
Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment

 , ,
, ,  , ,
, , 
 and
and
Abstract
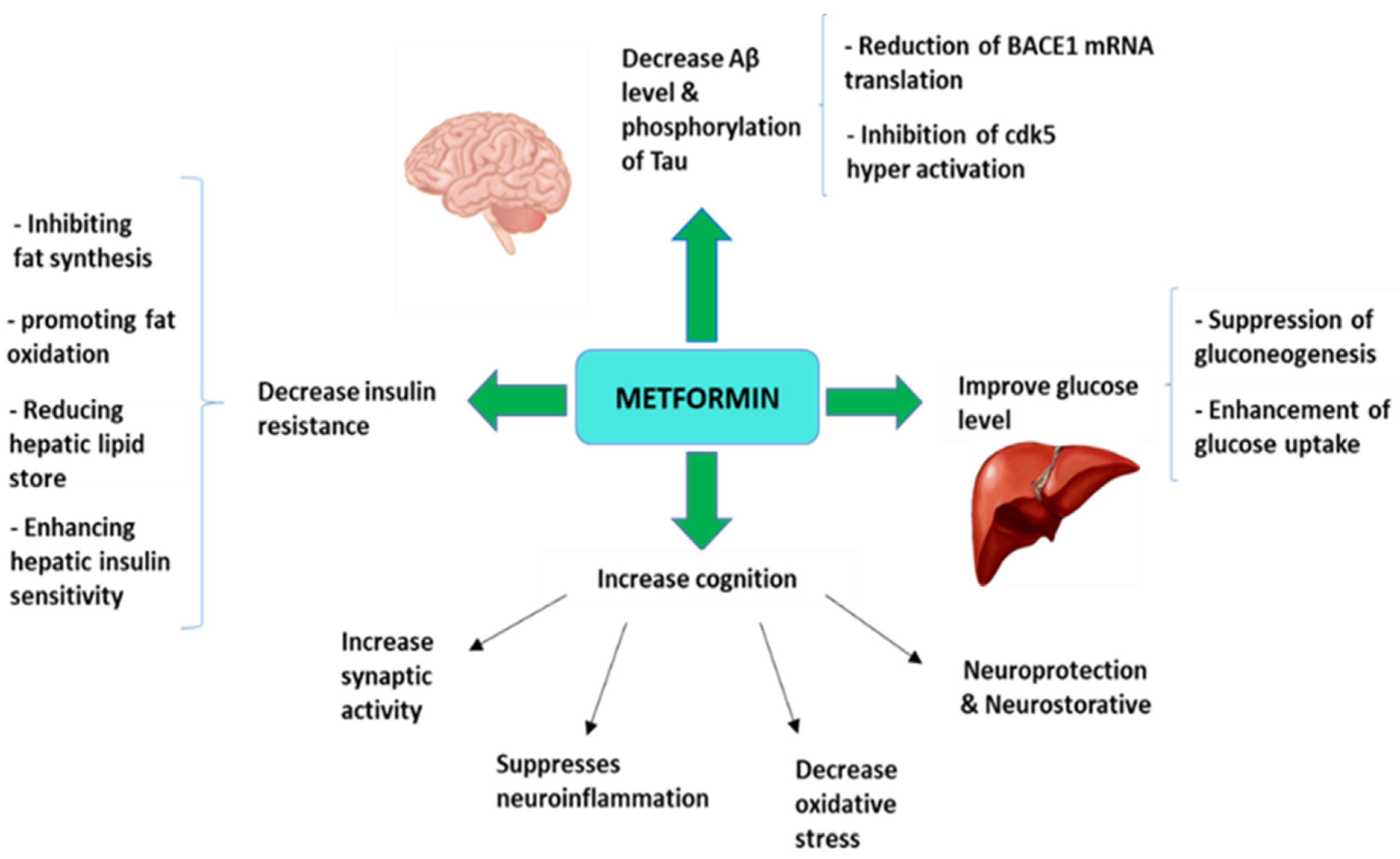
1. Introduction
2. Type 2 Diabetes Mellitus Related with Alzheimer’s Disease
3. Metformin as an Antidiabetic Drug Strategy for Alzheimer’s Disease Treatment
3.1. Preclinical Animal Studies with Metformin

{kind=link}
| Row | Reference | Animal Model/Gender | Starting Age | Metformin Dose | Duration of Therapy | Main Finding |
|---|---|---|---|---|---|---|
| 1 | [82] | B6C3-Tg (APPswe, PS1dE9) 85Dbo-fAD/F | 26 weeks old | 200 mg/kg/d | 14 days | Neuroprotection, Enhanced memory, reduced inflammation, regulation of AMPK/mTOR/S6K/Bace1 pathway. |
| 2 | [91] | SAMP8 mouse model of random onset- AD/M | 12 months old | 20–200 mg/kg/d | 8 weeks | Increased PKC, improved pGSK-3ser9,reduced pTau404 and APPc99, enhanced learning and memory. |
| 3 | [90] | PDAPP (J9) mice-AD/M&F | 6–8 weeks | 350 mg/kg/d | Until 14–16 months-old | increases insulin sensitivity in male, lifespan extension and delayed degradation of the estrous cycle in female |
| 4 | [94] | C57BL/6 mice-PD/M | 10-weeks | 200 mg/kg/d | 10 days. | Stimulate AMPK, mediating the pleiotropy |
| 5 | [95] | Wistar rats-AD/M | Five-month old | 50, 100–200 mg/kg/d | 3 weeks | Decreasing Memory loss, preserved the pAMPK and CREB levels, Improved TAS & SOD levels, increased antioxidant function |
| 6 | [96] | Wistar rats-AD/M | Adult | 100 mg/kg/d | 8 weeks | Enhances neuronal activity and neuropathological modifications, prevent synaptic plasticity impairment |
| 7 | [83] | Wistar rats-sAD/M | 9 weeks | 75–100 mg/kg/d | 21 days | Modulation of glucose delivery and uptake, anti-neuroinflammatory function, maintenance of synaptic plasticity |
| 8 | [86] | C57BL/6 mice-sAD/M | 12–14 weeks | 200 mg/kg/d | 21 days | Suppress glycemic levels and cognitive dysfunction, increases insulin receptor sensitivity, facilitate neuronal survival |
| 9 | [76] | APP/PS1 transgenic mice/F | 9 months old | 4 mg/mL in drinking water | 2 months | Promoted the phagocytosis of Aβ and tau proteins by enhancing microglial autophagy capability |
3.2. Metformin in Clinical Studies
4. Molecular Mechanism Involved in Neuroprotective Effects of Metformin in Alzheimer’s Disease
4.1. Metformin Effects on Amyloid and Tau
4.2. Metformin Effects on Mitochondria
4.3. Metformin Effects on Neurogenesis: The AMPK/aPKC/CBP Signaling Pathway
4.4. Metformin Effects on Learning and Memory
4.5. Metformin Effects on Synaptic Density and Dendritic Spines
4.6. Metformin Effects on Neuroinflammation
4.7. Neuroprotective and Neurorestorative Potential of Metformin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021, 7, e12179. [Google Scholar]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Perry, G.A. Multilevel View of the Development of Alzheimer’s Disease. Neuroscience 2021, 457, 283–293. [Google Scholar] [CrossRef]
- McMurtray, A.; Clark, D.G.; Christine, D.; Mendez, M.F. Early-onset dementia: Frequency and causes compared to late-onset dementia. Dement. Geriatr. Cogn. Disord. 2006, 21, 59–64. [Google Scholar] [CrossRef]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef]
- Chen, Y.; Hong, T.; Chen, F.; Sun, Y.; Wang, Y.; Cui, L. Interplay between Microglia and Alzheimer’s Disease-Focus on the Most Relevant Risks: APOE Genotype, Sex and Age. Front. Aging Neurosci. 2021, 13, 631827. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Emrani, S.; Arain, H.A.; De Marshall, C.; Nuriel, T. APOE4 is associated with cognitive and pathological heterogeneity in patients with Alzheimer’s disease: A systematic review. Alzheimers Res. Ther. 2020, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- Xue, F.; Du, H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells 2021, 10, 321. [Google Scholar] [CrossRef]
