
Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways

 , ,
, ,  ,
,  , , and
, , and
Abstract
Highlights
- Epilepsy is the second most prevalent neurological disease and appears in patients with neurodegenerative diseases, thus indicating a molecular link between them;
- There is growing evidence that relates the appearance of β-amyloid plaques, neurofibrillary tangles, α-synuclein, or mutations in the huntingtin protein to increased neuronal excitability that precedes seizures;
- Several approved drugs, such as atorvastatin, ceftriaxone, losartan, anakinra, rapamycin, and fingolimod, have been studied in animal models for antiseizure applications;
- Commonly used antiseizure drugs, such as levetiracetam, zonisamide, and valproate, are being investigated in other neurodegenerative diseases.
1. Introduction
2. Epilepsy in Neurodegenerative Diseases
2.1. Epilepsy and Alzheimer’s Disease
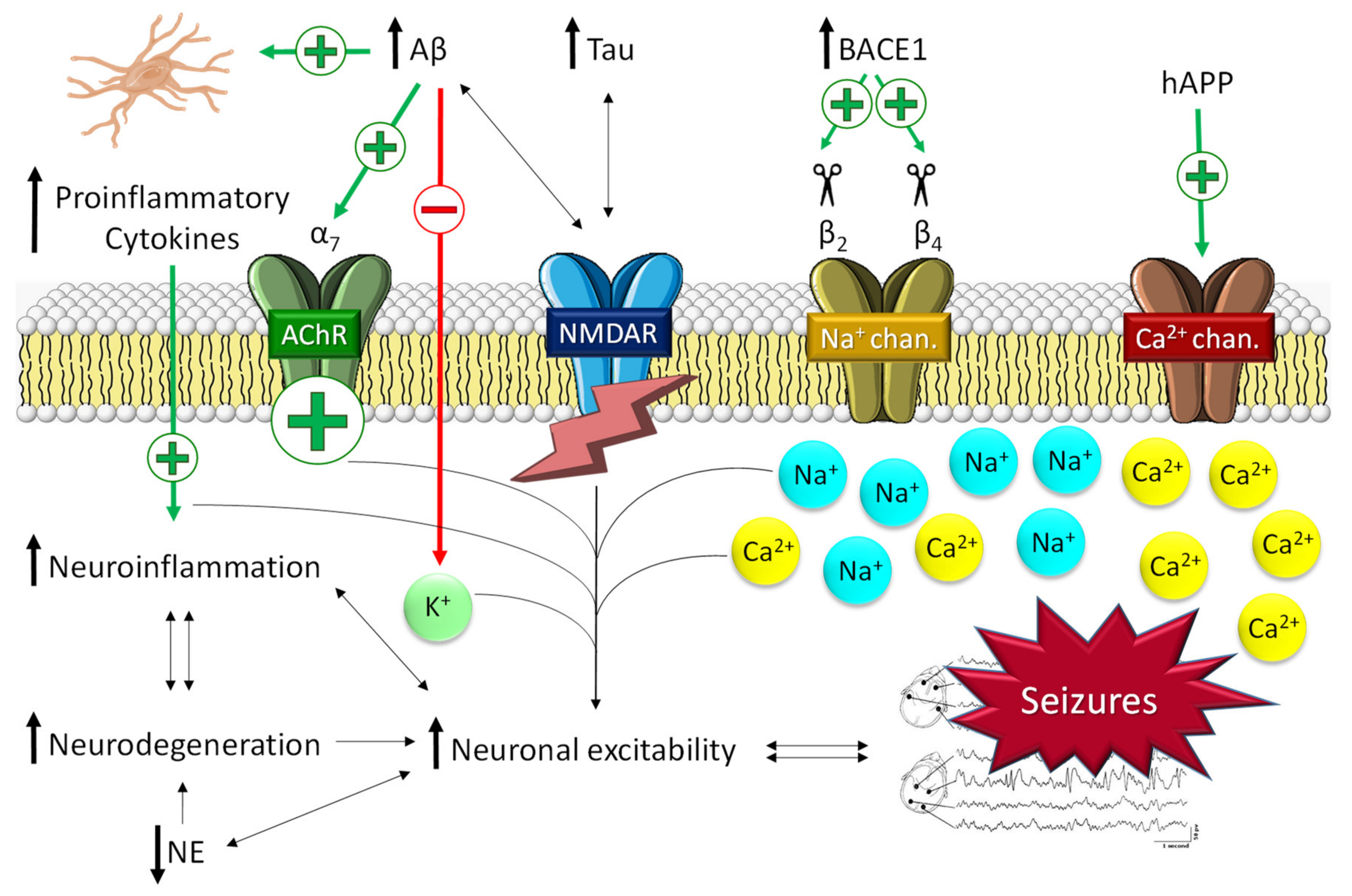
2.1.1. The Role of Aβ in Epilepsy
