
The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies


 , and
, and
Abstract
1. Introduction
2. Seizure Occurrence in the Context of Demyelinating Disorders
2.1. Seizures as A Clinical Manifestation of Multiple Sclerosis (MS)
2.2. Possible Pathophysiological Processes Underlying Seizure Development in Patients with MS
2.3. Cuprizone Induced Demyelination as a Model for Epilepsy
2.4. Common Inflammatory Molecules/Pathways Underlying Demyelination and Seizuregenesis: Role of Glial Cells

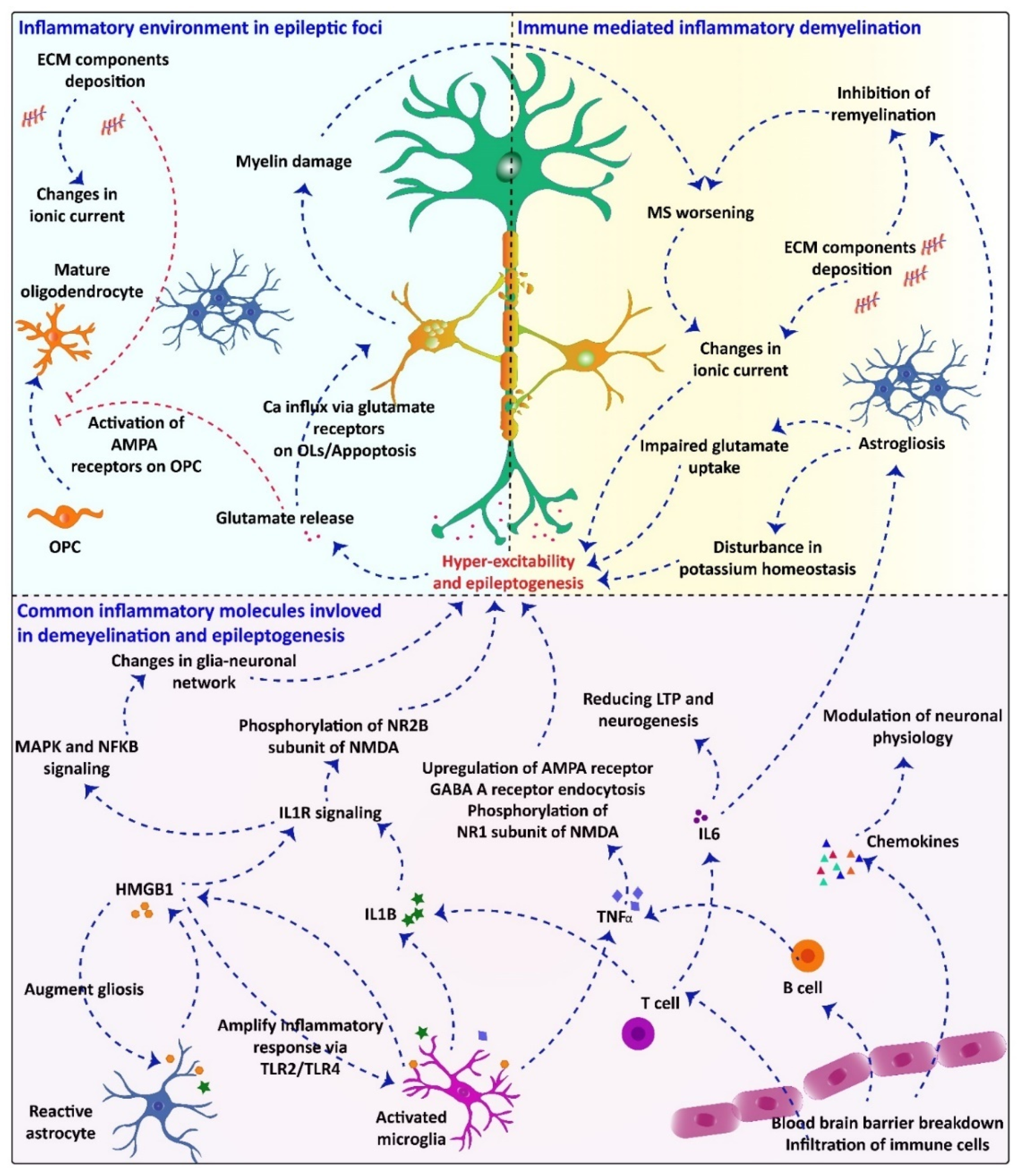{kind=link}
| Molecules/Receptor/Pathway | Source | Role in Demyelination | Role in Seizuregenesis | Ref. |
|---|---|---|---|---|
| HMGB1 | Astrocytes Microglia Neurons | Microglial proinflammatory response by production of proinflammatory factors (TNF-α, nitric oxide, interleukin-1b (IL-1b), IL-6, CXCL10 and CCL2 | Microglial activation via the TLR4/NF-κB signaling pathway Activation of IL-1R/TLR signaling in neurons Phosphorylation of the NR2B subunit of NMDA receptor, which leads to enhanced NMDA activity and Ca2+ influx into the neurons | [88,103,104] |
| TGF-β | Microglia | Induces astrogliosis Promotes Th17 cell differentiation | Astrocyte activation Inflammation Reduced inhibitory transmission | [105,106,107] |
| TLRs | Microglia | Production of IL-1, IL-6, and IL-12 Induce differentiation of naïve T cells into Th1 and Th17 cells | Hippocampal hyper-excitation via upregulating proinflammatory cytokines such as IFN-β in microglia and astrocytes Up regulation of proinflammatory cytokine such as IL1B, TNF-α, IL-6 | [91,108,109] |
| Hyaluronan/CD44 | Astrocytes/ Microglia | Activation of NFƙB to produce pro-IL1B and other proinflammatory cytokines Inhibition of OPC maturation | Hyaluronan plays a permissive role in MFS | [110,111,112] |
| mTOR | Astrocytes Microglia | Microglial proinflammatory activation Regulation of adaptive and innate immune response T cell proliferation | mTOR pathway regulation depends on glutamate receptor activation A strong link between neuronal hyper-excitability and aberrant mTOR activation | [113,114,115,116] |
| IL-1B | Macrophages T cells | Proinflammatory response through activation of IL-1R and NF-κB pathway BBB breakdown | Activation of neuronal sphingo myelinaseand Src kinases Phosphorylation of the NR2B subunit of NMDA receptor Reduction in GABA-mediated inhibition of glutamate uptake by astrocytes | [90,117,118,119,120] |
| TNF-α | Macrophages T cells B cells Neurons | Mediate apoptosis and chronic inflammation through TNFR 1 | Inhibition of glutamate uptake by astrocyte Up-regulation of AMPA receptors Phosphorylation of the NR1 subunit of the NMDA receptor Induction of GABA A receptor endocytosis | [91,106,117,121,122] |
3. White Matter Abnormalities following Epileptic Seizures
3.1. White Matter Disorders in Animal Models of Epilepsy
3.2. White Matter Alterations in Patients with Epilepsy
3.3. Pathophysiological Mechanisms Underlying White Matter Disruption in Epilepsy
3.4. Efficacy of MS and Epilepsy Modifying Drugs in Modulating Myelin Damage in the Context of MS and Epilepsy
4. Conclusions
5. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Korczyn, A.D.; Manouchehri, N.; Stüve, O. The temporal and causal relationship between inflammation and neurodegeneration in multiple sclerosis. Mult. Scler. J. 2019, 26, 876–886. [Google Scholar] [CrossRef]
- Solana, E.; Martinez-Heras, E.; Martinez-Lapiscina, E.H.; Sepulveda, M.; Sola-Valls, N.; Bargalló, N.; Berenguer, J.; Blanco, Y.; Andorra, M.; Pulido-Valdeolivas, I. Magnetic resonance markers of tissue damage related to connectivity disruption in multiple sclerosis. NeuroImage Clin. 2018, 20, 161–168. [Google Scholar] [CrossRef]
- Schoonheim, M.M.; Geurts, J.J.; Landi, D.; Douw, L.; van der Meer, M.L.; Vrenken, H.; Polman, C.H.; Barkhof, F.; Stam, C.J. Functional connectivity changes in multiple sclerosis patients: A graph analytical study of MEG resting state data. Hum. Brain Mapp. 2013, 34, 52–61. [Google Scholar] [CrossRef]
- Beghi, E. The epidemiology of epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef]
- De Boer, H.M.; Mula, M.; Sander, J. The global burden and stigma of epilepsy. Epilepsy Behav. 2008, 12, 540–546. [Google Scholar] [CrossRef]
- Jiruska, P.; de Curtis, M.; Jefferys, J.; Schevon, C.A.; Schiff, S.J.; Schindler, K. Synchronization and desynchronization in epilepsy: Controversies and hypotheses. J. Physiol. 2013, 591, 787–797. [Google Scholar] [CrossRef]
- Sirven, J.I. Epilepsy: A Spectrum Disorder. Cold Spring Harb. Perspect. Med. 2015, 5, a022848. [Google Scholar] [CrossRef]
- Löscher, W.; Brandt, C. Prevention or Modification of Epileptogenesis after Brain Insults: Experimental Approaches and Translational Research. Pharmacol. Rev. 2010, 62, 668–700. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Overview of drugs used for epilepsy and seizures: Etiology, diagnosis, and treatment. Pharm. Ther. 2010, 35, 392. [Google Scholar]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, D.-W. Focal Cortical Dysplasia and Epilepsy Surgery. J. Epilepsy Res. 2013, 3, 43–47. [Google Scholar] [CrossRef]
- Tuchman, R.; Rapin, I. Epilepsy in autism. Lancet Neurol. 2002, 1, 352–358. [Google Scholar] [CrossRef]
- Pack, A. Is There a Relationship between Multiple Sclerosis and Epilepsy? If So What Does it Tell Us about Epileptogenesis? Epilepsy Curr. 2018, 18, 95–96. [Google Scholar] [CrossRef]
- Leube, W. Ueber multiple inselförmige Sklerose des Gehirns und Rückenmarks. Nach Beobachtungen aus der Erlanger medicinischen Klinik. Dtsch. Arch. Klin. Med. 1871, 8, 1–29. [Google Scholar]
