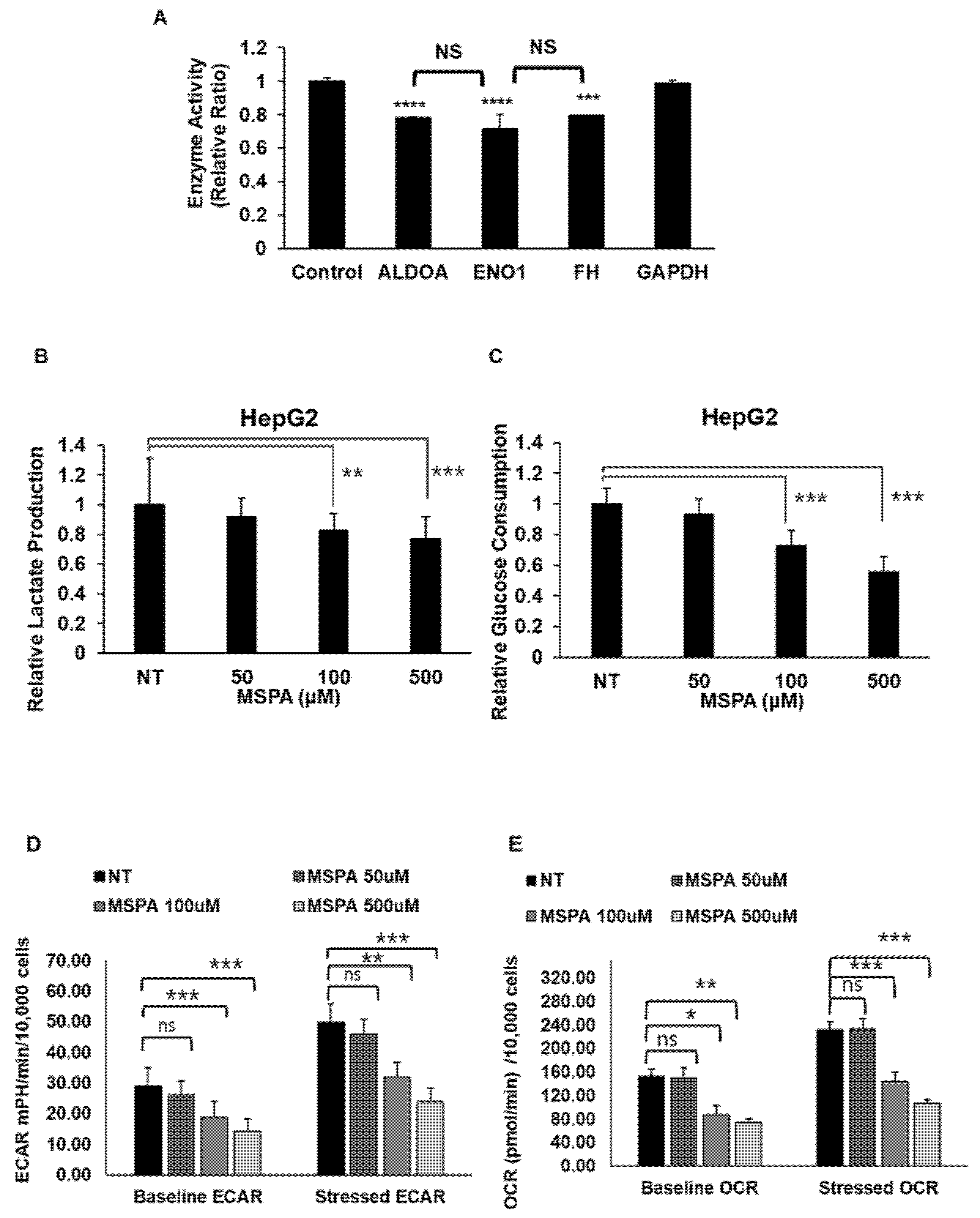
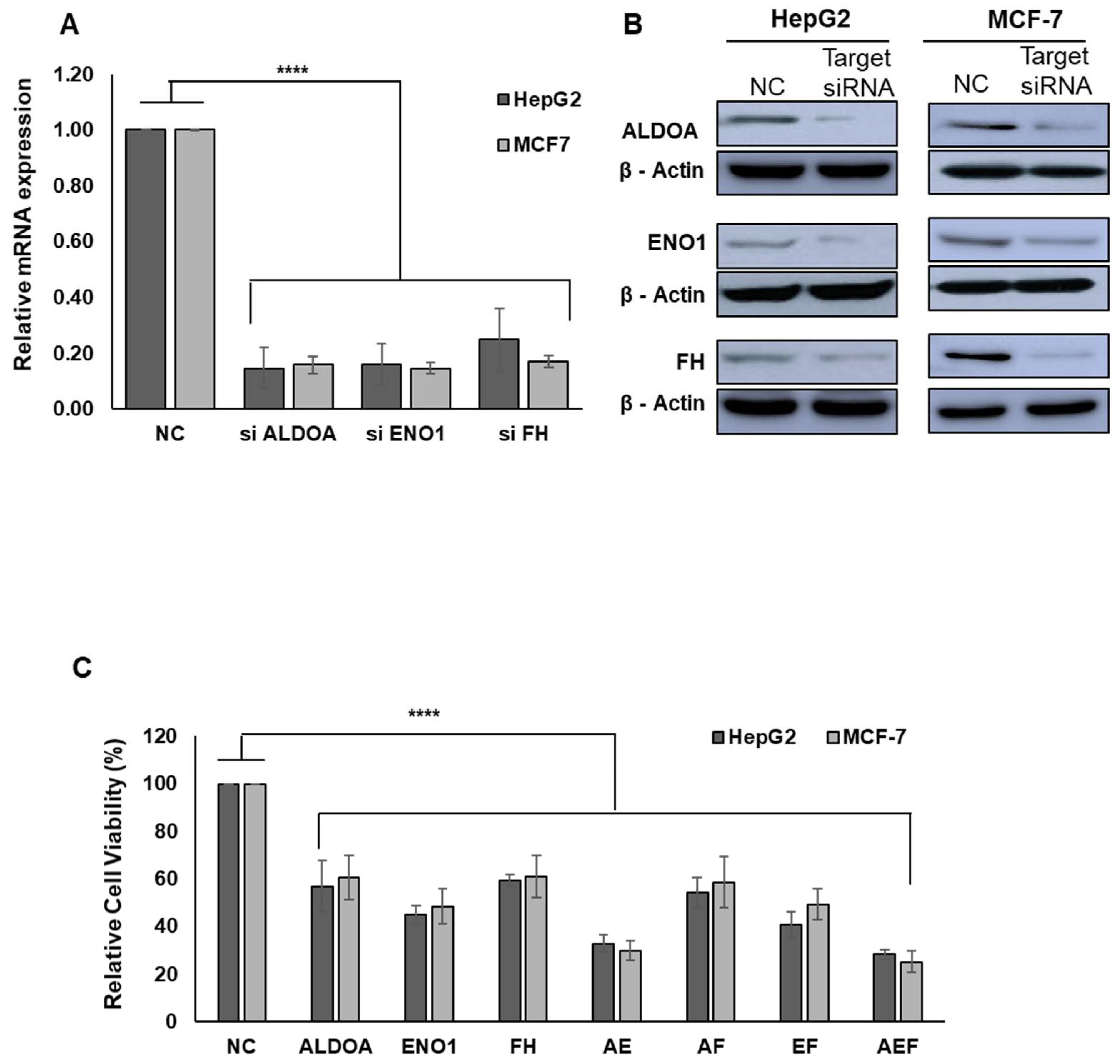
2.4. MSPA Inhibits Target-Enzyme Activities
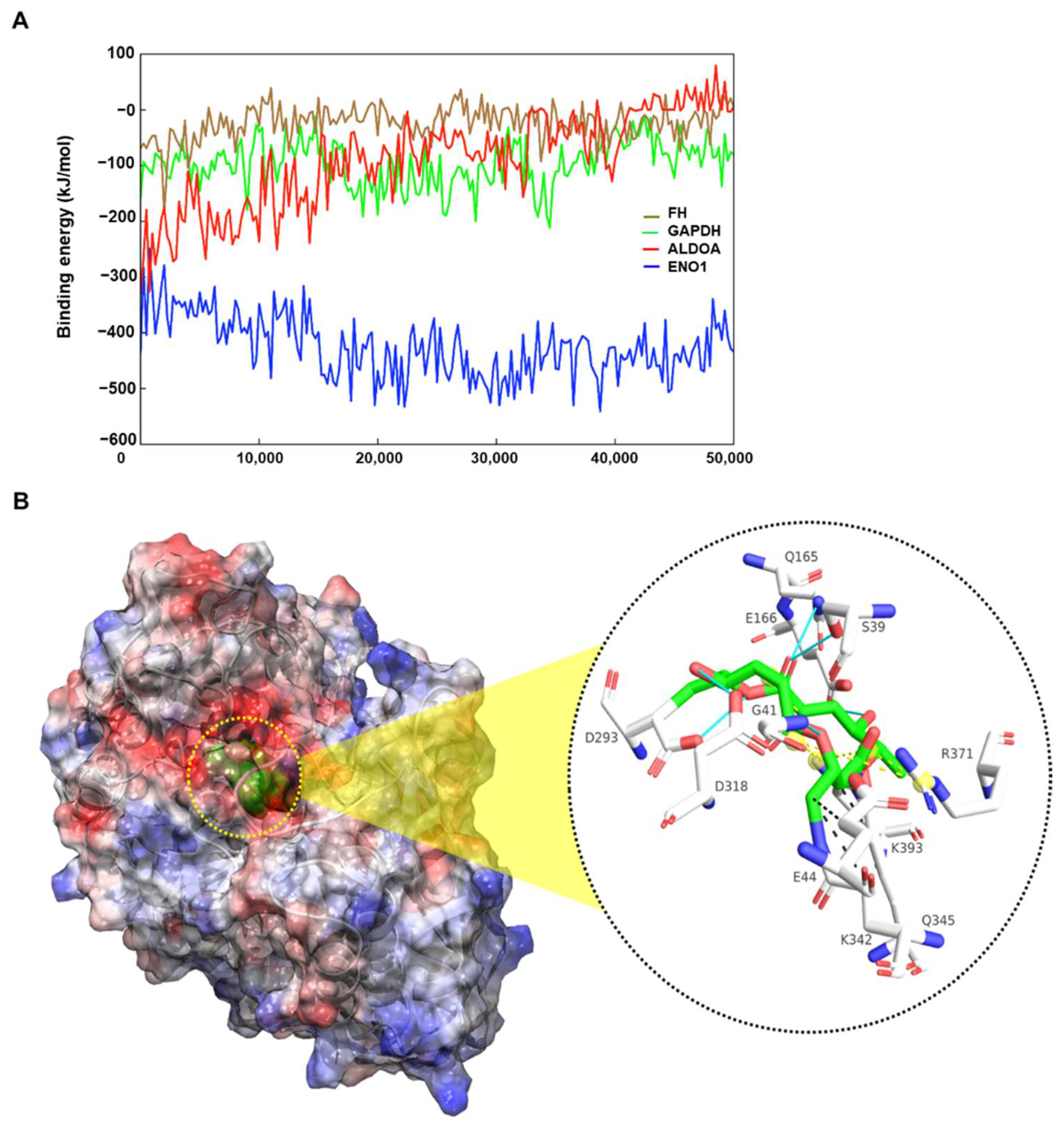
2.5. Molecular Docking and Molecular Dynamics Simulation
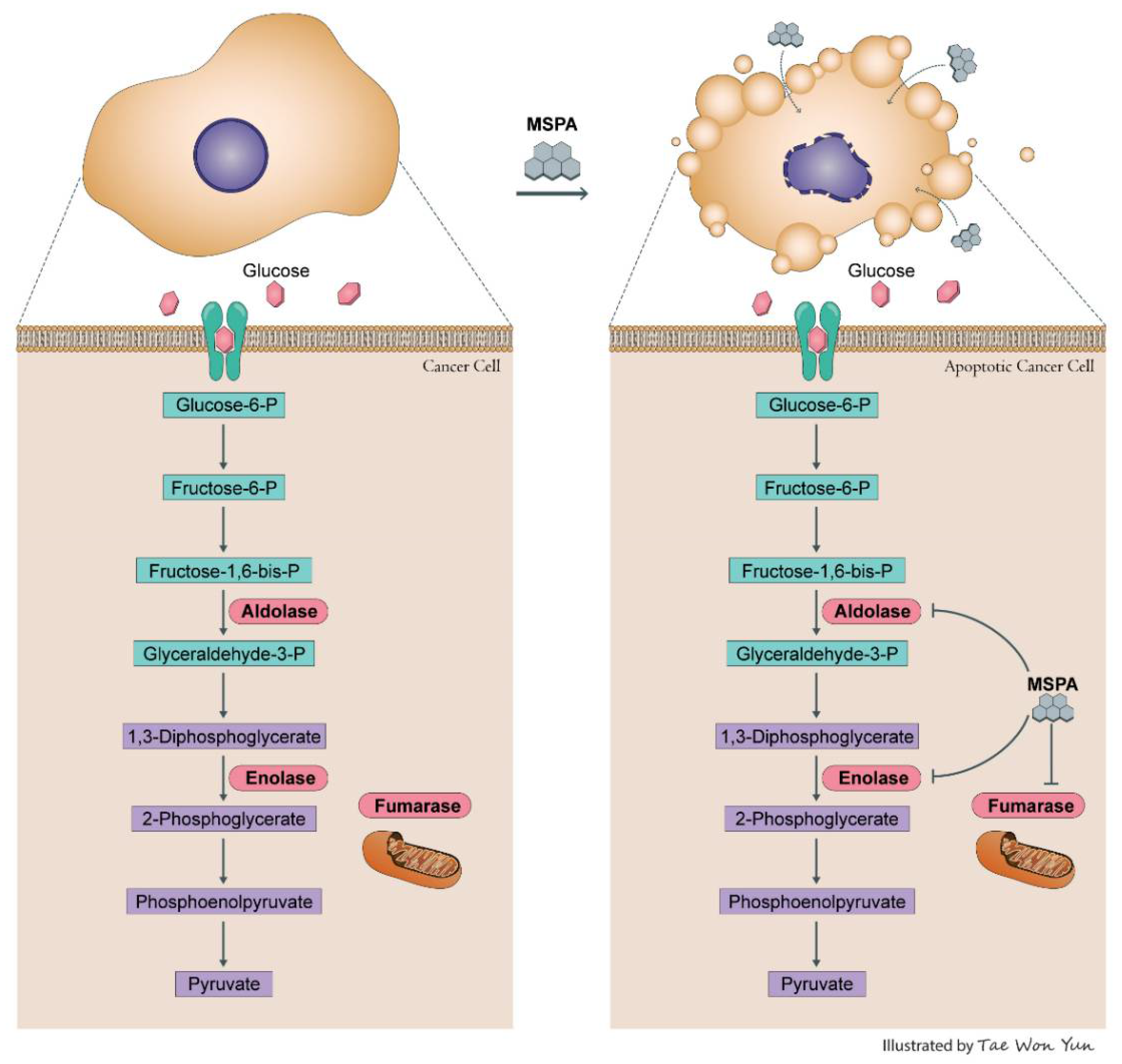
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cytotoxicity Assay
4.3. Apoptosis Assay and Immunofluorescence
4.4. Mass Spectrometry
4.5. Immunoblotting, Immunoprecipitation, and In Vitro Binding Assay
4.5.1. Immunoblotting
4.5.2. Immunoprecipitation
4.5.3. In Vitro Binding Assay
4.6. Enzyme Activity Assay
4.7. Enolase Activity Assay
4.8. Glucose Consumption and Lactate Production
4.9. Seahorse Energy Phenotype Assay
4.10. Molecular Docking and Molecular Dynamics Simulation
4.11. siRNA Sequences of Target Proteins
4.12. RNA Preparation and qRT-PCR Analysis
4.13. Statistical Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Origin of cancer cells. Oncologia 1956, 9, 75–83. [Google Scholar] [CrossRef]
- Chen, X.S.; Li, L.Y.; Guan, Y.D.; Yang, J.M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Matsuya, Y.; Nemoto, H.; Zaidi, S.F.; Sugiyama, T.; Yoshihisa, Y.; Shimizu, T.; Kondo, T. Mechanism of apoptosis induced by a newly synthesized derivative of macrosphelides with a thiazole side chain. Chem. Biol. Interact. 2009, 177, 218–226. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2016, 7, 5598–5612. [Google Scholar] [CrossRef]