2.1.2. The Role of Tau in Epilepsy
2.1.3. The Role of Allopregnanolone in AD and Epilepsy
2.2. Epilepsy and Parkinson’s Disease
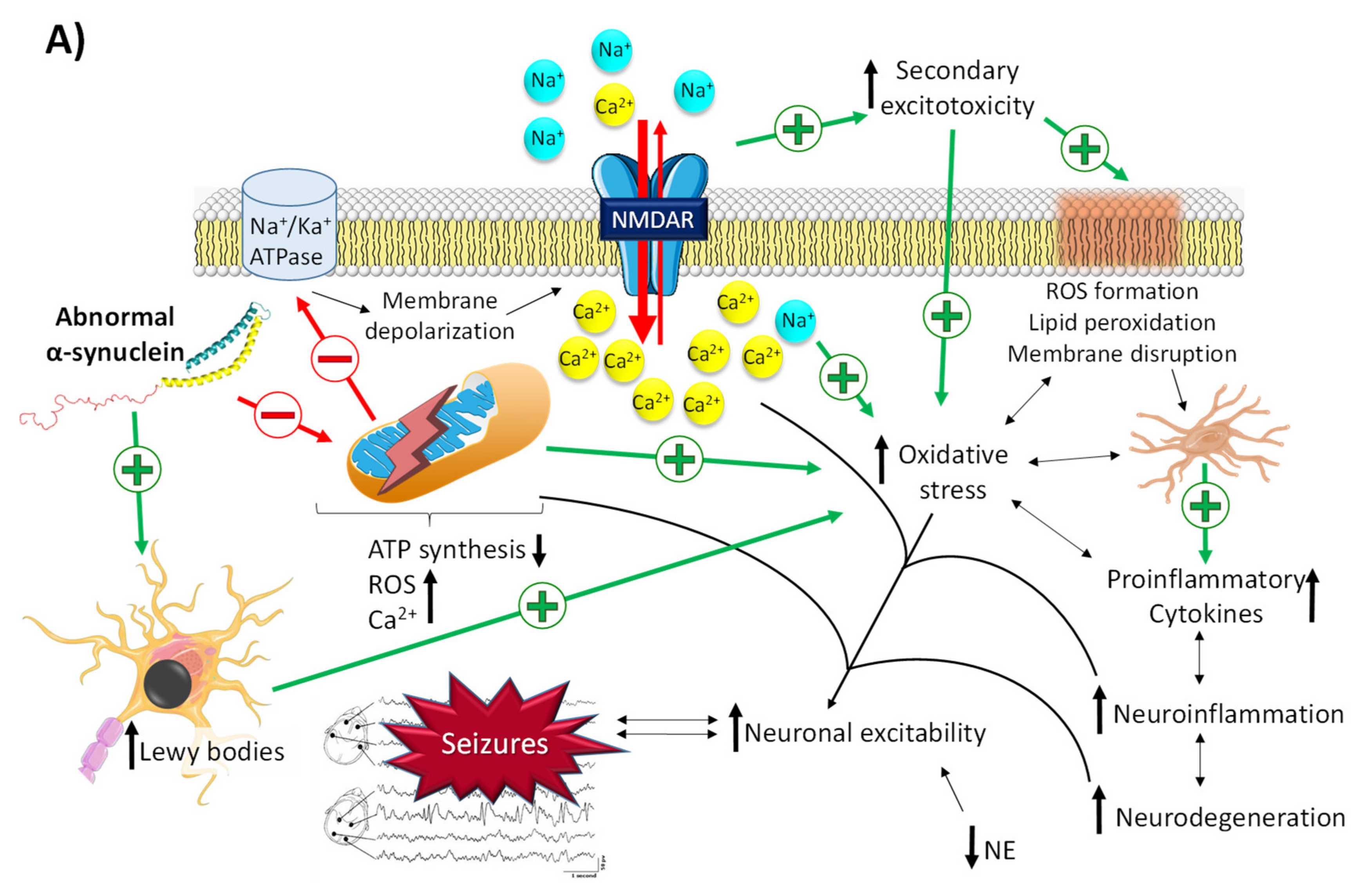
2.2.1. The Role of α-Synuclein in Epilepsy
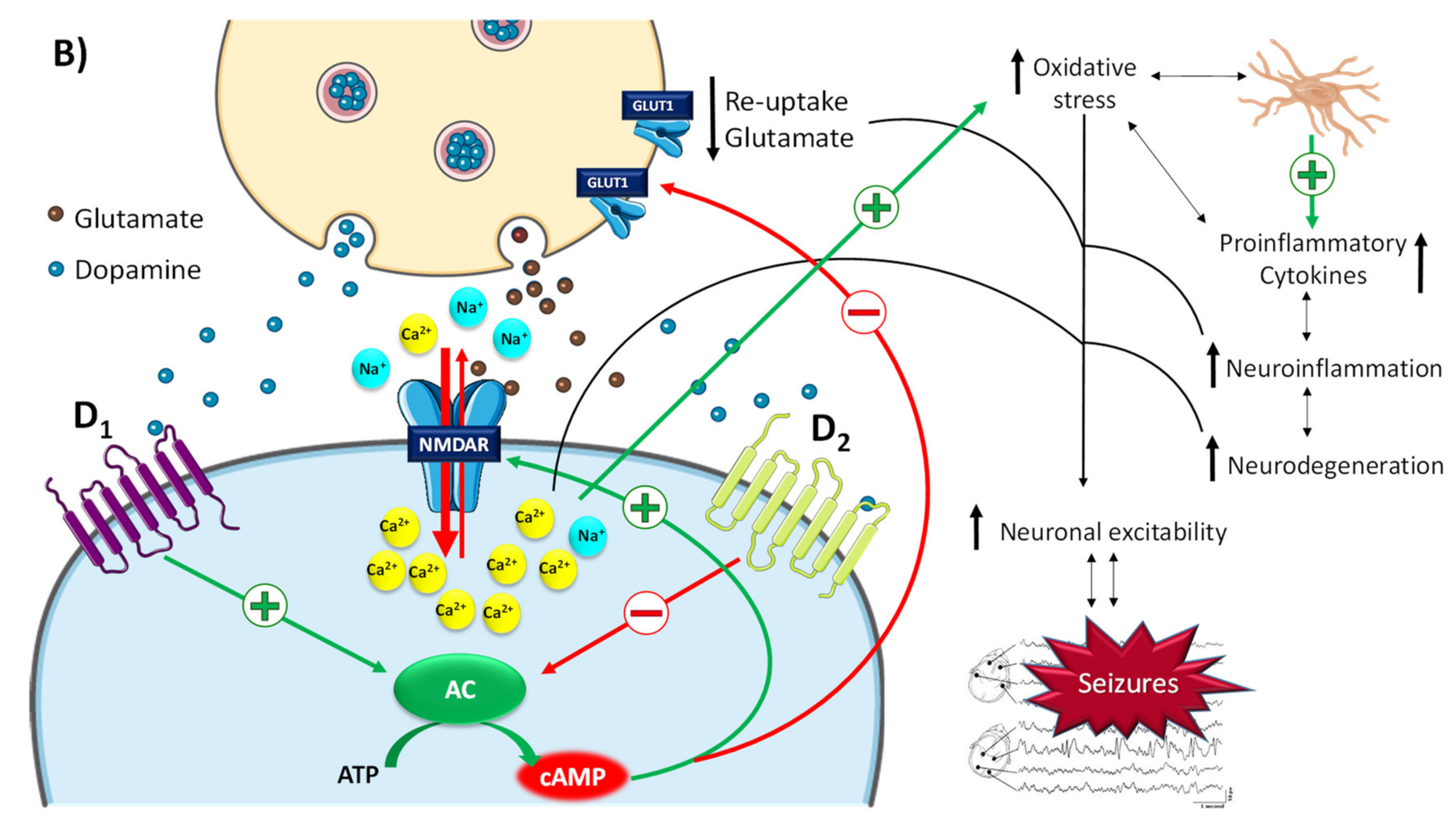
2.2.2. The Role of Dopamine and Norepinephrine in Epilepsy
2.2.3. The Role of Allopregnanolone in PD and Epilepsy
2.3. Epilepsy and Huntington’s Disease
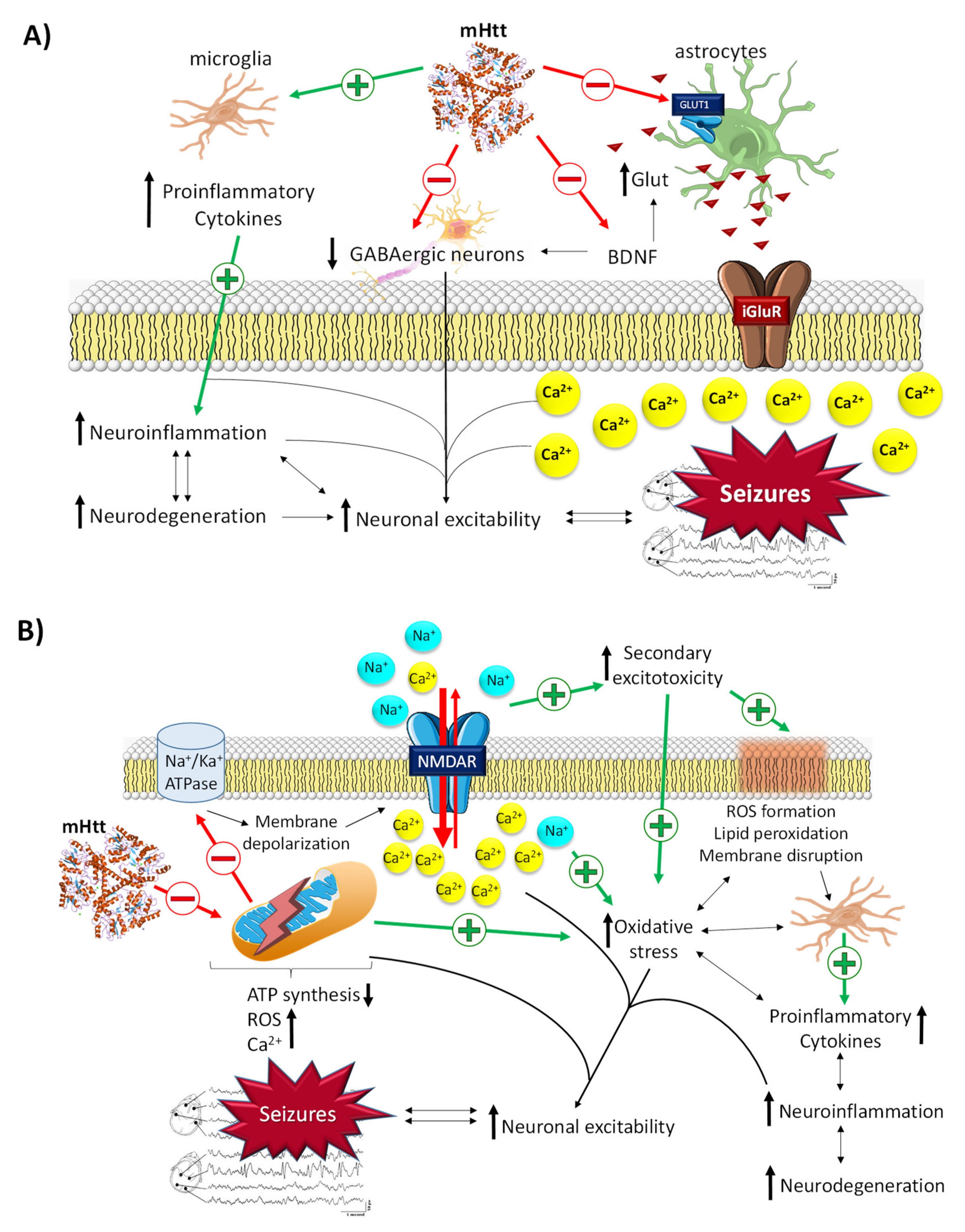
2.3.1. The Role of mHtt in Epilepsy
2.3.2. The Role of BDNF in Epilepsy
2.4. Epilepsy and Multiple Sclerosis
The Role of Allopregnanolone in MS and Epilepsy
3. Current Standards for Epilepsy Treatment and Refractory Epilepsy
4. Antiseizure Drugs in Neurodegenerative Diseases
4.1. ASDs in Alzheimer’s Disease
4.2. ASDs for Parkinson’s Disease
4.3. ASDs for Huntington’s Disease
4.4. ASDs for Multiple Sclerosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Hesdorffer, D.C.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsy 2014, 55, 475–482. [Google Scholar] [CrossRef]
- Galovic, M.; van Dooren, V.Q.H.; Postma, T.S.; Vos, S.B.; Caciagli, L.; Borzì, G.; Cueva Rosillo, J. Progressive Cortical Thinning in Patients with Focal Epilepsy. JAMA Neurol. 2019, 76, 1230–1239. [Google Scholar] [CrossRef]
- Whelan, C.D.; Altmann, A.; Botía, J.A.; Jahanshad, N.; Hibar, D.P.; Absil, J.; Alhusaini, S. Structural brain abnormalities in the common epilepsies assessed in a worldwide ENIGMA study. Brain 2018, 141, 391–408. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Nichols, E.; Abd Allah, F.; Abdela, J.; Abdelalim, A.; Niguse, H.; Abraha, M.G.A. Global, regional, and national burden of epilepsy, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 357–375. [Google Scholar] [CrossRef]
- Brainstorm Consortium, O. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Epilepsy. 2021. Available online: https://www.who.int/en/news-room/fact-sheets/detail/epilepsy (accessed on 14 September 2021).