- Adan, G.; Mitchell, J.W.; Ziso, B.; Larner, A.J. Diagnosis and Management of Seizures in Neurodegenerative Diseases. Curr. Treat. Options Neurol. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Baker, J.; Libretto, T.; Henley, W.; Zeman, A. A Longitudinal Study of Epileptic Seizures in Alzheimer’s Disease. Front. Neurol. 2019, 10, 1266. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Tai, X.Y.; Koepp, M.; Duncan, J.S.; Fox, N.; Thompson, P.; Baxendale, S.; Liu, J.; Reeves, C.; Michalak, Z.; Thom, M. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: A study of temporal lobe resections. Brain 2016, 139, 2441–2455. [Google Scholar] [CrossRef]
- Doucet, G.E.; He, X.; Sperling, M.; Sharan, A.; Tracy, J.I. Gray Matter Abnormalities in Temporal Lobe Epilepsy: Relationships with Resting-State Functional Connectivity and Episodic Memory Performance. PLoS ONE 2016, 11, e0154660. [Google Scholar] [CrossRef]
- Scanlon, C.; Mueller, S.G.; Cheong, I.; Hartig, M.; Weiner, M.W.; Laxer, K.D. Grey and white matter abnormalities in temporal lobe epilepsy with and without mesial temporal sclerosis. J. Neurol. 2013, 260, 2320–2329. [Google Scholar] [CrossRef]
- Drenthen, G.S.; Backes, W.H.; Aldenkamp, A.P.; Vermeulen, R.J.; Klinkenberg, S.; Jansen, J.F. On the merits of non-invasive myelin imaging in epilepsy, a literature review. J. Neurosci. Methods 2020, 338, 108687. [Google Scholar] [CrossRef]
- Abboud, H.; Yu, X.X.; Knusel, K.; Fernandez, H.H.; Cohen, J.A. Movement disorders in early MS and related diseases: A prospective observational study. Neurol. Clin. Pract. 2019, 9, 24–31. [Google Scholar] [CrossRef]
- Calabrese, M.; De Stefano, N.; Atzori, M.; Bernardi, V.; Mattisi, I.; Barachino, L.; Rinaldi, L.; Morra, A.; McAuliffe, M.M.J.; Perini, P.; et al. Extensive cortical inflammation is associated with epilepsy in multiple sclerosis. J. Neurol. 2008, 255, 581–586. [Google Scholar] [CrossRef]
- Uribe-San-Martín, R.; Ciampi-Díaz, E.; Suarez-Hernández, F.; Vásquez-Torres, M.; Godoy-Fernández, J.; Cárcamo-Rodríguez, C. Prevalence of epilepsy in a cohort of patients with multiple sclerosis. Seizure 2014, 23, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Uyttenboogaart, M.; Polman, S.; De Keyser, J. Seizures in multiple sclerosis. Epilepsia 2008, 49, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Catenoix, H.; Marignier, R.; Ritleng, C.; Dufour, M.; Mauguiere, F.; Confavreux, C.; Vukusic, S. Multiple sclerosis and epileptic seizures. Mult. Scler. J. 2010, 17, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Reider, N.; Cohen, J.; Trojano, M.; Sorensen, P.S.; Cutter, G.; Reingold, S.; Stuve, O. A systematic review of the incidence and prevalence of sleep disorders and seizure disorders in multiple sclerosis. Mult. Scler. J. 2014, 21, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Reindl, M.; Rostasy, K. MOG antibody-associated diseases. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e60. [Google Scholar] [CrossRef]
- Hacohen, Y.; Absoud, M.; Deiva, K.; Hemingway, C.; Nytrova, P.; Woodhall, M.; Palace, J.; Wassmer, E.; Tardieu, M.; Vincent, A.; et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e81. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Messina, S.; Woodhall, M.R.; Raza, N.; Everett, R.; Roca-Fernandez, A.; Tackley, G.; Hamid, S.; Sheard, A.; Reynolds, G.; et al. Clinical presentation and prognosis in MOG-antibody disease: A UK study. Brain 2017, 140, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.H.M.; Whittam, D.; Saviour, M.; Alorainy, A.; Mutch, K.; Linaker, S.; Solomon, T.; Bhojak, M.; Woodhall, M.; Waters, P.; et al. Seizures and Encephalitis in Myelin Oligodendrocyte Glycoprotein IgG Disease vs. Aquaporin 4 IgG Disease. JAMA Neurol. 2018, 75, 65–71. [Google Scholar] [CrossRef]
- Gutman, J.M.; Kupersmith, M.; Galetta, S.; Kister, I. Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies in patients with optic neuritis and seizures. J. Neurol. Sci. 2018, 387, 170–173. [Google Scholar] [CrossRef]
- Wang, L.; Zhangbao, J.; Zhou, L.; Zhang, Y.; Li, H.; Li, Y.; Huang, Y.; Wang, M.; Lu, C.; Lu, J.; et al. Encephalitis is an important clinical component of myelin oligodendrocyte glycoprotein antibody associated demyelination: A single-center cohort study in Shanghai, China. Eur. J. Neurol. 2019, 26, 168–174. [Google Scholar] [CrossRef]
- Foiadelli, T.; Gastaldi, M.; Scaranzin, S.; Franciotta, D.; Savasta, S. Seizures and myelin oligodendrocyte glycoprotein (MOG) antibodies: Two paradigmatic cases and a review of the literature. Mult. Scler. Relat. Disord. 2020, 41, 102011. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; O’Grady, G.L.; Malone, S.; Spooner, C.G.; Brown, D.A.; Gill, D.; Brilot, F.; Dale, R.C. Isolated seizures during the first episode of relapsing myelin oligodendrocyte glycoprotein antibody-associated demyelination in children. Dev. Med. Child Neurol. 2019, 61, 610–614. [Google Scholar] [CrossRef]
- Höftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Frontal Lobes 2018, 145, 263–283. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; De Seze, J.; Fujihara, K.; Greenberg, B.M.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Zhong, X.; Zhou, Y.; Chang, Y.; Wang, J.; Shu, Y.; Sun, X.; Peng, L.; Lau, A.; Kermode, A.G.; Qiu, W. Seizure and Myelin Oligodendrocyte Glycoprotein Antibody-Associated Encephalomyelitis in a Retrospective Cohort of Chinese Patients. Front. Neurol. 2019, 10, 415. [Google Scholar] [CrossRef]
- Lima, M.A.; Drislane, F.W.; Koralnik, I.J. Seizures and their outcome in progressive multifocal leukoencephalopathy. Neurology 2006, 66, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Orefice, G.; Morra, V.B.; Boccella, P.; Sarappa, C.; Lanzillo, R.; Vacca, G. Epileptic seizures in multiple sclerosis: Clinical and EEG correlations. Neurol. Sci. 2003, 24, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lapiscina, E.H.; Ayuso, T.; Lacruz, F.; Gurtubay, I.G.; Soriano, G.; Otano, M.; Bujanda, M.; Bacaicoa, M.C. Cortico-juxtacortical involvement increases risk of epileptic seizures in multiple sclerosis. Acta Neurol. Scand. 2013, 128, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Castellaro, M.; Bertoldo, A.; De Luca, A.; Pizzini, F.B.; Ricciardi, G.; Pitteri, M.; Zimatore, S.; Magliozzi, R.; Benedetti, M.D.; et al. Epilepsy in multiple sclerosis: The role of temporal lobe damage. Mult. Scler. J. 2016, 23, 473–482. [Google Scholar] [CrossRef]