- Gizak, A.; Wisniewski, J.; Heron, P.; Mamczur, P.; Sygusch, J.; Rakus, D. Targeting a moonlighting function of aldolase induces apoptosis in cancer cells. Cell Death Dis. 2019, 10, 712. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, Y.P.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Paek, S.M.; Seo, S.Y.; Kim, S.H.; Jung, J.W.; Lee, Y.S.; Jung, J.K.; Suh, Y.G. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.M.; Yun, H.; Kim, N.J.; Jung, J.W.; Chang, D.J.; Lee, S.; Yoo, J.; Park, H.J.; Suh, Y.G. Concise syntheses of (+)-macrosphelides A and B: Studies on the macro-ring closure strategy. J. Org. Chem. 2009, 74, 554–561. [Google Scholar] [CrossRef]
- Heo, Y.M.; Lee, H.; Shin, Y.K.; Paek, S.M. Development of an advanced synthetic route to macrosphelides and its application to the discovery of a more potent macrosphelide derivative. Molecules 2014, 19, 15572–15583. [Google Scholar] [CrossRef]
- Takamatsu, S.; Kim, Y.P.; Hayashi, M.; Hiraoka, H.; Natori, M.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. II. Physiochemical properties and structural elucidation. J. Antibiot. 1996, 49, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Zhao, Q.L.; Matsuya, Y.; Yu, D.Y.; Feril, L.B., Jr.; Nemoto, H.; Kondo, T. Rapid and transient intracellular oxidative stress due to novel macrosphelides trigger apoptosis via Fas/caspase-8-dependent pathway in human lymphoma U937 cells. Chem. Biol. Interact. 2007, 170, 86–99. [Google Scholar] [CrossRef]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.M.; Suh, Y.G. Design and synthesis of a macrosphelide A-biotin chimera. Org. Biomol. Chem. 2014, 12, 7127–7135. