
Inhibition of Autophagy In Vivo Extends Methamphetamine Toxicity to Mesencephalic Cell Bodies


 ,
,  , ,
, , 
Abstract
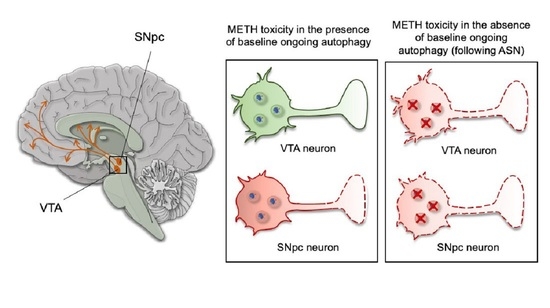
1. Introduction
2. Results
2.1. ASN (or GLN) Suppresses Autophagy and Induces Bax Overexpression When Combined with METH
2.2. ASN (or GLN)-Induced Autophagy Inhibition Dramatically Worsens METH-Induced Toxicity to Meso-Striatal DA Axons
2.3. Inhibition of Autophagy by ASN (or GLN) Extends METH Toxicity to Midbrain DA Cell Bodies
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Treatments and Experimental Design
4.3. HPLC Assay
4.4. Sample Preparation for Light and Confocal Microscopy
4.5. Haematoxylin and Eosin
4.6. Immunofluorescence
4.7. Immunohistochemistry
4.8. Stereology-like Neuronal Count
4.9. Statistical Analysis
4.10. Experiments Related to GLN and METH Treatments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fornai, F.; Puglisi-Allegra, S. Autophagy status as a gateway for stress-induced catecholamine interplay in neurodegeneration. Neurosci. Biobehav. Rev. 2021, 123, 238–256. [Google Scholar] [CrossRef]
- Hemmerle, A.M.; Herman, J.P.; Seroogy, K.B. Stress, depression and Parkinson’s disease. Exp. Neurol. 2012, 233, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.W.; Ameringer, S.W.; Cloud, L.J. An Integrated Review of Psychological Stress in Parkinson’s Disease: Biological Mechanisms and Symptom and Health Outcomes. Parkinson’s Dis. 2016, 2016, 9869712. [Google Scholar] [CrossRef]
- Matuszewich, L.; Yamamoto, B.K. Effects of chronic stress on methamphetamine-induced dopamine depletions in the striatum. Ann. N. Y. Acad. Sci. 2004, 1032, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Quinton, M.S.; Yamamoto, B.K. Neurotoxic effects of chronic restraint stress in the striatum of methamphetamine-exposed rats. Psychopharmacology 2007, 193, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, X.; Zhang, Y.; Zhang, L.; Feng, L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T α-synuclein transgenic mice. Exp. Neurol. 2016, 285, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Jablonski, S.A.; Amos-Kroohs, R.M.; Barnes, A.C.; Williams, M.T.; Vorhees, C.V. Effects of Housing on Methamphetamine-Induced Neurotoxicity and Spatial Learning and Memory. ACS Chem. Neurosci. 2017, 8, 1479–1489. [Google Scholar] [CrossRef]
- O’Dell, S.J.; Weihmuller, F.B.; Marshall, J.F. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991, 564, 256–260. [Google Scholar] [CrossRef]
- Hutchison, J.B.; Strupish, J.; Nahorski, S.R. Release of endogenous dopamine and cholecystokinin from rat striatal slices: Effects of amphetamine and dopamine antagonists. Brain Res. 1986, 370, 310–314. [Google Scholar] [CrossRef]
- Hurd, Y.L.; Ungerstedt, U. Cocaine: An in vivo microdialysis evaluation of its acute action on dopamine transmission in rat striatum. Synapse 1989, 3, 48–54. [Google Scholar] [CrossRef]
- Guix, T.; Hurd, Y.L.; Ungerstedt, U. Amphetamine enhances extracellular concentrations of dopamine and acetylcholine in dorsolateral striatum and nucleus accumbens of freely moving rats. Neurosci. Lett. 1992, 138, 137–140. [Google Scholar] [CrossRef]
- Miczek, K.A.; Mutschler, N.H.; van Erp, A.M.; Blank, A.D.; McInerney, S.C. d-Amphetamine “cue” generalizes to social defeat stress: Behavioral sensitization and attenuated accumbens dopamine. Psychopharmacology 1999, 147, 190–199. [Google Scholar] [CrossRef]
- Marsteller, D.A.; Gerasimov, M.R.; Schiffer, W.K.; Geiger, J.M.; Barnett, C.R.; Schaich Borg, J.; Scott, S.; Ceccarelli, J.; Volkow, N.D.; Molina, P.E.; et al. Acute handling stress modulates methylphenidate-induced catecholamine overflow in the medial prefrontal cortex. Neuropsychopharmacology 2002, 27, 163–170. [Google Scholar] [CrossRef][Green Version]