- Nicholas, R.; Magliozzi, R.; Campbell, G.; Mahad, D.; Reynolds, R. Temporal lobe cortical pathology and inhibitory GABA interneuron cell loss are associated with seizures in multiple sclerosis. Mult. Scler. J. 2015, 22, 25–35. [Google Scholar] [CrossRef]
- Cao, G.; Edden, R.A.E.; Gao, F.; Li, H.; Gong, T.; Chen, W.; Liu, X.; Wang, G.; Zhao, B. Reduced GABA levels correlate with cognitive impairment in patients with relapsing-remitting multiple sclerosis. Eur. Radiol. 2018, 28, 1140–1148. [Google Scholar] [CrossRef]
- Chen, C.; Liu, C.; Fang, L.; Zou, Y.; Ruan, H.; Wang, Y.; Cui, C.; Sun, X.; Peng, L.; Qiu, W. Different magnetic resonance imaging features between MOG antibody- and AQP4 antibody-mediated disease: A Chinese cohort study. J. Neurol. Sci. 2019, 405, 116430. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Zheng, Y.; Cai, M.-T.; Yang, F.; Fang, W.; Zhang, Y.-X.; Ding, M.-P. Seizure occurrence in myelin oligodendrocyte glycoprotein antibody-associated disease: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2020, 42, 102057. [Google Scholar] [CrossRef]
- Verkman, A.S.; Phuan, P.-W.; Asavapanumas, N.; Tradtrantip, L. Biology of AQP4 and Anti-AQP4 Antibody: Therapeutic Implications for NMO. Brain Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef]
- Schorner, A.; Weissert, R. Patients with Epileptic Seizures and Multiple Sclerosis in a Multiple Sclerosis Center in Southern Germany Between 2003–2015. Front. Neurol. 2019, 10, 613. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.; Zelano, J. Epilepsy in multiple sclerosis: A nationwide population-based register study. Neurology 2017, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Juárez, I.E.; Lopez-Meza, E.; González-Aragón, M.D.C.F.; Ramírez-Bermúdez, J.; Corona, T. Epilepsy and multiple sclerosis: Increased risk among progressive forms. Epilepsy Res. 2009, 84, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Hussona, M.A.; Kearney, H.; Fisher, A.; Lynch, J.; Looby, S.; Delanty, N. New onset seizures as a sole clinical presentation of multiple sclerosis. Mult. Scler. J. 2018, 25, 295–299. [Google Scholar] [CrossRef]
- Durmus, H.; Kurtuncu, M.; Tuzun, E.; Pehlivan, M.; Akman-Demir, G.; Yapıcı, Z.; Eraksoy, M. Comparative clinical characteristics of early- and adult-onset multiple sclerosis patients with seizures. Acta Neurol. Belg. 2013, 113, 421–426. [Google Scholar] [CrossRef]
- Benjaminsen, E.; Myhr, K.-M.; Alstadhaug, K.B. The prevalence and characteristics of epilepsy in patients with multiple sclerosis in Nordland county, Norway. Seizure 2017, 52, 131–135. [Google Scholar] [CrossRef]
- Calabrese, M.; Favaretto, A.; Martini, V.; Gallo, P. Grey matter lesions in MS: From histology to clinical implications. Prion 2013, 7, 20–27. [Google Scholar] [CrossRef]
- Geurts, J.J.; Barkhof, F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008, 7, 841–851. [Google Scholar] [CrossRef]
- Calabrese, M.; Grossi, P.; Favaretto, A.; Romualdi, C.; Atzori, M.; Rinaldi, F.; Perini, P.; Saladini, M.; Gallo, P. Cortical pathology in multiple sclerosis patients with epilepsy: A 3 year longitudinal study. J. Neurol. Neurosurg. Psychiatry 2011, 83, 49–54. [Google Scholar] [CrossRef]
- Thompson, A.; Kermode, A.G.; Moseley, I.F.; MacManus, D.; McDonald, W.I. Seizures due to multiple sclerosis: Seven patients with MRI correlations. J. Neurol. Neurosurg. Psychiatry 1993, 56, 1317–1320. [Google Scholar] [CrossRef]
- Zoupi, L.; Booker, S.A.; Eigel, D.; Werner, C.; Kind, P.C.; Spires-Jones, T.L.; Newland, B.; Williams, A.C. Selective vulnerability of inhibitory networks in multiple sclerosis. Acta Neuropathol. 2021, 141, 415–429. [Google Scholar] [CrossRef]
- Folbergrová, J.; Kunz, W.S. Mitochondrial dysfunction in epilepsy. Mitochondrion 2012, 12, 35–40. [Google Scholar] [CrossRef]
- Waxman, S.G. Acquired channelopathies in nerve injury and MS. Neurology 2001, 56, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Sontheimer, H. Glia as drivers of abnormal neuronal activity. Nat. Neurosci. 2016, 19, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Kesterson, J.W.; Carlton, W.W. Aqueductal stenosis as the cause of hydrocephalus in mice fed the substituted hydrazine, cuprizone. Exp. Mol. Pathol. 1970, 13, 281–294. [Google Scholar] [CrossRef]
- Kesterson, J.W.; Carlton, W.W. Cuprizone toxicosis in mice—Attempts to antidote the toxicity. Toxicol. Appl. Pharmacol. 1972, 22, 6–13. [Google Scholar] [CrossRef]
- Hoffmann, K.; Lindner, M.; Gröticke, I.; Stangel, M.; Löscher, W. Epileptic seizures and hippocampal damage after cuprizone-induced demyelination in C57BL/6 mice. Exp. Neurol. 2008, 210, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Lapato, A.; Szu, J.I.; Hasselmann, J.; Khalaj, A.J.; Binder, D.K.; Tiwari-Woodruff, S.K. Chronic demyelination-induced seizures. Neuroscience 2017, 346, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Micheva, K.D.; Wolman, D.; Mensh, B.D.; Pax, E.; Buchanan, J.; Smith, S.J.; Bock, D.D. A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. eLife 2016, 5, e15784. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Martinez, G.A.G.; Sharma, A.; Franklin, R.J. The role of astrocytes in remyelination. Trends Neurosci. 2020, 43, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Yeo, S.-I.; Ryu, H.J.; Kim, M.-J.; Kim, D.-S.; Jo, S.-M.; Kang, T.-C. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2010, 518, 4612–4628. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Nagelhus, E.A.; Ottersen, O.P. Aquaporin-4 and epilepsy. Glia 2012, 60, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2010, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Wekerle, H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: From pipe dreams to (therapeutic) pipelines. Proc. Natl. Acad. Sci. USA 2004, 101, 14599–14606. [Google Scholar] [CrossRef] [PubMed]