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.; Pisaneschi, F.; Link, T.M.; Lee, G.R.; Sun, D.; et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Satani, N.; Lin, Y.H.; Hammoudi, N.; Raghavan, S.; Georgiou, D.K.; Muller, F.L. ENOblock Does Not Inhibit the Activity of the Glycolytic Enzyme Enolase. PLoS ONE 2016, 11, e0168739. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chiou, J.; Yang, Y.F.; Su, C.Y.; Lin, Y.F.; Yang, C.N.; Lu, P.J.; Huang, M.S.; Yang, C.J.; Hsiao, M. Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs Survival. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef]
- Lin, Y.H.; Satani, N.; Hammoudi, N.; Yan, V.C.; Barekatain, Y.; Khadka, S.; Ackroyd, J.J.; Georgiou, D.K.; Pham, C.D.; Arthur, K.; et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat. Metab. 2020, 2, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Shriwas, P.; Roberts, D.; Li, Y.; Wang, L.; Qian, Y.; Bergmeier, S.; Hines, J.; Adhicary, S.; Nielsen, C.; Chen, X. A small-molecule pan-class I glucose transporter inhibitor reduces cancer cell proliferation in vitro and tumor growth in vivo by targeting glucose-based metabolism. Cancer Metab. 2021, 9, 14. [Google Scholar] [CrossRef]
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.M.; Kuasne, H.; et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wu, C.; Yang, K.; Yang, Y.; Liu, Y.; Gao, S.; Wang, Q.; Li, C.; Chen, L.; Li, H. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. 2021, 164, 105367. [Google Scholar] [CrossRef] [PubMed]