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L. Prevalence and incidence of epilepsy A systematic review and meta-analysis of international studies. Am. Acad. Neurol. 2016, 88, 296–303. [Google Scholar]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bhavnani, B. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006, 22, 1–22. [Google Scholar]
- Jackson, J. The Lumleian lectures on convulsive seizures. Br. Med. J. 1890, 1, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J. The Lumleian lectures on convulsive seizures. Br. Med. J. 1890, 1, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J. The Lumleian lectures on convulsive seizures. Br. Med. J. 1890, 1, 821–827. [Google Scholar] [CrossRef]
- Paz, J.T.; Huguenard, J.R. Microcircuits and their interactions in epilepsy: Is the focus out of focus? Nat. Neurosci. 2015, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Dewar, S.R.; Heilemann, M.V.; Engel, J.; Lee, E.E.; Pieters, H.C. Perceptions of illness severity in adults with drug-resistant temporal lobe epilepsy. Epilepsy. Behav. 2020, 109, 107091. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C. Hippocampal Sclerosis: Causes and Prevention. Semin. Neurol. 2015, 35, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Santulli, L.; Coppola, A.; Balestrini, S.; Striano, S. The challenges of treating epilepsy with 25 antiepileptic drugs. Pharmacol. Res. 2016, 107, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Scheffer, I.E.; Kiley, M. The management of epilepsy in children and adults. Med. J. Aust. 2018, 208, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Cano, A.; Turowski, P.; Ettcheto, M.; Duskey, J.T.; Tosi, G.; López, E.S.; García, M.L.; Camins, A.; Souto, E.B.; Ruiz, A.; et al. Nanomedicine—Based technologies and novel biomarkers for the diagnosis and treatment of Alzheimer’s disease: From current to future challenges. J. Nanobiotechnol. 2021, 19, 122. [Google Scholar] [CrossRef]
- ILAE. Alzheimer’s and epilepsy: Intimate connections. Epigraph 2018, 20. [Google Scholar]
- Stefanidou, M.; Beiser, A.S.; Himali, J.J.; Peng, T.J.; Devinsky, O.; Seshadri, S.; Friedman, D. Bi-directional association between epilepsy and dementia. Neurology 2020, 95, e3241–e3247. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Fornai, F.; Saccaro, L.F.; Busceti, C.L.; Biagioni, F. Epilepsy and Alzheimer ’s Disease: Potential mechanisms for an association. Brain Res. Bull. 2020, 160, 107–120. [Google Scholar] [CrossRef]
- Friedman, D.; Honig, L.S.; Scarmeas, N. Seizures and Epilepsy in Alzheimer’s Disease. CNS Neurosci. Ther. 2012, 18, 285–294. [Google Scholar] [CrossRef]
- Buda, O.; Arsene, D.; Ceausu, M.; Dermengiu, D.; Curca, G.C. Georges Marinesco and the early research in neuropathology. Neurology 2009, 72, 88–91. [Google Scholar] [CrossRef]
- Sjogren, T.; Sjogren, H.; Lindgren, G. Morbus Alzheimer and morbus Pick; A genetic, clinical and patho-anatomical study. Acta Psychiatr. Neurol. Scand. Suppl. 1952, 82, 1–152. [Google Scholar]
- Letemendia, F.; Pampiglione, G. Clinical and electroencephalographic observations in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiat. 1958, 21, 167. [Google Scholar] [CrossRef]
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent Hippocampal Seizures and Spikes Identified by Foramen Ovale Electrodes in Alzheimer’s Disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, F.; Bruce, L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Ye, C.P.; Selkoe, D.J.; Hartley, D.M. Protofibrils of amyloid β-protein inhibit specific K+ currents in neocortical cultures. Neurobiol. Dis. 2003, 13, 177–190. [Google Scholar] [CrossRef]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O’Malley, T.T.; Zhang, D.; Walsh, D.M. Soluble Aβ Oligomers Impair Hippocampal LTP by Disrupting Glutamatergic/GABAergic Balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- Kam, K.; Duffy, Á.M.; Moretto, J.; LaFrancois, J.J.; Scharfman, H.E. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of β-amyloid neuropathology. Sci. Rep. 2016, 28, 20119. [Google Scholar] [CrossRef]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.M.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar] [CrossRef]
- Lehnert, S.; Hartmann, S.; Hessler, S.; Adelsberger, H.; Huth, T.; Alzheimer, C. Ion channel regulation by β-secretase BACE1-enzymatic and non-enzymatic effects beyond Alzheimer’s disease. Channels 2016, 10, 365–378. [Google Scholar] [CrossRef]
- Kim, D.Y.; Gersbacher, M.T.; Inquimbert, P.; Kovacs, D.M. Reduced Sodium Channel Nav1.1 Levels in BACE1-null Mice. J. Biol. Chem. 2011, 286, 8106–8116. [Google Scholar] [CrossRef] [PubMed]