- Salou, M.; Nicol, B.; Garcia, A.; Laplaud, D.-A. Involvement of CD8+ T Cells in Multiple Sclerosis. Front. Immunol. 2015, 6, 604. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.-A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Lassmann, H.; Van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.-I.; Kim, S.U. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.-F. Understanding the neurobiology of CD200 and the CD200 receptor: A therapeutic target for controlling inflammation in human brains? Futur. Neurol. 2013, 8, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Teng, J.; Miao, W. HMGB1 mediates microglia activation via the TLR4/NF-κB pathway in coriaria lactone induced epilepsy. Mol. Med. Rep. 2018, 17, 5125–5131. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Abraham, C.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Vezzani, A.; Baram, T.Z. New Roles for Interleukin-1 Beta in the Mechanisms of Epilepsy. Epilepsy Curr. 2007, 7, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocr. 2012, 33, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, D.V.; Barish, M.E. Regulation of the hyperpolarization-activated cationic current Ih in mouse hippocampal pyramidal neurones by vitronectin, a component of extracellular matrix. J. Physiol. 2004, 560, 659–675. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vasilyev, D.V.; Barish, M.E. Regulation of an inactivating potassium current (IA) by the extracellular matrix protein vitronectin in embryonic mouse hippocampal neurones. J. Physiol. 2003, 547, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Avoli, M.; Louvel, J.; Pumain, R.; Köhling, R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog. Neurobiol. 2005, 77, 166–200. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. HCN channels: Function and clinical implications. Neurology 2013, 80, 304–310. [Google Scholar] [CrossRef]
- Catterall, W.A. Sodium Channel Mutations and Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Oxford University Press (OUP): Bethesda, MA, USA, 2012; pp. 675–687. [Google Scholar]
- Henshall, D.C.; Kobow, K. Epigenetics and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022731. [Google Scholar] [CrossRef]
- Eyo, U.; Murugan, M.; Wu, L.-J. Microglia-Neuron Communication in Epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Löscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58, 27–38. [Google Scholar] [CrossRef]
- Librizzi, L.; Noè, F.; Vezzani, A.; de Curtis, M.; Ravizza, T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann. Neurol. 2012, 72, 82–90. [Google Scholar] [CrossRef]
- Fabene, P.F.; Mora, G.N.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.M. Chronic inflammation in multiple sclerosis—Seeing what was always there. Nat. Rev. Neurol. 2019, 15, 582–593. [Google Scholar] [CrossRef]
- Block, M.L. Modulating mighty microglia. Nat. Chem. Biol. 2014, 10, 988–989. [Google Scholar] [CrossRef]
- Zurolo, E.; Iyer, A.; Maroso, M.; Carbonell, C.; Anink, J.J.; Ravizza, T.; Fluiter, K.; Spliet, W.G.M.; Van Rijen, P.C.; Vezzani, A.; et al. Activation of toll-like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 2011, 134, 1015–1032. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef]
- Göbel, K.; Ruck, T.; Meuth, S.G. Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. J. 2018, 24, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, S.; Guo, Z.; Bi, Y.; Zhou, M.; Li, P.; Seyedsadr, M.; Xu, X.; Li, J.-L.; Markovic-Plese, S.; et al. The TGF-β superfamily cytokine Activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation. Immunity 2021, 54, 308–323.e6. [Google Scholar] [CrossRef]
- Costello, D.A.; Lynch, M.A. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-β. Hippocampus 2013, 23, 696–707. [Google Scholar] [CrossRef]
- Miranda-Hernandez, S.; Baxter, A.G. Role of toll-like receptors in multiple sclerosis. Am. J. Clin. Exp. Immunol. 2013, 2, 75–93. [Google Scholar] [PubMed]
- Bausch, S. Potential roles for hyaluronan and CD44 in kainic acid-induced mossy fiber sprouting in organotypic hippocampal slice cultures. Neuroscience 2006, 143, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Hauser-Kawaguchi, A.; Luyt, L.G.; Turley, E. Design of peptide mimetics to block pro-inflammatory functions of HA fragments. Matrix Biol. 2019, 78–79, 346–356. [Google Scholar] [CrossRef]
- Back, S.A.; Tuohy, T.M.F.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Russo, C.D.; Lisi, L.; Tringali, G.; Navarra, P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem. Pharmacol. 2009, 78, 1242–1251. [Google Scholar] [CrossRef]
- Russo, C.D.; Lisi, L.; Feinstein, D.L.; Navarra, P. mTOR kinase, a key player in the regulation of glial functions: Relevance for the therapy of multiple sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-H.; Rensing, N.R.; Wong, M. The Mammalian Target of Rapamycin Signaling Pathway Mediates Epileptogenesis in a Model of Temporal Lobe Epilepsy. J. Neurosci. 2009, 29, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Avruch, J. Glutamatergic Regulation of the p70S6 Kinase in Primary Mouse Neurons. J. Biol. Chem. 2005, 280, 38121–38124. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine Effects on Glutamate Uptake by Human Astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Roseti, C.; van Vliet, E.; Cifelli, P.; Ruffolo, G.; Baayen, J.C.; Di Castro, M.A.; Bertollini, C.; Limatola, C.; Aronica, E.; Vezzani, A.; et al. GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: Implications for ictogenesis. Neurobiol. Dis. 2015, 82, 311–320. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.-L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1β Regulates Blood-Brain Barrier Permeability via Reactivation of the Hypoxia-Angiogenesis Program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Edelson, B.T. New Insights into the Role of IL-1β in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. J. Immunol. 2017, 198, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef]