- Antelman, S.M.; Eichler, A.J.; Black, C.A.; Kocan, D. Interchangeability of stress and amphetamine in sensitization. Science 1980, 207, 329–331. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Cabib, S.; Puglisi-Allegra, S. The mesoaccumbens dopamine in coping with stress. Neurosci. Biobehav. Rev. 2012, 36, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Post, R.M. Epigenetic basis of sensitization to stress, affective episodes, and stimulants: Implications for illness progression and prevention. Bipolar Disord. 2016, 18, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Douma, E.H.; de Kloet, E.R. Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neurosci. Biobehav. Rev. 2020, 108, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shang, X.; Zhai, B.; Zhang, H.; Zhang, T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem. Int. 2018, 114, 58–70. [Google Scholar] [CrossRef]
- Puri, D.; Subramanyam, D. Stress-(self) eating: Epigenetic regulation of autophagy in response to psychological stress. FEBS J. 2019, 286, 2447–2460. [Google Scholar] [CrossRef]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Seiden, L.S.; Fischman, M.W.; Schuster, C.R. Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend. 1976, 1, 215–219. [Google Scholar] [CrossRef]
- Schmidt, C.J.; Ritter, J.K.; Sonsalla, P.K.; Hanson, G.R.; Gibb, J.W. Role of dopamine in the neurotoxic effects of methamphetamine. J. Pharmacol. Exp. Ther. 1985, 233, 539–544. [Google Scholar] [PubMed]
- Sonsalla, P.K.; Heikkila, R.E. Neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and methamphetamine in several strains of mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 1988, 12, 345–354. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Leonido-Yee, M.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 2001, 158, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G.; Zigmond, M.J. Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]dopamine: Impact of ascorbic acid and glutathione. J. Neurochem. 1994, 63, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Hastings, T.G. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: Evidence against a role for extracellular dopamine. J. Neurosci. 1999, 19, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B.; Poli, A. Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim. Biophys. Acta 1980, 630, 103–118. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B.; Grinde, B.; Solheim, A.; Kovács, A.L.; Poli, A. Inhibitors and pathways of hepatocytic protein degradation. Acta Biol. Med. Ger. 1981, 40, 1587–1598. [Google Scholar]
- Kopitz, J.; Kisen, G.O.; Gordon, P.B.; Bohley, P.; Seglen, P.O. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef]
- Høyvik, H.; Gordon, P.B.; Berg, T.O.; Strømhaug, P.E.; Seglen, P.O. Inhibition of autophagic-lysosomal delivery and autophagic lactolysis by asparagine. J. Cell Biol. 1991, 113, 1305–1312. [Google Scholar] [CrossRef]
- Gordon, P.B.; Høyvik, H.; Seglen, P.O. Prelysosomal and lysosomal connections between autophagy and endocytosis. Biochem. J. 1992, 283, 361–369. [Google Scholar] [CrossRef]
- Fengsrud, M.; Roos, N.; Berg, T.; Liou, W.; Slot, J.W.; Seglen, P.O. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: Effects of vinblastine and asparagine on vacuole distributions. Exp. Cell Res. 1995, 221, 504–519. [Google Scholar] [CrossRef]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 inde-pendently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Kim, S.G.; Hoffman, G.R.; Poulogiannis, G.; Buel, G.R.; Jang, Y.J.; Lee, K.W.; Kim, B.Y.; Erikson, R.L.; Cantley, L.C.; Choo, A.Y.; et al. Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol. Cell 2013, 49, 172–185. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Cantini, F.; Madonna, M.; Bucci, D.; Limanaqi, F.; Soldani, P.; Fornai, F. In search for a gold-standard procedure to count motor neurons in the spinal cord. Histol. Histopathol. 2018, 33, 1021–1046. [Google Scholar] [CrossRef]