- Li, L.; Cavuoto, M.; Biddiscombe, K.; Pike, K.E. Diabetes Mellitus Increases Risk of Incident Dementia in APOEɛ4 Carriers: A Meta-Analysis. J. Alzheimers Dis. 2020, 74, 1295–1308. [Google Scholar] [CrossRef]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, M., Jr.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 89–109. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83. [Google Scholar]
- de la Monte, S.M.; Tong, M.; Wands, J.R. The 20-Year Voyage Aboard the Journal of Alzheimer’s Disease: Docking at ‘Type 3 Diabetes’, Environmental/Exposure Factors, Pathogenic Mechanisms, and Potential Treatments. J. Alzheimers Dis. 2018, 62, 1381–1390. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Carranza-Naval, M.J.; Vargas-Soria, M.; Hierro-Bujalance, C.; Baena-Nieto, G.; Garcia-Alloza, M.; Infante-Garcia, C.; Del Marco, A. Alzheimer’s Disease and Diabetes: Role of Diet, Microbiota and Inflammation in Preclinical Models. Biomolecules 2021, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.E. Metformin and Alzheimer’s disease risk. Am. J. Psychiatry 2014, 171, 119. [Google Scholar] [CrossRef]
- Vinuesa, A.; Pomilio, C.; Gregosa, A.; Bentivegna, M.; Presa, J.; Bellotto, M.; Saravia, F.; Beauquis, J. Inflammation and Insulin Resistance as Risk Factors and Potential Therapeutic Targets for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 653651. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 4981814. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Rhea, E.M.; Banks, W.A.; Hanson, A.J. Insulin resistance, dyslipidemia, and apolipoprotein E interactions as mechanisms in cognitive impairment and Alzheimer’s disease. Exp. Biol. Med. 2016, 241, 1676–1683. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Xu, G.; Johnson, S.C.; Birdsill, A.C.; Jonaitis, E.M.; Sager, M.A.; Hermann, B.P.; La Rue, A.; Asthana, S.; Bendlin, B.B. Insulin resistance, brain atrophy, and cognitive performance in late middle-aged adults. Diabetes Care 2013, 36, 443–449. [Google Scholar] [CrossRef]
- Rebelos, E.; Rinne, J.O.; Nuutila, P.; Ekblad, L.L. Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline-Does Insulin Have Anything to Do with It? A Narrative Review. J. Clin. Med. 2021, 10, 1532. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodríguez-Rodríguez, N.; Bringas-Vega, M.L.; García-Del-Barco-Herrera, D.; Berlanga-Saez, J.O.; García-Ojalvo, A.; Valdés-Sosa, M.J.; Valdés-Sosa, P.A. Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review. Front. Endocrinol. 2020, 11, 560375. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wang, H.Y.; Capuano, A.W.; Khan, A.; Taïb, B.; Anokye-Danso, F.; Schneider, J.A.; Bennett, D.A.; Ahima, R.S.; Arnold, S.E. Brain Insulin Signaling, Alzheimer Disease Pathology, and Cognitive Function. Ann. Neurol. 2020, 88, 513–525. [Google Scholar] [CrossRef]
- Barber, T.M.; Kyrou, I.; Randeva, H.S.; Weickert, M.O. Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction. Int. J. Mol. Sci. 2021, 22, 546. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Carvalho, C.; Cardoso, S.M.; Correia, S.C.; Moreira, P.I. Tortuous Paths of Insulin Signaling and Mitochondria in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1128, 161–183. [Google Scholar]