- Wheeler, D.; Knapp, E.; Bandaru, V.V.; Wang, Y.; Knorr, D.; Poirier, C.; Mattson, M.P.; Geiger, J.D.; Haughey, N.J. TNFα-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J. Neurochem. 2009, 109, 1237–1249. [Google Scholar] [CrossRef]
- Kim, J.H.; Bin Kim, J.; Suh, S.-I.; Kim, D.W. Subcortical grey matter changes in juvenile myoclonic epilepsy. NeuroImage Clin. 2018, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Mathern, G.W. Animal Model Development Based on the Human Epilepsies: Which Causes and Syndromes Should Be Modeled? In Models of Seizures and Epilepsy; Elsevier BV: Amsterdam, The Netherlands, 2006; pp. 653–658. [Google Scholar]
- Zare, M.; Nazari, M.; Shojaei, A.; Raoufy, M.R.; Mirnajafi-Zadeh, J. Online analysis of local field potentials for seizure detection in freely moving rats. Iran J. Basic Med. Sci. 2020, 23, 173–177. [Google Scholar] [PubMed]
- Ghasemi-Kasman, M.; Baharvand, H.; Javan, M. Enhanced neurogenesis in degenerated hippocampi following pretreatment with miR-302/367 expressing lentiviral vector in mice. Biomed. Pharmacother. 2017, 96, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xiong, J.; Hu, J.; Kong, M.; Cheng, L.; Chen, H.; Li, T.; Jiang, L. Altered hippocampal myelinated fiber integrity in a lithium-pilocarpine model of temporal lobe epilepsy: A histopathological and stereological investigation. Brain Res. 2013, 1522, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-J.; Han, W.; He, R.; Li, T.-Y.; Xie, L.-L.; Cheng, L.; Chen, H.-S.; Jiang, L. Alterations of Hippocampal Myelin Sheath and Axon Sprouting by Status Convulsion and Regulating Lingo-1 Expression with RNA Interference in Immature and Adult Rats. Neurochem. Res. 2018, 43, 721–735. [Google Scholar] [CrossRef]
- Otte, W.M.; Bielefeld, P.; Dijkhuizen, R.M.; Braun, K.P. Focal neocortical epilepsy affects hippocampal volume, shape, and structural integrity: A longitudinal MRI and immunohistochemistry study in a rat model. Epilepsia 2012, 53, 1264–1273. [Google Scholar] [CrossRef]
- You, Y.; Bai, H.; Wang, C.; Chen, L.-W.; Liu, B.; Zhang, H.; Gao, G.-D. Myelin damage of hippocampus and cerebral cortex in rat pentylenetetrazol model. Brain Res. 2011, 1381, 208–216. [Google Scholar] [CrossRef]
- Sherafat, M.A.; Ronaghi, A.; Ahmad-Molaei, L.; Nejadhoseynian, M.; Ghasemi, R.; Hosseini, A.; Naderi, N.; Motamedi, F. Kindling-induced learning deficiency and possible cellular and molecular involved mechanisms. Neurol. Sci. 2012, 34, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Laitinen, T.; Lehtimäki, K.; Rieppo, L.; Pitkänen, A.; Gröhn, O. Diffusion tensor MRI with tract-based spatial statistics and histology reveals undiscovered lesioned areas in kainate model of epilepsy in rat. Brain Struct. Funct. 2011, 216, 123–135. [Google Scholar] [CrossRef]
- Sharma, P.; Powell, K.L.; Wlodek, M.E.; O’Brien, T.J.; Gilby, K.L. Delayed myelination and neurodevelopment in male seizure-prone versus seizure-resistant rats. Epilepsia 2018, 59, 753–764. [Google Scholar] [CrossRef]
- Danış, Ö.; Demir, S.; Günel, A.; Aker, R.G.; Gülçebi, M.; Onat, F.; Ogan, A. Changes in intracellular protein expression in cortex, thalamus and hippocampus in a genetic rat model of absence epilepsy. Brain Res. Bull. 2011, 84, 381–388. [Google Scholar] [CrossRef]
- Sharma, P.; Wright, D.; Johnston, L.A.; Powell, K.; Wlodek, M.; Shultz, S.R.; O’Brien, T.; Gilby, K.L. Differences in white matter structure between seizure prone (FAST) and seizure resistant (SLOW) rat strains. Neurobiol. Dis. 2017, 104, 33–40. [Google Scholar] [CrossRef]
- Sharma, P.; Dedeurwaerdere, S.; Vandenberg, M.A.; Fang, K.; Johnston, L.A.; Shultz, S.R.; O’Brien, T.J.; Gilby, K.L. Neuroanatomical differences in FAST and SLOW rat strains with differential vulnerability to kindling and behavioral comorbidities. Epilepsy Behav. 2016, 65, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Chahboune, H.; Mishra, A.; DeSalvo, M.; Staib, L.; Purcaro, M.; Scheinost, D.; Papademetris, X.; Fyson, S.; Lőrincz, M.; Crunelli, V.; et al. DTI abnormalities in anterior corpus callosum of rats with spike–wave epilepsy. NeuroImage 2009, 47, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Unterberger, I.; Bauer, R.; Walser, G.; Bauer, G. Corpus callosum and epilepsies. Seizure 2016, 37, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Inui, T.; Yamamura, T.; Yuasa, H.; Kawai, Y.; Okaniwa, A.; Serikawa, T.; Yamada, J. The spontaneously epileptic rat (SER), a zitter* tremor double mutant rat: Histopathological findings in the central nervous system. Brain Res. 1990, 517, 123–133. [Google Scholar] [CrossRef]
- Hopkins, K.J.; Wang, G.-J.; Schmued, L.C. Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res. 2000, 864, 69–80. [Google Scholar] [CrossRef]
- Aung, W.Y.; Mar, S.; Benzinger, T.L. Diffusion tensor MRI as a biomarker in axonal and myelin damage. Imaging Med. 2013, 5, 427–440. [Google Scholar] [CrossRef]
- Salmenpera, T.M.; Simister, R.J.; Bartlett, P.; Symms, M.R.; Boulby, P.A.; Free, S.L.; Barker, G.J.; Duncan, J.S. High-resolution diffusion tensor imaging of the hippocampus in temporal lobe epilepsy. Epilepsy Res. 2006, 71, 102–106. [Google Scholar] [CrossRef]
- Assaf, B.A.; Mohamed, F.B.; Abou-Khaled, K.J.; Williams, J.M.; Yazeji, M.S.; Haselgrove, J.; Faro, S.H. Diffusion Tensor Imaging of the Hippocampal Formation in Temporal Lobe Epilepsy. Am. J. Neuroradiol. 2003, 24, 1857–1862. [Google Scholar] [PubMed]