- Weihmuller, F.B.; O’Dell, S.J.; Marshall, J.F. MK-801 protection against methamphetamine-induced striatal dopamine terminal injury is associated with attenuated dopamine overflow. Synapse 1992, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Eisch, A.J.; Gaffney, M.; Weihmuller, F.B.; O’Dell, S.J.; Marshall, J.F. Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res. 1992, 598, 321–326. [Google Scholar] [CrossRef]
- Baumann, M.H.; Ayestas, M.A.; Sharpe, L.G.; Lewis, D.B.; Rice, K.C.; Rothman, R.B. Persistent antagonism of methamphetamine-induced dopamine release in rats pretreated with GBR12909 decanoate. J. Pharmacol. Exp. Ther. 2002, 301, 1190–1197. [Google Scholar] [CrossRef]
- Riddle, E.L.; Fleckenstein, A.E.; Hanson, G.R. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006, 8, E413–E418. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Ali, S.; Epstein, C. Involvement of oxygen-based radicals in methamphetamine-induced neurotoxicity: Evidence from the use of CuZnSOD transgenic mice. Ann. N. Y. Acad. Sci. 1994, 738, 388–391. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Diaz-Corrales, F.J.; Fukuda, M.; Kitaichi, K.; Miyoshi, K.; Ogawa, N. Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone-formation-related molecules. FASEB J. 2006, 20, 571–573. [Google Scholar] [CrossRef]
- Sulzer, D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 2011, 69, 628–649. [Google Scholar] [CrossRef]
- Ingram, S.L.; Prasad, B.M.; Amara, S.G. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 2002, 5, 971–978. [Google Scholar] [CrossRef]
- John, C.E.; Jones, S.R. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology 2007, 52, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.G.; Forbes, B.; Cheung, P.Y.; Martini, A.; Burrell, M.H.; Freestone, P.S.; Lipski, J. Action potential and calcium dependence of tonic somatodendritic dopamine release in the Substantia Nigra pars compacta. J. Neurochem. 2019, 148, 462–479. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Jochnowitz, N.D.; Zeevalk, G.D.; Oostveen, J.A.; Hall, E.D. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996, 738, 172–175. [Google Scholar] [CrossRef]
- Ladenheim, B.; Krasnova, I.N.; Deng, X.; Oyler, J.M.; Polettini, A.; Moran, T.H.; Huestis, M.A.; Cadet, J.L. Methamphetamine-induced neurotoxicity is attenuated in transgenic mice with a null mutation for interleukin-6. Mol. Pharmacol. 2000, 58, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Ares-Santos, S.; Granado, N.; Espadas, I.; Martinez-Murillo, R.; Moratalla, R. Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 2014, 39, 1066–1080. [Google Scholar] [CrossRef]
- Seiden, L.S. Methamphetamine: Toxicity to dopaminergic neurons. NIDA Res. Monogr. 1985, 62, 100–116. [Google Scholar]
- Kataoka, Y.; Gomita, Y.; Fukuda, T.; Eto, K.; Araki, Y. Effects of aggregation on methamphetamine toxicity in mice. Acta Med. 1986, 40, 121–126. [Google Scholar] [CrossRef]
- Cragg, S.J.; Greenfield, S.A. Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J. Neurosci. 1997, 17, 5738–5746. [Google Scholar] [CrossRef]
- Kogan, F.J.; Nichols, W.K.; Gibb, J.W. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur. J. Pharmacol. 1976, 36, 363–371. [Google Scholar] [CrossRef]
- Wagner, G.C.; Ricaurte, G.A.; Seiden, L.S.; Schuster, C.R.; Miller, R.J.; Westley, J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980, 181, 151–160. [Google Scholar] [CrossRef]
- Fleckenstein, A.E.; Metzger, R.R.; Wilkins, D.G.; Gibb, J.W.; Hanson, G.R. Rapid and reversible effects of methamphetamine on dopamine transporters. J. Pharmacol. Exp. Ther. 1997, 282, 834–838. [Google Scholar]