- De Felice, F.G. Connecting type 2 diabetes to Alzheimer’s disease. Expert Rev. Neurother. 2013, 13, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Benedict, C. A Key Role of Insulin Receptors in Memory. Diabetes 2015, 64, 3653–3655. [Google Scholar] [CrossRef] [PubMed]
- Kulstad, J.J.; Green, P.S.; Cook, D.G.; Watson, G.S.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Rhoads, K.; Mehta, P.D. Craft Differential modulation of plasma beta-amyloid by insulin in patients with Alzheimer disease. Neurology 2006, 66, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N. Advances on the understanding of the origins of synaptic pathology in AD. Curr. Genom. 2007, 8, 486–508. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid- oligomer hypothesis: Beginning of the third decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef]
- Fishel, M.A.; Watson, G.S.; Montine, T.J.; Wang, Q.; Green, P.S.; Kulstad, J.J.; Cook, D.G.; Peskind, E.R.; Baker, L.D.; Goldgaber, D.; et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch. Neurol. 2005, 62, 1539–1544. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Elevation of plasma beta-amyloid level by glucose loading in Alzheimer mouse models. Biochem. Biophys. Res. Commun. 2009, 385, 193–197. [Google Scholar] [CrossRef]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, B.; Zhang, F.; Wu, J.; Hu, Y.; Liu, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance by activating JAK2/STAT3/SOCS-1 signaling pathway. Diabetes 2012, 61, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Tong, M.; Daiello, L.A.; Ott, B.R. Early-Stage Alzheimer’s Disease Is Associated with Simultaneous Systemic and Central Nervous System Dysregulation of Insulin-Linked Metabolic Pathways. J. Alzheimers Dis. 2019, 68, 657–668. [Google Scholar] [CrossRef] [PubMed]
- El Massry, M.; Alaeddine, L.M.; Ali, L.; Saad, C.; Eid, A.A. Metformin: A Growing Journey from Glycemic Control to the Treatment of Alzheimer’s Disease and Depression. Curr. Med. Chem. 2021, 28, 2328–2345. [Google Scholar] [CrossRef] [PubMed]
- Plucińska, K.; Dekeryte, R.; Koss, D.; Shearer, K.; Mody, N.; Whitfield, P.D.; Doherty, M.K.; Mingarelli, M.; Welch, A.; Riedel, G.; et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 2016, 59, 1513–1523. [Google Scholar] [CrossRef]
- Gratuze, M.; Joly-Amado, A.; Vieau, D.; Buée, L.; Blum, D. Mutual Relationship between Tau and Central Insulin Signalling: Consequences for AD and Tauopathies? Neuroendocrinology 2018, 107, 181–195. [Google Scholar] [CrossRef]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, P.; Sandebring-Matton, A.; Merino-Serrais, P.; Parrado-Fernandez, C.; Rabano, A.; Winblad, B.; Ávila, J.; Ferrer, I.; Cedazo-Minguez, A. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain 2017, 140, 3269–3285. [Google Scholar] [CrossRef]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef]
- Gonçalves, R.A.; Wijesekara, N.; De Felice, F.G. The Link between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front. Cell Neurosci. 2019, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin–A future therapy for neurodegenerative diseases. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.; Reynolds, C.D.; Yang, S.H. Metformin and cognition from the perspectives of sex, age, and disease. Geroscience 2020, 42, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar]