- Muronetz, V.I.; Melnikova, A.K.; Barinova, K.V.; Schmalhausen, E.V. Inhibitors of Glyceraldehyde 3-Phosphate Dehydrogenase and Unexpected Effects of Its Reduced Activity. Biochemistry 2019, 84, 1268–1279. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S. Evolution of GAPDH as a druggable target of tumor glycolysis? Expert Opin. Ther. Targets 2018, 22, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wang, L.; Liu, H.L. ENO1 Overexpression in Pancreatic Cancer Patients and Its Clinical and Diagnostic Significance. Gastroenterol. Res. Pract. 2018, 2018, 3842198. [Google Scholar] [CrossRef]
- Zhu, W.; Li, H.; Yu, Y.; Chen, J.; Chen, X.; Ren, F.; Ren, Z.; Cui, G. Enolase-1 serves as a biomarker of diagnosis and prognosis in hepatocellular carcinoma patients. Cancer Manag. Res. 2018, 10, 5735–5745. [Google Scholar] [CrossRef]
- Sun, L.; Guo, C.; Cao, J.; Burnett, J.; Yang, Z.; Ran, Y.; Sun, D. Over-Expression of Alpha-Enolase as a Prognostic Biomarker in Patients with Pancreatic Cancer. Int. J. Med. Sci. 2017, 14, 655–661. [Google Scholar] [CrossRef]
- White-Al Habeeb, N.M.; Di Meo, A.; Scorilas, A.; Rotondo, F.; Masui, O.; Seivwright, A.; Gabril, M.; Girgis, A.H.; Jewett, M.A.; Yousef, G.M. Alpha-enolase is a potential prognostic marker in clear cell renal cell carcinoma. Clin. Exp. Metastasis 2015, 32, 531–541. [Google Scholar] [CrossRef]
- Yang, T.; Shu, X.; Zhang, H.W.; Sun, L.X.; Yu, L.; Liu, J.; Sun, L.C.; Yang, Z.H.; Ran, Y.L. Enolase 1 regulates stem cell-like properties in gastric cancer cells by stimulating glycolysis. Cell Death Dis. 2020, 11, 870. [Google Scholar] [CrossRef]