- Nash, A.; Gijsen, H.J.M.; Hrupka, B.J.; Teng, K.S.-L.; Lichtenthaler, S.F.; Takeshima, H.; Gunnersen, J.M.; Munro, K.M. BACE inhibitor treatment of mice induces hyperactivity in a seizure-related gene 6 family dependent manner without altering learning and memory. Sci. Rep. 2021, 11, 15084. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer. Res. 2017, 14, 1140–1448. [Google Scholar] [CrossRef]
- Meng, F.; Yao, L. The role of inflammation in epileptogenesis. Acta Epileptol. 2020, 2, 15. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Zubareva, O.E.; Kovalenko, A.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status Epilepticus Impairs Synaptic Plasticity in Rat Hippocampus and Is Followed by Changes in Expression of NMDA Receptors. Biochemistry 2017, 82, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E.-M. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.-F.; Zeng, C.; Ma, Y.-F.; Guo, T.-H.; Chen, J.-M.; Chen, Y.; Cai, X.-F. Potential roles of Cdk5/p35 and tau protein in hippocampal mossy fiber sprouting in the PTZ kindling model. Clin. Lab. 2010, 56, 127–136. [Google Scholar] [PubMed]
- Thom, M.; Liu, J.Y.W.; Thompson, P.; Phadke, R.; Narkiewicz, M.; Martinian, L.; Marsdon, D. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: A post-mortem study. Brain 2011, 134, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-J.; Zheng, P.; Wright, D.K.; Dezsi, G.; Braine, E.; Nguyen, T.; Corcoran, N.M. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 2016, 139, 1919–1938. [Google Scholar] [CrossRef]
- Luchetti, S.; Huitinga, I.; Swaab, D.F. Neurosteroid and GABA-A receptor alterations in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis. Neuroscience 2011, 191, 6–21. [Google Scholar] [CrossRef]
- Luchetti, S.; Bossers, K.; van de Bilt, S.; Agrapart, V.; Morales, R.R.; Frajese, G.V.; Swaab, D.F. Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol. Aging. 2011, 31, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Bill, F.; Foundation, M.G. Global, regional and national burden of Parkinson’ s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 19. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Fedotova, E.I.; Abramov, A.Y.; Berezhnov, A.V. Alpha-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Biochem. Moscow Suppl. Ser. A 2018, 12, 10–19. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Sánchez, A.M.; Levine, M.S.; Cepeda, C. Epilepsy in Other Neurodegenerative Disorders: Huntington’s and Parkinson’s Diseases, 2nd ed.; Models of Seizures and Epilepsy; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Chapter 17; ISBN 9780128040669. [Google Scholar]
- Feddersen, B.; Jan Einhellig, M.R.; Stoyke, C.; Krauss, P.; Noachtar, S. Parkinson’s disease: Less epileptic seizures more status epilepticus. Epilepsy Res. 2014, 18, 349–354. [Google Scholar] [CrossRef]
- Bodenmann, P.; Ghika, J.; van Melle, G.; Bogousslavsky, J. Neurological comorbidity in parkinsonism. Rev. Neurol. 2001, 157, 45–54. [Google Scholar] [PubMed]
- Gruntz, K.; Bloechliger, M.; Becker, C.; Jick, S.S.; Fuhr, P.; Meier, C.R.; Rüegg, S. Parkinson disease and the risk of epileptic seizures. Ann. Neurol. 2018, 83, 363–374. [Google Scholar] [CrossRef]
- Clinckers, R.; Smolders, I.; Meurs, A.; Ebingerand, G.; Michotte, Y. Anticonvulsant action of hippocampal dopamine and serotoninis independently mediated by D2 and 5-HT1A receptors. J. Neurochem. 2004, 89, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Borrelli, E. The role of dopamine signaling in epileptogenesis. Front. Cell Neurosci. 2013, 7, 157. [Google Scholar] [CrossRef]
- Rong, H.; Jin, L.; Wei, W.; Wang, X.; Xi, Z. Alpha-synuclein is a potential biomarker in the serum and CSF of patients with intractable epilepsy. Seizure 2015, 27, 6–9. [Google Scholar] [CrossRef]
- Park, J.; Burgess, J.D.; Faroqi, A.H.; Demeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; Mclean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef]
- Gardoni, F.; Bellone, C. Modulation of the glutamatergic transmission by Dopamine: A focus on Parkinson, Huntington and Addiction diseases. Front. Cell Neurosci. 2015, 9, 25. [Google Scholar] [CrossRef]
- Cepeda, C.; Li, Z.; Cromwell, H.; Altemus, K.; Crawford, C.; Nansen, E.; Ariano, M. Electrophysiological and morphological analyses of cortical neurons obtained from children with catastrophic epilepsy: Dopamine receptor modulation of glutamatergic responses. Dev. Neurosci. 1999, 21, 223–235. [Google Scholar] [CrossRef]
- Buddhala, C.; Loftin, S.K.; Kuley, B.M.; Cairns, N.J.; Campbell, M.C.; Perlmutter, J.S.; Kotzbauer, P.T. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2015, 2, 949–959. [Google Scholar] [CrossRef]