- De La Roque, A.D.; Oppenheim, C.; Chassoux, F.; Rodrigo, S.; Daumas-Duport, C.; Devaux, B. Diffusion tensor imaging of partial intractable epilepsy. Eur. Radiol. 2004, 15, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riley, J.D.; Juranek, J.; Cramer, S.C. Vulnerability of the frontal-temporal connections in temporal lobe epilepsy. Epilepsy Res. 2008, 82, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Coste, S.; Ryvlin, P.; Hermier, M.; Ostrowsky, K.; Adeleine, P.; Froment, J.C.; Mauguiere, F. Temporopolar changes in temporal lobe epilepsy: A quantitative MRI-based study. Neurology 2002, 59, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Otte, W.M.; Van Eijsden, P.; Sander, J.; Duncan, J.S.; Dijkhuizen, R.; Braun, K.P.J. A meta-analysis of white matter changes in temporal lobe epilepsy as studied with diffusion tensor imaging. Epilepsia 2012, 53, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Seidenberg, M.; Kelly, K.G.; Parrish, J.; Geary, E.; Dow, C.; Rutecki, P.; Hermann, B. Ipsilateral and Contralateral MRI Volumetric Abnormalities in Chronic Unilateral Temporal Lobe Epilepsy and their Clinical Correlates. Epilepsia 2005, 46, 420–430. [Google Scholar] [CrossRef]
- McMillan, A.B.; Hermann, B.P.; Johnson, S.C.; Hansen, R.R.; Seidenberg, M.; Meyerand, M.E. Voxel-based morphometry of unilateral temporal lobe epilepsy reveals abnormalities in cerebral white matter. NeuroImage 2004, 23, 167–174. [Google Scholar] [CrossRef]
- Ahmadi, M.E.; Hagler, D.J.; McDonald, C.R.; Tecoma, E.S.; Iragui, V.J.; Dale, A.M.; Halgren, E. Side Matters: Diffusion Tensor Imaging Tractography in Left and Right Temporal Lobe Epilepsy. Am. J. Neuroradiol. 2009, 30, 1740–1747. [Google Scholar] [CrossRef]
- Kemmotsu, N.; Girard, H.M.; Bernhardt, B.C.; Bonilha, L.; Lin, J.J.; Tecoma, E.S.; Iragui, V.J.; Hagler, D.J., Jr.; Halgren, E.; McDonald, C.R. MRI analysis in temporal lobe epilepsy: Cortical thinning and white matter disruptions are related to side of seizure onset. Epilepsia 2011, 52, 2257–2266. [Google Scholar] [CrossRef]
- Concha, L.; Beaulieu, C.; Wheatley, B.M.; Gross, D.W. Bilateral White Matter Diffusion Changes Persist after Epilepsy Surgery. Epilepsia 2007, 48, 931–940. [Google Scholar] [CrossRef]
- Gross, D.W.; Concha, L.; Beaulieu, C. Extratemporal White Matter Abnormalities in Mesial Temporal Lobe Epilepsy Demonstrated with Diffusion Tensor Imaging. Epilepsia 2006, 47, 1360–1363. [Google Scholar] [CrossRef]
- Diniz, P.B.; Salmon, C.E.G.; Velasco, T.; Sakamoto, A.C.; Leite, J.P.; Santos, A.C. Diffusivity alterations in Temporal Lobe Epilepsy. Proc. Int. Soc. Mag. Reson. Med. 2011, 19, 4225. [Google Scholar]
- Corrêa, D.G.; Ventura, N.; Zimmermann, N.; Doring, T.M.; Tukamoto, G.; Leme, J.; Pereira, M.; D’Andrea, I.; Rêgo, C.; Alves-Leon, S.V.; et al. Evaluation of deep gray matter volume, cortical thickness and white matter integrity in patients with typical absence epilepsy: A study using voxelwise-based techniques. Neuroradiology 2017, 59, 237–245. [Google Scholar] [CrossRef]
- Pulsipher, D.T.; Seidenberg, M.; Morton, J.J.; Geary, E.; Parrish, J.; Hermann, B. MRI volume loss of subcortical structures in unilateral temporal lobe epilepsy. Epilepsy Behav. 2007, 11, 442–449. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, R.; Wehner, T.; LaPresto, E.; Ping, L.; Tkach, J.; Noachtar, S.; Diehl, B. Differences in corpus callosum volume and diffusivity between temporal and frontal lobe epilepsy. Epilepsy Behav. 2010, 19, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Chang, K.H.; Suh, D.C.; Cheon, J.E.; Jeong, S.W.; Han, M.H.; Lee, S.K. Focal lesion in the splenium of the corpus callosum in epileptic patients: Antiepileptic drug toxicity? Am. J. Neuroradiol. 1999, 20, 125–129. [Google Scholar] [PubMed]
- Oster, J.; Doherty, C.; Grant, P.E.; Simon, M.; Cole, A.J. Diffusion-weighted Imaging Abnormalities in the Splenium after Seizures. Epilepsia 2003, 44, 852–854. [Google Scholar] [CrossRef]
- Atkinson, D.S., Jr.; Abou-Khalil, B.; Charles, P.D.; Welch, L. Midsagittal corpus callosum area, intelligence and language in epilepsy. J. Neuroimaging 1996, 6, 235–239. [Google Scholar] [CrossRef]
- Hermann, B.; Hansen, R.; Seidenberg, M.; Magnotta, V.; O’Leary, D. Neurodevelopmental vulnerability of the corpus callosum to childhood onset localization-related epilepsy. NeuroImage 2003, 18, 284–292. [Google Scholar] [CrossRef]
- Riley, J.D.; Franklin, D.L.; Choi, V.; Kim, R.C.; Binder, D.K.; Cramer, S.C.; Lin, J.J. Altered white matter integrity in temporal lobe epilepsy: Association with cognitive and clinical profiles. Epilepsia 2010, 51, 536–545. [Google Scholar] [CrossRef]
- Hutchinson, E.; Pulsipher, D.; Dabbs, K.; Gutierrez, A.M.Y.; Sheth, R.; Jones, J.; Seidenberg, M.; Meyerand, E.; Hermann, B. Children with new-onset epilepsy exhibit diffusion abnormalities in cerebral white matter in the absence of volumetric differences. Epilepsy Res. 2010, 88, 208–214. [Google Scholar] [CrossRef]
- Arfanakis, K.; Hermann, B.P.; Rogers, B.; Carew, J.D.; Seidenberg, M.; Meyerand, M.E. Diffusion tensor MRI in temporal lobe epilepsy. Magn. Reson. Imaging 2002, 20, 511–519. [Google Scholar] [CrossRef]
- Weber, B.; Luders, E.; Faber, J.; Richter, S.; Quesada, C.M.; Urbach, H.; Thompson, P.M.; Toga, A.W.; Elger, C.E.; Helmstaedter, C. Distinct regional atrophy in the corpus callosum of patients with temporal lobe epilepsy. Brain 2007, 130, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, E.; Kis, A.; Go, C.; Raybaud, C.; Snead, O.; Smith, M. Abnormal white matter on diffusion tensor imaging in children with new-onset seizures. Epilepsy Res. 2013, 104, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, D.; Go, C.; Rutka, J.T.; Rydenhag, B.; Mabbott, D.J.; Snead III, O.C.; Raybaud, C.R.; Widjaja, E. Bilateral diffusion tensor abnormalities of temporal lobe and cingulate gyrus white matter in children with temporal lobe epilepsy. Epilepsy Res. 2008, 81, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Hermann, B.; Seidenberg, M.; Bell, B.; Rutecki, P.; Sheth, R.; Ruggles, K.; Wendt, G.; O’Leary, D.; Magnotta, V. The Neurodevelopmental Impact of Childhood-onset Temporal Lobe Epilepsy on Brain Structure and Function. Epilepsia 2002, 43, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.-Y.; Gu, R.; Qu, H.; Li, M.; Chen, L.; Liu, R.; Yuan, P. The relationship between the occurrence of intractable epilepsy with glial cells and myelin sheath—An experimental study. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4516–4524. [Google Scholar]