- Xu, W.; Yang, W.; Wu, C.; Ma, X.; Li, H.; Zheng, J. Enolase 1 Correlated with Cancer Progression and Immune-Infiltrating in Multiple Cancer Types: A Pan-Cancer Analysis. Front. Oncol. 2020, 10, 593706. [Google Scholar] [CrossRef]
- Leshets, M.; Ramamurthy, D.; Lisby, M.; Lehming, N.; Pines, O. Fumarase is involved in DNA double-strand break resection through a functional interaction with Sae2. Curr. Genet. 2018, 64, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Leshets, M.; Silas, Y.B.H.; Lehming, N.; Pines, O. Fumarase: From the TCA Cycle to DNA Damage Response and Tumor Suppression. Front. Mol. Biosci. 2018, 5, 68. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure 2005, 13, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Konieczna, A.; Szczepanska, A.; Sawiuk, K.; Wegrzyn, G.; Lyzen, R. Effects of partial silencing of genes coding for enzymes involved in glycolysis and tricarboxylic acid cycle on the enterance of human fibroblasts to the S phase. BMC Cell Biol. 2015, 16, 16. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins Identified from around the 55 kDa Band (First Arrow in Figure 2A) | |||
|---|---|---|---|
| Proteins | Peptide | Coverage (%) | Function |
| ENO1 (Enolase 1) | RHIADLAGNSEVILPVPAFNVINGGSHAGNK.L KLAMQEFMILPVGAANFR.E KYGKDATNVGDEGGFAPNILENKEGLELLK.T KDATNVGDEGGFAPNILENKEGLELLK.T KAGYTDKVVIGMDVAASEFFR.S KVVIGMDVAASEFFR.S RSGKYDLDFK.S RSGKYDLDFKSPDDPSR.Y KYDLDFKSPDDPSR.Y KSFIKDYPVVSIEDPFDQDDWGAWQK.F KDYPVVSIEDPFDQDDWGAWQK.F KFTASAGIQVVGDDLTVTNPK.R KFTASAGIQVVGDDLTVTNPKR.I KSCNCLLLK.V RNFRNPLAK. | 43.1 | Glycolytic enzyme Auto-antigen |