- Lv, W.S.; Wen, J.P.; Li, L.; Sun, R.X.; Wang, J.; Xian, Y.X.; Cao, C.X.; Wang, Y.L.; Gao, Y.Y. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012, 1444, 11–19. [Google Scholar] [CrossRef]
- Syal, C.; Kosaraju, J.; Hamilton, L.; Aumont, A.; Chu, A.; Sarma, S.N.; Thomas, J.; Seegobin, M.; Dilworth, F.J.; He, L.; et al. Dysregulated expression of monoacylglycerol lipase is a marker for anti-diabetic drug metformin-targeted therapy to correct impaired neurogenesis and spatial memory in Alzheimer’s disease. Theranostics 2020, 10, 6337–6360. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Mostafa, D.K.; Ismail, C.A.; Ghareeb, D.A. Differential metformin dose-dependent effects on cognition in rats: Role of Akt. Psychopharmacology 2016, 233, 2513–2524. [Google Scholar] [CrossRef]
- Kodali, M.; Attaluri, S.; Madhu, L.N.; Shuai, B.; Upadhya, R.; Gonzalez, J.J.; Rao, X.; Shetty, A.K. Metformin treatment in late middle age improves cognitive function with alleviation of microglial activation and enhancement of autophagy in the hippocampus. Aging Cell 2021, 20, e13277. [Google Scholar] [CrossRef] [PubMed]
- Laurijssens, B.; Aujard, F.; Rahman, A. Animal models of Alzheimer’s disease and drug development. Drug. Discov. Today Technol. 2013, 10, e319–e327. [Google Scholar] [CrossRef]
- Nakai, T.; Yamada, K.; Mizoguchi, H. Alzheimer’s Disease Animal Models: Elucidation of Biomarkers and Therapeutic Approaches for Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 5549. [Google Scholar] [CrossRef]
- Correia, S.; Carvalho, C.; Santos, M.S.; Proença, T.; Nunes, E.; Duarte, A.I.; Monteiro, P.; Seiça, R.; Oliveira, C.R.; Moreira, P.I. Metformin protects the brain against the oxidative imbalance promoted by type 2 diabetes. Med. Chem. 2008, 4, 358–364. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z.; et al. Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimer Res. Ther. 2021, 13, 40. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Sanchez-Mejias, E.; Sanchez-Varo, R.; Garcia-Leon, J.A.; Nuñez-Diaz, C.; Davila, J.C.; Vitorica, J.; LaFerla, F.M.; Moreno-Gonzalez, I.; Gutierrez, A.; et al. Animal and Cellular Models of Alzheimer’s Disease: Progress, Promise, and Future Approaches. Neuroscientist 2021, 26, 10738584211001753. [Google Scholar]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Obafemi, T.O.; Olasehinde, O.R.; Olaoye, O.A.; Jaiyesimi, K.F.; Adewumi, F.D.; Adewale, O.B.; Afolabi, B.A. Metformin/Donepezil combination modulates brain antioxidant status and hippocampal endoplasmic reticulum stress in type 2 diabetic rats. J. Diabetes Metab. Disord. 2020, 19, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Esmaeili, M.H.; Esmaeili, M. Metformin improves learning and memory in streptozotocin-induced rat model of sporadic Alzheimer’s disease. Indian J. Fund. App. Life Sci. 2016, 6 (Suppl. 1), 270–281. [Google Scholar]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Oliveira, W.H.; Nunes, A.K.; França, M.E.; Santos, L.A.; Lós, D.B.; Rocha, S.W.; Barbosa, K.P.; Rodrigues, G.B.; Peixoto, C.A. Effects of metformin on inflammation and short-term memory in streptozotocin-induced diabetic mice. Brain Res. 2016, 1644, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, V.; Narbute, K.; Pupure, J.; Langrate, I.K.; Muceniece, R.; Kluša, V. Neuroprotective potential of antihyperglycemic drug metformin in streptozotocin-induced rat model of sporadic Alzheimer’s disease. Eur. J. Pharmacol. 2020, 881, 173290. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm. 2013, 120, 233–252. [Google Scholar] [CrossRef]