- Luo, Y.; Hu, Q.; Zhang, Q.; Hong, S.; Tang, X.; Cheng, L.; Jiang, L. Alterations in hippocampal myelin and oligodendrocyte precursor cells during epileptogenesis. Brain Res. 2015, 1627, 154–164. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Chen, H.; Cheng, L.; Jiang, L. Influence of the epileptiform discharge microenvironment on the differentiation of oligodendrocyte precursor cells. Brain Res. 2017, 1679, 53–63. [Google Scholar] [CrossRef]
- Lin, T.-K.; Chen, S.-D.; Lin, K.-J.; Chuang, Y.-C. Seizure-Induced Oxidative Stress in Status Epilepticus: Is Antioxidant Beneficial? Antioxidants 2020, 9, 1029. [Google Scholar] [CrossRef]
- Vezzani, A.; Granata, T. Brain Inflammation in Epilepsy: Experimental and Clinical Evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Mattson, M.P. Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 125–134. [Google Scholar]
- Matute, C.; Alberdi, E.; Domercq, M.; Gomez, M.V.S.; Samartin, A.L.P.; Rodríguez-Antigüedad, A.; Cerda, F.P. Excitotoxic damage to white matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef]
- De Lanerolle, N.C.; Kim, J.H.; Williamson, A.; Spencer, S.S.; Zaveri, H.P.; Eid, T.; Spencer, D.D. A Retrospective Analysis of Hippocampal Pathology in Human Temporal Lobe Epilepsy: Evidence for Distinctive Patient Subcategories. Epilepsia 2003, 44, 677–687. [Google Scholar] [CrossRef]
- Tasch, E.; Cendes, F.; Li, L.; Dubeau, F.; Andermann, F.; Arnold, D.L. Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann. Neurol. 1999, 45, 568–576. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and How Do Seizures Kill Neurons, and Is Cell Death Relevant to Epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122. [Google Scholar] [CrossRef]
- Barres, B.; Jacobson, M.; Schmid, R.; Sendtner, M.; Raff, M. Does oligodendrocyte survival depend on axons? Curr. Biol. 1993, 3, 489–497. [Google Scholar] [CrossRef]
- Fernandez, P.-A.; Tang, D.; Cheng, L.; Prochiantz, A.; Mudge, A.W.; Raff, M.C. Evidence that Axon-Derived Neuregulin Promotes Oligodendrocyte Survival in the Developing Rat Optic Nerve. Neuron 2000, 28, 81–90. [Google Scholar] [CrossRef]
- You, Y.; Zhao, Y.; Bai, H.; Liu, Z.; Meng, F.; Zhang, H.; Xu, R. Glatiramer acetate, an anti-demyelination drug, reduced rats’ epileptic seizures induced by pentylenetetrazol via protection of myelin sheath. Eur. J. Pharm. Sci. 2013, 49, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.; Baharvand, H.; Javan, M. Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015, 311, 34–44. [Google Scholar] [CrossRef]
- Gol, M.; Ghorbanian, D.; Hassanzadeh, S.; Javan, M.; Mirnajafi-Zadeh, J.; Ghasemi-Kasman, M. Fingolimod enhances myelin repair of hippocampus in pentylenetetrazol-induced kindling model. Eur. J. Pharm. Sci. 2017, 96, 72–83. [Google Scholar] [CrossRef]
- Gao, F.; Liu, Y.; Li, X.; Wang, Y.; Wei, D.; Jiang, W. Fingolimod (FTY720) inhibits neuroinflammation and attenuates spontaneous convulsions in lithium-pilocarpine induced status epilepticus in rat model. Pharmacol. Biochem. Behav. 2012, 103, 187–196. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Gnatkovsky, V.; Othman, I.; Shaikh, M.F. From the Molecular Mechanism to Pre-clinical Results: Anti-epileptic Effects of Fingolimod. Curr. Neuropharmacol. 2020, 18, 1126–1137. [Google Scholar] [CrossRef]
- Pourabdolhossein, F.; Mozafari, S.; Morvan-Dubois, G.; Mirnajafi-Zadeh, J.; López-Juárez, A.; Pierre-Simons, J.; Demeneix, B.A.; Javan, M. Nogo Receptor Inhibition Enhances Functional Recovery following Lysolecithin-Induced Demyelination in Mouse Optic Chiasm. PLoS ONE 2014, 9, e106378. [Google Scholar] [CrossRef]
- Sun, J.-J.; Ren, Q.-G.; Xu, L.; Zhang, Z.-J. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci. Rep. 2015, 5, srep14235. [Google Scholar] [CrossRef]
- Sotgiu, S.; Murrighile, M.R.; Constantin, G. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report. BMC Neurol. 2010, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Du, C.; Wei, W.; Wu, Z.; Zhao, G.; Li, Z.; Xie, X. The Antiepileptic Drug Valproic Acid Restores T Cell Homeostasis and Ameliorates Pathogenesis of Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2012, 287, 28656–28665. [Google Scholar] [CrossRef] [PubMed]
- Pazhoohan, S.; Satarian, L.; Asghari, A.-A.; Salimi, M.; Kiani, S.; Mani, A.; Javan, M. Valproic Acid Attenuates Disease Symptoms and Increases Endogenous Myelin Repair by Recruiting Neural Stem Cells and Oligodendrocyte Progenitors in Experimental Autoimmune Encephalomyelitis. Neurodegener. Dis. 2013, 13, 45–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Y.; Schluesener, H. Valproic acid ameliorates inflammation in experimental autoimmune encephalomyelitis rats. Neuroscience 2012, 221, 140–150. [Google Scholar] [CrossRef]
- Lo, A.C.; Saab, C.Y.; Black, J.A.; Waxman, S.G. Phenytoin Protects Spinal Cord Axons and Preserves Axonal Conduction and Neurological Function in a Model of Neuroinflammation In Vivo. J. Neurophysiol. 2003, 90, 3566–3571. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, R.; Hickman, S.J.; Toosy, A.; Sharrack, B.; Mallik, S.; Paling, D.; Altmann, D.R.; Yiannakas, M.C.; Malladi, P.; Sheridan, R.; et al. Phenytoin for neuroprotection in patients with acute optic neuritis: A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 259–269. [Google Scholar] [CrossRef]
- Ramsaransing, G.; Zwanikken, C.; De Keyser, J. Drug points: Worsening of symptoms of multiple sclerosis associated with carbamazepine. BMJ 2000, 320, 1113. [Google Scholar] [CrossRef] [PubMed]