- Fumagalli, F.; Gainetdinov, R.R.; Valenzano, K.J.; Caron, M.G. Role of dopamine transporter in methamphetamine-induced neurotoxicity: Evidence from mice lacking the transporter. J. Neurosci. 1998, 18, 4861–4869. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.E.; Volz, T.J.; Riddle, E.L.; Gibb, J.W.; Hanson, G.R. New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.J.; Hanson, G.R.; Fleckenstein, A.E. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J. Neurochem. 2007, 101, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Kokoshka, J.M.; Vaughan, R.A.; Hanson, G.R.; Fleckenstein, A.E. Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters. Eur. J. Pharmacol. 1998, 361, 269–275. [Google Scholar] [CrossRef]
- Brown, J.M.; Hanson, G.R.; Fleckenstein, A.E. Methamphetamine rapidly decreases vesicular dopamine uptake. J. Neurochem. 2000, 74, 2221–2223. [Google Scholar] [CrossRef]
- Riddle, E.L.; Topham, M.K.; Haycock, J.W.; Hanson, G.R.; Fleckenstein, A.E. Differential traffi cking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur. J. Pharmacol. 2002, 449, 71–74. [Google Scholar] [CrossRef]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate redistributes vesicular monoamine transporter-2: Role of dopamine receptors. J. Neurosci. 2002, 22, 8705–8710. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J. Pharmacol. Exp. Ther. 2003, 304, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Rayport, S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: A mechanism of action. Neuron 1990, 5, 797–808. [Google Scholar] [CrossRef]
- Sulzer, D.; Pothos, E.; Sung, H.M.; Maidment, N.T.; Hoebel, B.G.; Rayport, S. Weak base model of amphetamine action. Ann. N. Y. Acad. Sci. 1992, 654, 525–528. [Google Scholar] [CrossRef]
- Green, A.L.; El Hait, M.A. p-Methoxyamphetamine, a potent reversible inhibitor of type-A monoamine oxidase in vitro and in vivo. J. Pharm. Pharmacol. 1980, 32, 262–266. [Google Scholar] [CrossRef]
- Suzuki, O.; Hattori, H.; Asano, M.; Oya, M.; Katsumata, Y. Inhibition of monoamine oxidase by d-methamphetamine. Biochem. Pharmacol. 1980, 29, 2071–2073. [Google Scholar] [CrossRef]
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The toxicity of melanin precursors. J. Invest. Dermatol. 1978, 70, 113–116. [Google Scholar] [CrossRef]
- Cohen, G. Oxy-radical toxicity in catecholamine neurons. Neurotoxicology 1984, 5, 77–82. [Google Scholar] [PubMed]
- Schlüter, O.M.; Fornai, F.; Alessandrí, M.G.; Takamori, S.; Geppert, M.; Jahn, R.; Südhof, T.C. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 2003, 118, 985–1002. [Google Scholar] [CrossRef]
- Cabib, S. Strain-dependent behavioural sensitization to amphetamine: Role of environmental influences. Behav. Pharmacol. 1993, 4, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Paku, S.; Takahashi, M.; Kubo, K.; Wagner, G.C.; Nakashima, T. Methamphetamine-induced neurotoxicity in BALB/c, DBA/2N and C57BL/6N mice. Neuropharmacology 1998, 37, 1177–1184. [Google Scholar] [CrossRef]
- Fornai, F. Experimental Models in Parkinson’s Disease. In Proceedings of the LIMPE Seminars, Alghero, Italy, 23–25 September 2007. [Google Scholar]
- Ferrucci, M.; Castino, R.; Lazzeri, G.; Cantafora, E.; Lenzi, P.; Isidoro, C.; Longone, P.; Paparelli, A.; Fornai, F.E. Impairment of autophagy removes the resistance of dopaminergic cell bodies to methamphetamine toxicity. In Session: Neurotoxicity of Amphetamine and Related Addictive Drugs; November 15–19, poster #57; Society for Neuroscience: Washington, DC, USA, 2008. [Google Scholar]
- Lowry, O.H.; Rosembrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: San Diego, CA, USA, 2004. [Google Scholar]
- Vidyadhara, D.J.; Yarreiphang, H.; Raju, T.R.; Alladi, P.A. Admixing of MPTP-Resistant and Susceptible Mice Strains Augments Nigrostriatal Neuronal Correlates to Resist MPTP-Induced Neurodegeneration. Mol. Neurobiol. 2017, 54, 6148–6162. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef]