| FH (Fumarate hydratase) | RAAAEVNQDYGLDPK.I AIEMLGGELGSK.I KSKEFAQIIK.I RTHTQDAVPLTLGQEFSGYVQQVK.Y RIYELAAGGTAVGTGLNTR.I | 14.9 | Mitochondrial isoenzyme Involved in the TCA cycle |
| PDIA6 (Protein disulfide isomerase family A member 6) | RTGEAIVDAALSALR.Q KLAAVDATVNQVLASR.Y RTCEEHQLCVVAVLPHILDTGAAGR.N RGSTAPVGGGAFPTIVER.E | 14.2 | A member of the disulfide isomerase (PDI) family Chaperone activity |
| ENO2 (Enolase 2) | RAAVPSGASTGIYEALELR.D KFGANAILGVSLAVCK.A RSGETEDTFIADLVVGLCTGQIK.T | 12.7 | Glucose metabolism Neurotrophic and neuroprotective properties |
| EEF1A1 (Eukaryotic translation elongation factor 1 alpha 1) | KYYVTIIDAPGHR.D RYEEIVKEVSTYIK.K RVETGVLKPGMVVTFAPVNVTTEVK.S | 11.1 | Isoform of the alpha subunit of the elongation factor-1 complex Enzymatic delivery of aminoacyl tRNAs to the ribosome |
| Proteins Identified from around the 40 kDa Band (Second Arrow in Figure 2A) | |||
| Proteins | Peptide | Coverage (%) | Function |
| GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) | KVDIVAINDPFIDLNYMVYMFQYDSTHGK.F KAENGKLVINGNPITIFQER.D KLVINGNPITIFQER.D KLVINGNPITIFQERDPSK.I KWGDAGAEYVVESTGVFTTMEK.A KRVIISAPSADAPMFVMGVNHEK.Y RVIISAPSADAPMFVMGVNHEK.Y KVIHDNFGIVEGLMTTVHAITATQK.T RGALQNIIPASTGAAK.A RVPTANVSVVDLTCR.L RVVDLMAHMASK.E RVVDLMAHMASKE. | 47.5 | Glycolysis Uracil DNA glycosylase activity |
| ALDOA (Aldolase) | KGILAADESTGSIAK.R KIGEHTPSALAIMENANVLAR.Y RALANSLACQGKYTPSGQAGAAASESLFVSNHAY. KYTPSGQAGAAASESLFVSNHAY. | 18.4 | Glycolytic enzyme Gluconeogenesis |
| TALDO1 (Transaldolase 1) | LIELYK.E KLSSTWEGIQAGK.E KSYEPLEDPGVK.S KIYNYYK.K KLLGELLQDNAK.L RWLHNEDQMAVEK.L | 17.2 | A key enzyme of the nonoxidative pentose phosphate pathway Lipid biosynthesis |