- Plaschke, K.; Kopitz, J.; Siegelin, M.; Schliebs, R.; Salkovic-Petrisic, M.; Riederer, P.; Hoyer, S. Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J. Alzheimers Dis. 2010, 19, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Grünblatt, E.; Salkovic-Petrisic, M.; Osmanovic, J.; Riederer, P.; Hoyer, S.J. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Salkovic-Petrisic, M.; Hoyer, S.J. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: An experimental approach. Neuropsychiatr. Disord. Integr. Approach 2007, 217–233. [Google Scholar]
- Saffari, P.M.; Alijanpour, S.; Takzaree, N.; Sahebgharani, M.; Etemad-Moghadam, S.; Noorbakhsh, F.; Partoazar, A. Metformin loaded phosphatidylserine nanoliposomes improve memory deficit and reduce neuroinflammation in streptozotocin-induced Alzheimer’s disease model. Life Sci. 2020, 255, 117861. [Google Scholar] [CrossRef] [PubMed]
- Ditacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin improves learning and memory in the SAMP8 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef]
- Bao, J.; Mahaman, Y.A.R.; Liu, R.; Wang, J.Z.; Zhang, Z.; Zhang, B.; Wang, X. Sex Differences in the Cognitive and Hippocampal Effects of Streptozotocin in an Animal Model of Sporadic AD. Front. Aging Neurosci. 2017, 31, 347. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Huang, S.; Chen, Q.B.; Zhang, D.; Li, W.; Ao, R.; Leung, F.C.; Zhang, Z.; Huang, J.; Tang, Y.; et al. Metformin Ameliorates Abeta Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s disease. Oxid. Med. Cell Longev. 2020, 2315106. [Google Scholar]
- Katila, N.; Bhurtel, S.; Park, P.H.; Hong, J.T.; Choi, D.Y. Activation of AMPK/aPKCζ/CREB pathway by metformin is associated with upregulation of GDNF and dopamine. Biochem. Pharmacol. 2020, 180, 114193. [Google Scholar] [CrossRef] [PubMed]
- Aksoz, E.; Gocmez, S.S.; Sahin, T.D.; Aksit, D.; Aksit, H.; Utkan, T. The protective effect of metformin in scopolamine-induced learning and memory impairment in rats. Pharmacol. Rep. 2019, 71, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Asadbegi, M.; Yaghmaei, P.; Salehi, I.; Ebrahim-Habibi, A.; Komaki, A. Neuroprotective effects of metformin against Aβ-mediated inhibition of long-term potentiation in rats fed a high-fat diet. Brain Res. Bull. 2016, 121, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Chin-Hsiao, T. Metformin and the Risk of Dementia in Type 2 Diabetes Patients. Aging Dis. 2019, 10, 37–48. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in amnestic mild cognitive impairment: Results of a pilot randomized placebo controlled clinical trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Ma, Y.; Christophi, C.A.; Florez, H.; Golden, S.H.; Hazuda, H.; Crandall, J.; Venditti, E.; Watson, K.; Jeffries, S.; et al. Diabetes Prevention Program Research Group. Metformin, Lifestyle Intervention, and Cognition in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2017, 40, 58–965. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in Alzheimer disease: Pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Madhu, L.N.; Kodali, M.; Shetty, A.K. Promise of metformin for preventing age-related cognitive dysfunction. Neural Regen Res. 2022, 17, 503–507. [Google Scholar]
- Scherrer, J.F.; Morley, J.E.; Salas, J.; Floyd, J.S.; Farr, S.A.; Dublin, S. Association between Metformin Initiation and Incident Dementia among African American and White Veterans Health Administration Patients. Ann. Fam. Med. 2019, 17, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.M.; Hartikainen, S. Metformin and Risk of Alzheimer’s Disease among Community-Dwelling people with Diabetes: A National Case-Control Study. J. Clin. Endocrinol. Metab. 2020, 105, dgz234. [Google Scholar] [CrossRef]