- Solaro, C.; Brichetto, G.; Battaglia, M.A.; Uccelli, M.M.; Mancardi, G.L. Antiepileptic medications in multiple sclerosis: Adverse effects in a three-year follow-up study. Neurol. Sci. 2005, 25, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Meletti, S.; Lucchi, C.; Monti, G.; Giovannini, G.; Bedin, R.; Trenti, T.; Rustichelli, C.; Biagini, G. Decreased allopregnanolone levels in cerebrospinal fluid obtained during status epilepticus. Epilepsia 2017, 58, e16–e20. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, M.; Biagini, G.; Avoli, M. Neurosteroids and focal epileptic disorders. Int. J. Mol. Sci. 2020, 21, 9391. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134, 2703–2721. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Baker, G.B.; Power, C. Allopregnanolone and neuroinflammation: A focus on multiple sclerosis. Front. Cell. Neurosci. 2014, 8, 134. [Google Scholar] [CrossRef]

| Demyelinating Disease | Epilepsy Prevalence | Clinical Manifestation | MRI Findings | Most Frequent Seizure Type | Electroencephalographic (EEG) Characteristics | Possible Pathophysiological Mechanism | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Multiple sclerosis | 0.5–8.3% with an average of 2.3% | Earlier onset of MS symptoms Worse cognitive performance in patients with frequent seizures or status epilepticus | Cortical and juxtacortical lesions Extensive cortical inflammation lower brain volumes Temporal lobe damage: Hippocampus, lateral temporal lobe, cingulate, and insula Cortical thinning and alteration of diffusion metrics in temporal lobe including insular cortex and cingulate gyrus | Partial secondary generalized | Diffuse asynchronous theta activity Synchronous rhythmic slow waves Focalized flattened EEG patterns Focal abnormalities | Temporal lobe cortical pathology Inhibitory GABA interneuron cell loss in layers IV and VI Reduced cortical thickness in the middle temporal gyrus Type I GMLs mostly in middle temporal gyrus Decreased GABA in left hippocampus and posterior cingulate cortex of RRMS Presence of cortical lesions Progressive brain atrophy | [25,26,27,28,42,43,44,45,46,47] | |
| Progressive multifocal leukoencephalopathy | 18% | New-onset seizures | Lesions adjacent to the hemispheric cortices | Simple and complex partial seizures Partial seizures with secondary generalization | - | - | [41] | |
| Antibody-associated demyelination | MOG-IgG * | 20.5% ** | Encephalopathy Younger onset age Higher EDSS score Meningeal irritation Fever, headache, nausea and vomiting CSF leukocytosis | Inflammatory cortical brain lesions Subcortical white matter lesions Deep white matter lesion including periventricular and corpus callosum Cerebral peduncle less optic nerve and spinal cord involvement | Generalized tonic clonic seizure | Background theta to delta rhythm Intermittent low amplitude fast waves Focal sharp-wave Complex and asymmetric focal slow waves | - | [33,40,48,49] |
| AQP4-IgG *** | 1% | - | - | - | - | Slow K+ clearance | [33,50] | |
| Type of Study | Number of Patient with MS | Patients with Seizures (Percentage) | Predominant Seizure Type | Seizure Occurrence at MS Onset | Seizure Occurrence before MS Onset | Seizure Occurrence after MS Onset | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Simple Partial | Complex Partial | Secondary Generalized (sGTCS) | Generalized Tonic Clonic | Status Epilepticus | |||||||
| Cohort | 5041 | 102 (2%) In 67 patients (1.3%), epileptic seizure could not be explained by any cause other than MS | 34 (50.7%) | Less frequent | 28 (41.8%) | 33 (49.3%) | 18 (26.9%) | 7 cases | 26 case | 69 case | [28] |
| Retrospective review of the records | 310 | 10 (3.2%) | 2 | 1 case of simple partial | 6 | 2 | 2 cases of sGTCS | 4 cases | Not reported | Not reported | [26] |
| Retrospective registered-based study | 14,545 | Cumulative incidence: 502 (3.5%)(CI 3.17–3.76) The 5-year prevalence: 1.7% (CI 1.54–1.98) | Single seizure was present in 3.0% (CI 2.77–3.32) of patients with MS | 0.48% | Not reported | Not reported | Not reported | [52] | |||
| Systematic review | 32 studies | Incidence: 2.28% (CI: 1.11–3.44%), at the range of 0.65–5.97% Prevalence: 3.09% (CI: 2.01–4.16%) at the range of 0.89–8.06% | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported | [29] |
| Retrospective review of the records | 1267 * | 22 (1.74%) | Focal onset in 17 patients (77.3%) | 14 out of 17 patients with MS (82.4%) | 5 (22.7%) | 3 (13%) | - | 2 (9.1%) | 16 (72.7%) | [51] | |
| Cohort | 428 | 13 (3%) | 10 (77%) | Half of patient with focal seizure (38.5%) | 3 (23%) | 0 | 4 (31%) | - | 8 ** | [44] | |
| Retrospective cross-sectional epidemiological study | 431 | 19 (4.4%) 14 cases with active epilepsy | 4 cases | 3 cases | 11 cases | - | 5 (36%) | 2 | 1 | 11 cases | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayatpour, A.; Farhangi, S.; Verdaguer, E.; Olloquequi, J.; Ureña, J.; Auladell, C.; Javan, M. The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies. Pharmaceuticals 2021, 14, 1031. https://doi.org/10.3390/ph14101031
Rayatpour A, Farhangi S, Verdaguer E, Olloquequi J, Ureña J, Auladell C, Javan M. The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies. Pharmaceuticals. 2021; 14(10):1031. https://doi.org/10.3390/ph14101031
Chicago/Turabian StyleRayatpour, Atefeh, Sahar Farhangi, Ester Verdaguer, Jordi Olloquequi, Jesus Ureña, Carme Auladell, and Mohammad Javan. 2021. "The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies" Pharmaceuticals 14, no. 10: 1031. https://doi.org/10.3390/ph14101031
APA StyleRayatpour, A., Farhangi, S., Verdaguer, E., Olloquequi, J., Ureña, J., Auladell, C., & Javan, M. (2021). The Cross Talk between Underlying Mechanisms of Multiple Sclerosis and Epilepsy May Provide New Insights for More Efficient Therapies. Pharmaceuticals, 14(10), 1031. https://doi.org/10.3390/ph14101031