| APEX1 (Apurinic/apyrimidinic endodeoxyribonuclease 1) | KGAVAEDGDELRTEPEAK.K KKNDKEAAGEGPALYEDPPDQK.T KVSYGIGDEEHDQEGR.V | 16.7 | Endodeoxyribonuclease activity Exonuclease activity |
| ALDOC (Aldolase) | KGVVPLAGTDGETTTQGLDGLSER.C RYASICQQNGIVPIVEPEILPDGDHDLKR.C | 14.0 | Glycolytic enzyme Involved in the innate immune system |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, K.; Rajasekaran, N.; Chelakkot, C.; Lee, H.S.; Paek, S.-M.; Yang, H.; Jia, L.; Park, H.G.; Son, W.S.; Kim, Y.-J.; et al. Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH. Pharmaceuticals 2021, 14, 1060. https://doi.org/10.3390/ph14101060
Song K, Rajasekaran N, Chelakkot C, Lee HS, Paek S-M, Yang H, Jia L, Park HG, Son WS, Kim Y-J, et al. Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH. Pharmaceuticals. 2021; 14(10):1060. https://doi.org/10.3390/ph14101060
Chicago/Turabian StyleSong, Kyoung, Nirmal Rajasekaran, Chaithanya Chelakkot, Hun Seok Lee, Seung-Mann Paek, Hobin Yang, Lina Jia, Hee Geon Park, Woo Sung Son, Yu-Jin Kim, and et al. 2021. "Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH" Pharmaceuticals 14, no. 10: 1060. https://doi.org/10.3390/ph14101060
APA StyleSong, K., Rajasekaran, N., Chelakkot, C., Lee, H. S., Paek, S.-M., Yang, H., Jia, L., Park, H. G., Son, W. S., Kim, Y.-J., Choi, J.-S., Jeong, H. M., Suh, Y.-G., Yun, H., & Shin, Y. K. (2021). Macrosphelide A Exhibits a Specific Anti-Cancer Effect by Simultaneously Inactivating ENO1, ALDOA, and FH. Pharmaceuticals, 14(10), 1060. https://doi.org/10.3390/ph14101060