- Koo, B.K.; Kim, L.K.; Lee, J.Y.; Moon, M.K. Taking metformin and cognitive function change in older patients with diabetes. Geriatr. Gerontol. Int. 2019, 19, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Makkar, S.; Crawford, J.D.; Kochan, N.A.; Wen, W.; Draper, B.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Metformin Use Is Associated with Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults with Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef]
- Ramamurthy, S.; Ronnett, G. AMP-activated protein kinase (AMPK) and energy-sensing in the brain. Exp. Neurobiol. 2012, 21, 52–60. [Google Scholar] [CrossRef]
- Domise, M.; Vingtdeux, V. AMPK in Neurodegenerative Diseases. Exp. Suppl. 2016, 107, 153–177. [Google Scholar]
- Seixas da Silva, G.S.; Melo, H.M.; Lourenco, M.V.; Lyra, E.S.N.M.; de Carvalho, M.B.; Alves-Leon, S.V.; de Souza, J.M.; Klein, W.L.; da-Silva, W.S.; Ferreira, S.T.; et al. Amyloid-β oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons. J. Biol. Chem. 2017, 292, 7395–7406. [Google Scholar] [CrossRef]
- Culmsee, C.; Monnig, J.; Kemp, B.E.; Mattson, M.P. AMP-activated kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J. Mol. Neurosci. 2001, 17, 45–58. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Wilson, C.M.; Magnaudeix, A.; Yardin, C.; Terro, F. Autophagy dysfunction and its link to Alzheimer’s disease and type II diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 226–246. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J.C. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol. Life Sci. 2010, 67, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, N.; Shi, F.X.; Xu, W.Q.; Cao, Y.; Lei, Y.; Wang, J.Z.; Tian, Q.; Zhou, X.W. Upregulation of AMPK Ameliorates Alzheimer’s Disease-Like Tau Pathology and Memory Impairment. Mol. Neurobiol. 2020, 57, 3349–3361. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef]
- Domise, M.; Didier, S.; Marinangeli, C.; Zhao, H.; Chandakkar, P.; Buée, L.; Viollet, B.; Davies, P.; Marambaud, P.; Vingtdeux, V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 2016, 6, 26758. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, W.; Li, H.; Pang, P.; Xue, F.; Wan, L.; Pei, L.; Yan, H. Metformin restores hippocampal neurogenesis and learning and memory via regulating gut microbiota in the obese mouse model. Brain Behav. Immun. 2021, 95, 68–83. [Google Scholar] [CrossRef]
- Peng, Y.; Gao, P.; Shi, L.; Chen, L.; Liu, J.; Long, J. Central and peripheral metabolic defects contribute to the pathogenesis of alzheimer’s disease: Targeting mitochondria for diagnosis and prevention. Antioxid. Redox Signal. 2020, 32, 1188–1236. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondra and mitochondrial cascades in alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.-M.; Chen, Y.L.; Pei, D.; Cheng, Y.C.; Sun, B.; Nicol, C.J.; Yen, C.H.; Chen, H.M.; Liang, Y.J.; Chiang, M.C. The neuroprotective role of metformin in advanced glycation end product treated human neural stem cells is AMPK-dependent. Biochim. Biophys. Acta 2015, 1852, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, A.; Hsu, K.; Niibori, Y.; Seegobin, M.; Cancino, G.I.; He, L.; Wondisford, F.E.; Bennett, S.; Lagace, D.; Frankland, P.W.; et al. The aPKC-CBP Pathway Regulates Adult Hippocampal Neurogenesis in an Age-Dependent Manner. Stem. Cell Rep. 2016, 7, 719–734. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanokashira, D.; Kurata, E.; Fukuokaya, W.; Kawabe, K.; Kashiwada, M.; Takeuchi, H.; Nakazato, M.; Taguchi, A. Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Biol. 2018, 8, 1104–1118. [Google Scholar] [CrossRef]