- Szot, P. Common factors among Alzheimer’s disease, Parkinson’s disease, and epilepsy: Possible role of the noradrenergic nervous system. Epilepsia 2012, 53, 61–66. [Google Scholar] [CrossRef]
- Weinshenker, D.; Szot, P. The role of catecholamines in seizure susceptibility: New results using genetically engineered mice. Pharmacol. Ther. 2002, 94, 213–233. [Google Scholar] [CrossRef]
- Bixo, M.; Andersson, A.; Winblad, B.; Purdy, R.H.; Bäckström, T. Progesterone, 5alpha-pregnane-3,20-dione and 3alpha-hydroxy-5alpha-pregnane-20-one in specific regions of the human female brain in different endocrine states. Brain Res. 1997, 74, 173–178. [Google Scholar] [CrossRef]
- Di Michele, F.; Longone, P.; Romeo, E.; Lucchetti, S.; Brusa, L.; Pierantozzi, M.; Bassi, A.; Bernardi, G.; Stanzione, P. Decreased plasma and cerebrospinal fluid content of neuroactive steroids in Parkinson’s disease. Neurol. Sci. 2003, 24, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Sánchez-López, E.; Ettcheto, M.; López-Machado, A.; Espina, M.; Souto, E.B.; Galindo, R.; Camins, A.; García, M.L.; Turowski, P. Current advances in the development of novel polymeric nanoparticles for the treatment of neurodegenerative diseases. Nanomedicine 2020, 15, 1239–1261. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Espina, M.; Auladell, C.; Folch, J.; Kühne, B.A.; Barenys, M.; Sánchez-López, E.; Souto, E.B.; García, M.L.; et al. Epigallocatechin-3-gallate PEGylated poly (lactic-co-glycolic) acid nanoparticles mitigate striatal pathology and motor deficits in 3-nitropropionic acid intoxicated mice. Nanomedicine 2021, 16, 19–35. [Google Scholar] [CrossRef]
- Andrew, S.; Goldberg, Y.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 1–8. [Google Scholar] [CrossRef]
- Cloud, L.J.; Rosenblatt, A.; Margolis, R.L.; Ross, C.A.; Pillai, J.A.; Corey-Bloom, J.; Tully, H.M.; Bird, T.; Panegyres, P.K.; Nichter, C.A.; et al. Seizures in Juvenile Huntington’s Disease: Frequency and Characterization in a Multicenter Cohort. Mov. Disord. 2012, 27, 1797–1800. [Google Scholar] [CrossRef]
- Sipilä, J.O.T.; Soilu-Hänninen, M.; Majamaa, K. Comorbid epilepsy in Finnish patients with adult-onset Huntington’s disease. BMC Neurol. 2016, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sánchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J. Impaired TrkB Receptor Signaling Underlies Corticostriatal Dysfunction in Huntington’s Disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef]
- Tanaka, T.; Saito, H.; Matsuki, N. Inhibition of GABAA Synaptic Responses by Brain-Derived Neurotrophic Factor (BDNF) in Rat Hippocampus. J. Neurosci. 1997, 17, 2959–2966. [Google Scholar] [CrossRef]
- Li, Y.-X.; Zhang, Y.; Lester, H.A.; Schuman, E.M.; Davidson, N. Enhancement of Neurotransmitter Release Induced by Brain-Derived Neurotrophic Factor in Cultured Hippocampal Neurons. J. Neurosci. 1998, 18, 10231–10240. [Google Scholar] [CrossRef]
- El Bahh, B.; Balosso, S.; Hamilton, T.; Herzog, H.; Beck-Sickinger, A.G.; Sperk, G.; Gehlert, D.R. The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y2 and not Y5 receptors. Eur. J. Neurosci. 2005, 22, 1417–1430. [Google Scholar] [CrossRef]
- Falcicchia, C.; Paolone, G.; Emerich, D.F.; Lovisari, F.; Bell, W.J.; Fradet, T.; Wahlberg, L.U.; Simonato, M. Seizure-Suppressant and Neuroprotective Effects of Encapsulated BDNF-Producing Cells in a Rat Model of Temporal Lobe Epilepsy. Mol. Ther. Methods Clin. Dev. 2018, 9, 211–224. [Google Scholar] [CrossRef]
- Chen, S.; Wu, C.; Hwang, W.; Yang, D. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Levin, M.; Douglas, J.; Meyers, L.; Lee, S.; Shin, Y.; Gardner, L. Neurodegeneration in multiple sclerosis involves multiple pathogenic mechanisms. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Marrodan, M.; Ysrraelit, M.C. Mechanisms of Neurodegeneration and Axonal Dysfunction in Progressive Multiple Sclerosis. Biomedicines 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Sandi, D.; Fricska-Nagy, Z.; Bencsik, K.; Vécsei, L. Neurodegeneration in Multiple Sclerosis: Symptoms of Silent Progression, Biomarkers and Neuroprotective Therapy—Kynurenines Are Important Players. Molecules 2021, 26, 3423. [Google Scholar] [CrossRef] [PubMed]
- Bhagavati, S. Autoimmune Disorders of the Nervous System: Pathophysiology, Clinical Features, and Therapy. Front. Neurol. 2021, 12, 664664. [Google Scholar] [CrossRef]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Poser, C.M.; Brinar, V.V. Epilepsy and multiple sclerosis. Epilepsy Behav. 2003, 4, 6–12. [Google Scholar] [CrossRef]
- Vlaicu, M.B. Epilepsy in multiple sclerosis as a network disease. Mult. Scler. Relat. Disord. 2019, 36, 101390. [Google Scholar] [CrossRef]