- Lu, J.; Wu, M.; Yue, Z. Autophagy and Parkinson’s Disease. Adv. Exp. Med. Biol. 2020, 1207, 21–51. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Wood, S.; Sage, J.R.; Shuman, T.; Anagnostaras, S.G. Psychostimulants and cognition: A continuum of behavioral and cognitive activation. Pharmacol. Rev. 2013, 66, 193–221. [Google Scholar] [CrossRef]
- Chen, C.K.; Lin, S.K.; Chen, Y.C.; Huang, M.C.; Chen, T.T.; Ree, S.C.; Wang, L.J. Persistence of psychotic symptoms as an indicator of cognitive impairment in methamphetamine users. Drug Alcohol Depend. 2015, 148, 158–164. [Google Scholar] [CrossRef]
- London, E.D.; Kohno, M.; Morales, A.M.; Ballard, M.E. Chronic methamphetamine abuse and corticostriatal deficits revealed by neuroimaging. Brain Res. 2015, 1628, 174–185. [Google Scholar] [CrossRef]
- Potvin, S.; Pelletier, J.; Grot, S.; Hébert, C.; Barr, A.M.; Lecomte, T. Cognitive deficits in individuals with methamphetamine use disorder: A meta-analysis. Addict. Behav. 2018, 80, 154–160. [Google Scholar] [CrossRef]
- Proebstl, L.; Krause, D.; Kamp, F.; Hager, L.; Manz, K.; Schacht-Jablonowsky, M.; Straif, M.; Riebschläger, M.; Neumann, S.; Schreiber, A.; et al. Methamphetamine withdrawal and the restoration of cognitive functions—A study over a course of 6 months abstinence. Psychiatry Res. 2019, 281, 112599. [Google Scholar] [CrossRef]
- D’Amelio, M.; Serra, L.; Bozzali, M. Ventral Tegmental Area in Prodromal Alzheimer’s Disease: Bridging the Gap between Mice and Humans. J. Alzheimer’s Dis. 2018, 63, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Serra, L.; D’Amelio, M.; Di Domenico, C.; Dipasquale, O.; Marra, C.; Mercuri, N.B.; Caltagirone, C.; Cercignani, M.; Bozzali, M. In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer’s disease. Neurobiol. Aging 2018, 72, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef]
- Vecchio, L.M.; Sullivan, P.; Dunn, A.R.; Bermejo, M.K.; Fu, R.; Masoud, S.T.; Gregersen, E.; Urs, N.M.; Nazari, R.; Jensen, P.H.; et al. Enhanced tyrosine hydroxylase activity induces oxidative stress, causes accumulation of autotoxic catecholamine metabolites, and augments amphetamine effects in vivo. J. Neurochem. 2021, 158, 960–979. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2020, 130, 422–437. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | METH | ASN | ASN + METH | |
|---|---|---|---|---|
| Immunofluorescent area (μm2)/cell | 2.67 ± 0.27 | 62.31 ± 10.17 * | 0.72 ± 0.03 | 23.47 ± 4.70 # |
| n° Tot cells | 26 | 24 | 25 | 20 |
| Controls | METH | GLN | GLN + METH | |
| Immunofluorescent area (μm2)/cell | 1.53 ± 0.30 | 78.14 ± 6.20 * | 0.86 ± 0.21 | 19.68 ± 2.21 # |
| n° Tot cells | 25 | 24 | 24 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrucci, M.; Biagioni, F.; Busceti, C.L.; Vidoni, C.; Castino, R.; Isidoro, C.; Ryskalin, L.; Frati, A.; Puglisi-Allegra, S.; Fornai, F. Inhibition of Autophagy In Vivo Extends Methamphetamine Toxicity to Mesencephalic Cell Bodies. Pharmaceuticals 2021, 14, 1003. https://doi.org/10.3390/ph14101003
Ferrucci M, Biagioni F, Busceti CL, Vidoni C, Castino R, Isidoro C, Ryskalin L, Frati A, Puglisi-Allegra S, Fornai F. Inhibition of Autophagy In Vivo Extends Methamphetamine Toxicity to Mesencephalic Cell Bodies. Pharmaceuticals. 2021; 14(10):1003. https://doi.org/10.3390/ph14101003
Chicago/Turabian StyleFerrucci, Michela, Francesca Biagioni, Carla L. Busceti, Chiara Vidoni, Roberta Castino, Ciro Isidoro, Larisa Ryskalin, Alessandro Frati, Stefano Puglisi-Allegra, and Francesco Fornai. 2021. "Inhibition of Autophagy In Vivo Extends Methamphetamine Toxicity to Mesencephalic Cell Bodies" Pharmaceuticals 14, no. 10: 1003. https://doi.org/10.3390/ph14101003
APA StyleFerrucci, M., Biagioni, F., Busceti, C. L., Vidoni, C., Castino, R., Isidoro, C., Ryskalin, L., Frati, A., Puglisi-Allegra, S., & Fornai, F. (2021). Inhibition of Autophagy In Vivo Extends Methamphetamine Toxicity to Mesencephalic Cell Bodies. Pharmaceuticals, 14(10), 1003. https://doi.org/10.3390/ph14101003