- Rajangam, J.; Bhatt, S.; Krishnan, N.; Sammeta, M.; Joshna, L. Influence of Metformin on learning and memory in experimental Amnesia model in Mice. Ann. Alzheimers Dement. Care 2020, 4, 5–9. [Google Scholar]
- Boccardi, V.; Murasecco, I.; Mecocci, P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res. Rev. 2019, 4, 100936. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jiménez, M.; Zaarkti, A.; García-Arnés, J.A.; García-Casares, N. Antidiabetic Drugs in Alzheimer’s disease and Mild Cognitive Impairment: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2020, 49, 423–434. [Google Scholar]
- Zhu, X.; Shen, J.; Feng, S.; Huang, C.; Liu, Z.; Sun, Y.E.; Liu, H. Metformin improves cognition of aged mice by promoting cerebral angiogenesis and neurogenesis. Aging 2020, 12, 17845–17862. [Google Scholar] [CrossRef]
- Chen, W.B.; Chen, J.; Liu, Z.Y.; Luo, B.; Zhou, T.; Fei, E.K. Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons. Brain Sci. 2020, 10, 706. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deng, J.; Sheng, W.; Zuo, Z. Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharm. Biochem. Behav. 2012, 101, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Boros, B.D.; Greathouse, K.M.; Gentry, E.G.; Curtis, K.A.; Birchall, E.L.; Gearing, M.; Herskowitz, J.H. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann. Neurol. 2017, 82, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Boros, B.D.; Greathouse, K.M.; Gearing, M.; Herskowitz, J.H. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol. Aging 2019, 73, 92–103. [Google Scholar] [CrossRef]
- Walker, C.K.; Herskowitz, J.H. Dendritic spines: Mediators of cognitive resilience in aging and Alzheimer’s disease. Neuroscientist 2020. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Baxter, M.G. Synaptic health. JAMA Psychiatry 2014, 71, 835–837. [Google Scholar] [CrossRef]
- Dumitriu, D.; Hao, J.; Hara, Y.; Kaufmann, J.; Janssen, W.G.; Lou, W.; Rapp, P.R.; Morrison, J.H. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J. Neurosci. 2010, 30, 7507–7515. [Google Scholar] [CrossRef]
- Pereira, A.C.; Lambert, H.K.; Grossman, Y.S.; Dumitriu, D.; Waldman, R.; Jannetty, S.K.; Calakos, K.; Janssen, W.G.; McEwen, B.S.; Morrison, J.H. Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc. Natl. Acad. Sci. USA 2014, 111, 18733–18738. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Scheff, S.W.; Neltner, J.H.; Nelson, P.T. Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem. Pharmacol. 2014, 88, 517–528. [Google Scholar] [CrossRef]
- Cardoso, S.; Moreira, P.I. Antidiabetic drugs for Alzheimer’s and Parkinson’s diseases: Repurposing insulin, metformin, and thiazolidinediones. Int. Rev. Neurobiol. 2020, 155, 37–64. [Google Scholar]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds that extend longevity are protective in neurodegenerative diseases and provide a novel treatment strategy for these devastating disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, J.; Guo, F.L.; Gao, X.; Xie, X.; Liu, S.; Yang, X.; Yang, X.; Zhang, L.; Ye, Y.; et al. Metformin Ameliorates Synaptic Defects in a Mouse Model of AD by Inhibiting Cdk5 Activity. Front. Cell Neurosci. 2020, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, Y.; Liang, R.; Zhang, Y. Inhibition of cyclin-dependent kinase 5 activity alleviates diabetes-related cognitive deficits. FASEB J. 2019, 33, 14506–14515. [Google Scholar] [CrossRef]