- Uribe-San-Martín, R.; Ciampi-Díaz, E.; Suarez-Hernández, F.; Vásquez-Torres, M.; Godoy-Fernández, J.; Cárcamo-Rodríguez, C. Prevalence of epilepsy in a cohort of patients with multiple sclerosis. Seizure 2014, 23, 81–83. [Google Scholar] [CrossRef]
- Lapato, A.S.; Szu, J.; Hasselmann, J.P.C.; Khalaj, A.J.; Binder, D.K.; Tiwari-Woodruff, S.K. Chronic demyelination-induced seizures. Neuroscience 2017, 346, 409–422. [Google Scholar] [CrossRef]
- Hamada, M.S.; Kole, M.H.P. Myelin Loss and Axonal Ion Channel Adaptations Associated with Gray Matter Neuronal Hyperexcitability. J. Neurosci. 2015, 35, 7272–7286. [Google Scholar] [CrossRef]
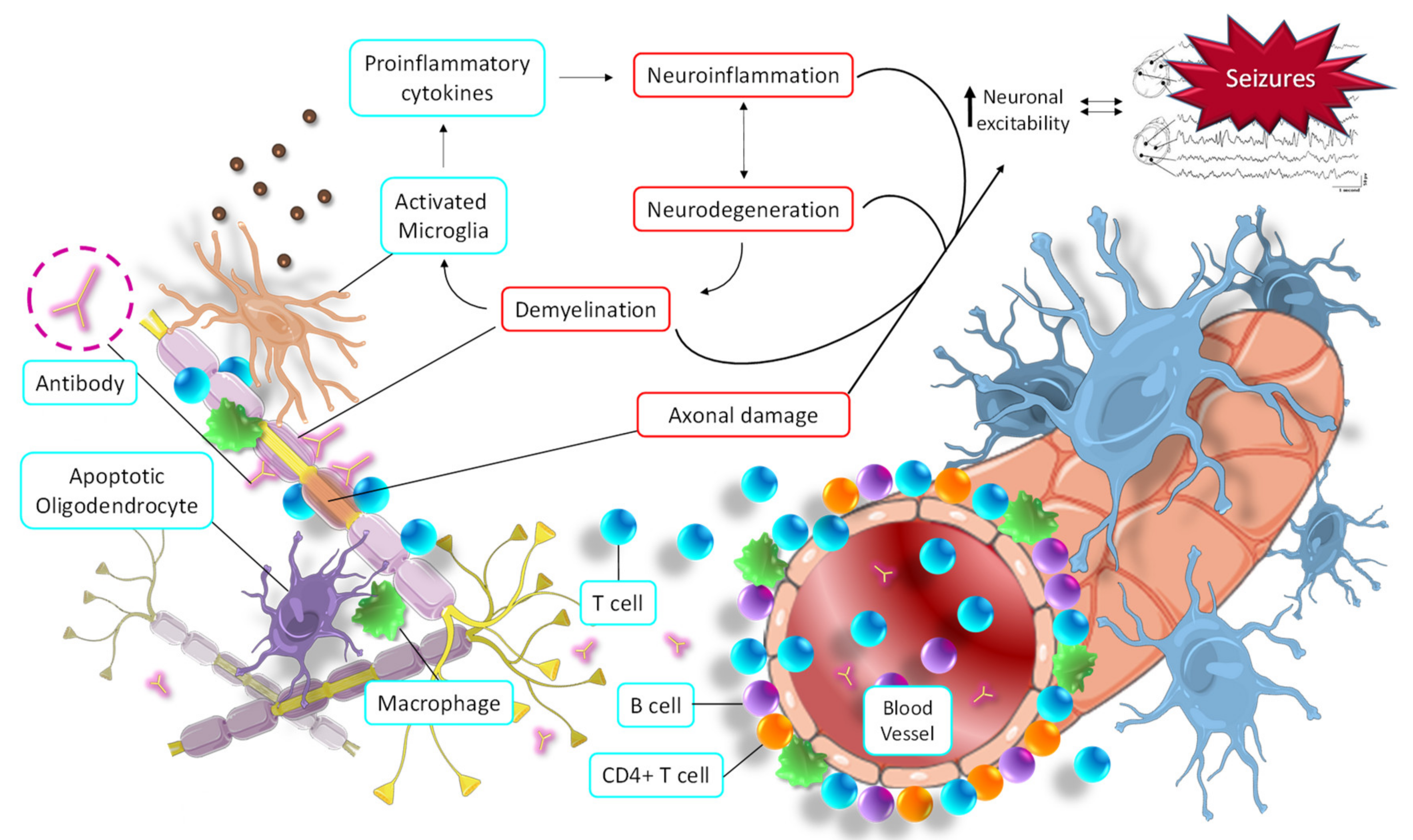
- Rayatpour, A.; Farhangi, S.; Verdaguer, E.; Olloquequi, J.; Ureña, J.; Auladell, C.; Javan, M. The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies. Pharmaceuticals 2021, 14, 1031. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134, 2703–2721. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, F.; Baker, G.B.; Power, C. Allopregnanolone and neuroinflammation: A focus on multiple sclerosis. Front. Cell Neurosci. 2014, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, M.; Biagini, G.; Avoli, M. Neurosteroids and focal epileptic disorders. Int. J. Mol. Sci. 2020, 21, 9391. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawskibc, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Dhir, A.; Macdonald, R.L.; Rogawski, M.A. Chapter 39—Mechanisms of action of antiseizure drugs. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 663–681. [Google Scholar]
- Shorvon, S.D.; Goodridge, D.M.G. Longitudinal cohort studies of the prognosis of epilepsy: Contribution of the National General Practice Study of Epilepsy and other studies. Brain 2013, 136, 3497–3510. [Google Scholar] [CrossRef] [PubMed]
- Krauss, G.L.; Klein, P.; Brandt, C.; Lee, S.K.; Milanov, I.; Milovanovic, M.; Steinhoff, B.J.; Kamin, M. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: A multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020, 19, 38–48. [Google Scholar] [CrossRef]
- Franz, D.N.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P. Everolimus for treatment-refractory seizures in TSC. Neurol. Clin. Pr. 2018, 8, 412–420. [Google Scholar] [CrossRef]
- Schulz, A.; Ajayi, T.; Specchio, N.; de los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- Beghi, E. Overview of Studies to Prevent Posttraumatic Epilepsy. Epilepsia 2003, 44, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Ura, H.; Takumi, I.; Kobayashi, S.; Maru, E.; Morita, A. The angiotensin II type I receptor antagonist losartan retards amygdala kindling-induced epileptogenesis. Brain Res. 2018, 1694, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bar-Klein, G.; Klee, R.; Brandt, C.; Bankstahl, M.; Bascuñana, P.; Töllner, K.; Dalipaj, H.; Bankstahl, J.P.; Friedman, A. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann. Neurol. 2016, 80, 896–908. [Google Scholar] [CrossRef]
- Noe, F.M.; Polascheck, N.; Frigerio, F.; Bankstahl, M.; Ravizza, T.; Marchini, S.; Beltrame, L. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 2013, 59, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2019, 142, e39. [Google Scholar] [CrossRef]
- Citraro, R.; Chimirri, S.; Aiello, R.; Gallelli, L.; Trimboli, F.; Britti, D.; de Sarro, G.; Russo, E. Protective effects of some statins on epileptogenesis and depressive-like behavior in WAG/Rij rats, a genetic animal model of absence epilepsy. Epilepsia 2014, 55, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, G.S.; Kabakov, A.Y.; Hameed, M.Q.; Dhamne, S.C.; Rosenberg, P.A.; Rotenberg, A. Ceftriaxone Treatment after Traumatic Brain Injury Restores Expression of the Glutamate Transporter, GLT-1, Reduces Regional Gliosis, and Reduces Post-Traumatic Seizures in the Rat. J. Neurotrauma 2013, 30, 1434–1441. [Google Scholar] [CrossRef]
- Pitsch, J.; Kuehn, J.C.; Gnatkovsky, V.; Müller, J.A.; van Loo, K.M.J.; de Curtis, M.; Vatter, H. Anti-epileptogenic and Anti-convulsive Effects of Fingolimod in Experimental Temporal Lobe Epilepsy. Mol. Neurobiol. 2019, 56, 1825–1840. [Google Scholar] [CrossRef]
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386. [Google Scholar] [CrossRef]
- Bertram, E. The Relevance of Kindling for Human Epilepsy. Epilepsia 2007, 48, 65–74. [Google Scholar] [CrossRef]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.; Albert, M.S.; Krauss, G.; Speck, C.L.; Gallagher, M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 2015, 7, 688–698. [Google Scholar] [CrossRef] [PubMed]
- NIH. Clinical Trials.gov: Levetirazetam & Alzheimer’s Disease. 2021. Available online: https://clinicaltrials.gov/ct2/results?cond=Alzheimer+Disease&term=levetiracetam&cntry=&state=&city=&dist= (accessed on 14 September 2021).