- Cai, H.B.; Fan, Z.Z.; Tian, T.; Li, Z.C.; Zhao, C.C.; Guo, W.T.; Ge, Z.M. Diabetes-induced H3K9 Hyperacetylation Promotes Development of Alzheimer’s Disease through CDK5. J. Alzheimers Dis. 2020, 77, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.S.; Yeom, Y.S.; Jang, J.H.; Kim, Y.H.; Im, J.I.; Kim, I.S.; Yang, S.J. Anti-inflammatory Effects of Metformin on Neuro-inflammation and NLRP3 Inflammasome Activation in BV-2 Microglial Cells. Biomed. Sci. Lett. 2019, 25, 92–98. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, G.; Zhang, Z.; Wang, Y.; Yang, G.Y. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci. Lett. 2014, 579, 46–57. [Google Scholar] [CrossRef]
- Hettich, M.M.; Matthes, F.; Ryan, D.P.; Griesche, N.; Schröder, S.; Dorn, S.; Krauβ, S.; Ehninger, D. The anti-diabetic drug metformin reduces BACE1 protein level by interfering with the MID1 complex. PLoS ONE 2014, 9, e102420. [Google Scholar] [CrossRef]
- Hasanpour Dehkordi, A.; Abbaszadeh, A.; Mir, S.; Hasanvand, A. Metformin and its anti-inflammatory and anti-oxidative effects; new concepts. J. Renal Inj. Prev. 2019, 8, 54–61. [Google Scholar] [CrossRef]
- Miziak, B.; Błaszczyk, B.; Czuczwar, S.J. Some Candidate Drugs for Pharmacotherapy of Alzheimer’s Disease. Pharmaceuticals 2021, 14, 458. [Google Scholar] [CrossRef] [PubMed]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef] [PubMed]
- Fatt, M.; Hsu, K.; He, L.; Wondisford, F.; Miller, F.D.; Kaplan, D.R.; Wang, J. Metformin Acts on Two Different Molecular Pathways to Enhance Adult Neural Precursor Proliferation/Self-Renewal and Differentiation. Stem. Cell Rep. 2015, 5, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Bijak, M. Metformin as a Potential Agent in the Treatment of Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 5957. [Google Scholar] [CrossRef]
- Esmaeilnejad, S.; Semnanian, S.; Javan, M. Metformin Protects Myelin from Degeneration in A Mouse Model of Iysophosphatidylcholine-Induced Demyelination in The Optic Chiasm. Cell J. 2021, 23, 119–128. [Google Scholar]
- Corcoran, C.; Jacobs, T.F. Metformin. In StatPearls [Internet]. Treasure Island (FL); StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poor, S.R.; Ettcheto, M.; Cano, A.; Sanchez-Lopez, E.; Manzine, P.R.; Olloquequi, J.; Camins, A.; Javan, M. Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals 2021, 14, 890. https://doi.org/10.3390/ph14090890
Poor SR, Ettcheto M, Cano A, Sanchez-Lopez E, Manzine PR, Olloquequi J, Camins A, Javan M. Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals. 2021; 14(9):890. https://doi.org/10.3390/ph14090890
Chicago/Turabian StylePoor, Saghar Rabiei, Miren Ettcheto, Amanda Cano, Elena Sanchez-Lopez, Patricia Regina Manzine, Jordi Olloquequi, Antoni Camins, and Mohammad Javan. 2021. "Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment" Pharmaceuticals 14, no. 9: 890. https://doi.org/10.3390/ph14090890
APA StylePoor, S. R., Ettcheto, M., Cano, A., Sanchez-Lopez, E., Manzine, P. R., Olloquequi, J., Camins, A., & Javan, M. (2021). Metformin a Potential Pharmacological Strategy in Late Onset Alzheimer’s Disease Treatment. Pharmaceuticals, 14(9), 890. https://doi.org/10.3390/ph14090890