- Cumbo, E.; Ligori, L.D. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010, 17, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Musaeus, C.S.; Shafi, M.M.; Santarnecchi, E.; Herman, S.T.; Press, D.Z. Levetiracetam Alters Oscillatory Connectivity in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1065–1076. [Google Scholar] [CrossRef]
- Alborghetti, M.; Nicoletti, F. Different generations of Type-B monoamine oxidase inhibitors in parkinson’s disease: From bench to bedside. Curr. Neuropharmacol. 2019, 17, 861–873. [Google Scholar] [CrossRef]
- Uemura, M.T.; Asano, T.; Hikawa, R.; Yamakado, H.; Takahashi, R. Zonisamide inhibits monoamine oxidase and enhances motor performance and social activity. Neurosci. Res. 2017, 124, 25–32. [Google Scholar] [CrossRef]
- Yang, Y.C.; Tai, C.H.; Pan, M.K.; Kuo, C.C. The T-type calcium channel as a new therapeutic target for Parkinson’s disease. Pflugers Arch. 2014, 466, 747–755. [Google Scholar] [CrossRef]
- Kunisawa, N.; Shimizu, S.; Kato, M.; Iha, H.A.; Iwai, C.; Hashimura, M.; Ogawa, M.; Kawaji, S. Pharmacological characterization of nicotine-induced tremor: Responses to anti-tremor and anti-epileptic agents. J. Pharmacol. Sci. 2018, 137, 162–169. [Google Scholar] [CrossRef]
- Nishijima, H.; Miki, Y.; Ueno, S.; Tomiyama, M. Zonisamide Enhances Motor Effects of Levodopa, Not of Apomorphine, in a Rat Model of Parkinson’s Disease. Park. Dis. 2018, 2018, 8626783. [Google Scholar] [CrossRef]
- Oki, M.; Kaneko, S.; Morise, S.; Takenouchi, N.; Hashizume, T.; Tsuge, A.; Nakamura, M. Zonisamide ameliorates levodopa-induced dyskinesia and reduces expression of striatal genes in Parkinson model rats. Neurosci. Res. 2017, 122, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xue, L.; Liu, Y.; Yang, Z.; Chi, S.; Xie, A. Zonisamide for the Treatment of Parkinson Disease: A Current Update. Front. Neurosci. 2020, 14, 574652. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Hanashiro, S.; Sawada, M.; Iwasaki, Y. Preliminary study of zonisamide monotherapy in de novo patients with early Parkinson’s disease. Neurolo. Clin. Neurosci. 2015, 3, 163–166. [Google Scholar] [CrossRef][Green Version]
- Murata, M.; Hasegawa, K.; Kanazawa, I.; Fukasaka, J.; Kochi, K.; Shimazu, R. Zonisamide improves wearing-off in Parkinson’s disease: A randomized, double-blind study. Mov. Disord. 2015, 30, 1343–1350. [Google Scholar] [CrossRef]
- Murata, M.; Hasegawa, K.; Kanazawa, I.; Shirakura, K.; Kochi, K.; Shimazu, R. Randomized placebo-controlled trial of zonisamide in patients with Parkinson’s disease. Neurol. Clin. Neurosci. 2016, 4, 10–15. [Google Scholar] [CrossRef]
- Murata, M.; Horiuchi, E.; Kanazawa, I. Zonisamide has beneficial effects on Parkinson’s disease patients. Neurosci. Res. 2001, 41, 397–399. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.-C.; Ferreira, J.; Massart, R.; Youssov, K.; de Michele, G.; Rae, D.; Squitieri, F.; Seppi, K. International Guidelines for the Treatment of Huntington’ s Disease. Front. Neurol. 2019, 10, 710. [Google Scholar] [CrossRef]
- Videnovic, A. Treatment of Huntington Disease. Curr. Treat. Options Neurol. 2014, 15, 424–438. [Google Scholar] [CrossRef]
- Foley, P.L.; Vesterinen, H.M.; Laird, B.J.; Sena, E.S.; Colvin, L.A.; Chandran, S.; MacLeod, M.R.; Fallon, M.T. Prevalence and natural history of pain in adults with multiple sclerosis: Systematic review and meta-analysis. Pain 2013, 154, 632–642. [Google Scholar] [CrossRef]
- Wiffen, P.J.; Derry, S.; Moore, R.A.; Aldington, D.; Cole, P.; Rice, A.S.; Lunn, M.P. Antiepileptic drugs for neuropathic pain and fibromyalgia—An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2013, 2013, CD010567. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; Frohman, E.; Racke, M. Levetiracetam for Phasic Spasticity in Multiple Sclerosis. Arch. Neurol. 2003, 60, 1772–1774. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Du, C.; Wei, W.; Wu, Z.; Zhao, G.; Li, Z.; Xie, X. The Antiepileptic Drug Valproic Acid Restores T Cell Homeostasis and Ameliorates Pathogenesis of Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2012, 287, 28656–28665. [Google Scholar] [CrossRef]
- Solaro, C.; Restivo, D.; Mancardi, G.L.; Tanganelli, P. Oxcarbazepine for treating paroxysmal painful symptoms in multiple sclerosis: A pilot study. Neurol. Sci. 2007, 28, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Furby, J.; Hayton, T.; Smith, K.J.; Altmann, D.R.; Brenner, R.; Chataway, J. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010, 9, 681–688. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Antiseizure Drugs | Proposed Mechanisms of Action | |
|---|---|---|---|
| Voltage-gated ion channels | Na+ channels | Phenytoin, fosphenytoin, carbamazepine, oxcarbazepine, eslicarbazepine acetate, lamotrigine, lacosamide, cenobamate *, rufinamide, topiramate, zonisamide | Enhancement of the rapid/slow inactivation of Na+ channels, inhibiting the propagation of action potentials |
| Ca2+ channels | Ethosuximide | Inhibits hyperexcitability by regulating Ca2+ currents | |
| K+ channels | Retigabine (ezogabine) | Generates a subthreshold K+ current that stabilizes the membrane potential | |
| GABA-mediated inhibition | Phenobarbital, primidone, benzodiazepines, stiripentol *, topiramate, felbamate, cenobamate, retigabine (ezogabine), tiagabine, vigabatrin, acetazolamide, topiramate, zonisamide, lacosamide * | Increased synaptic inhibition and reduced glutamate activity | |
| Synaptic release machinery | SV2A | Levetiracetam, brivaracetam | Inhibition of excitatory neurotransmitter release |
| α2δ subunit of voltage-gated Ca2+ channels | Gabapentin, pregabalin | Inhibition of excitatory neurotransmitter release | |
| AMPA receptor | Perampanel | Inhibits the extracellular Ca2+ concentration and neuronal excitability | |
| Mixed/unknown | Valproate, felbamate, cenobamate, topiramate, zonisamide, rufinamide, adrenocorticotrophin, cannabidiol | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano, A.; Fonseca, E.; Ettcheto, M.; Sánchez-López, E.; de Rojas, I.; Alonso-Lana, S.; Morató, X.; Souto, E.B.; Toledo, M.; Boada, M.; et al. Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways. Pharmaceuticals 2021, 14, 1057. https://doi.org/10.3390/ph14101057
Cano A, Fonseca E, Ettcheto M, Sánchez-López E, de Rojas I, Alonso-Lana S, Morató X, Souto EB, Toledo M, Boada M, et al. Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways. Pharmaceuticals. 2021; 14(10):1057. https://doi.org/10.3390/ph14101057
Chicago/Turabian StyleCano, Amanda, Elena Fonseca, Miren Ettcheto, Elena Sánchez-López, Itziar de Rojas, Silvia Alonso-Lana, Xavier Morató, Eliana B. Souto, Manuel Toledo, Mercè Boada, and et al. 2021. "Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways" Pharmaceuticals 14, no. 10: 1057. https://doi.org/10.3390/ph14101057
APA StyleCano, A., Fonseca, E., Ettcheto, M., Sánchez-López, E., de Rojas, I., Alonso-Lana, S., Morató, X., Souto, E. B., Toledo, M., Boada, M., Marquié, M., & Ruíz, A. (2021). Epilepsy in Neurodegenerative Diseases: Related Drugs and Molecular Pathways. Pharmaceuticals, 14(10), 1057. https://doi.org/10.3390/ph14101057