SAR Analysis of Small Molecules Interfering with Energy-Metabolism in Mycobacterium tuberculosis




Abstract
1. Introduction
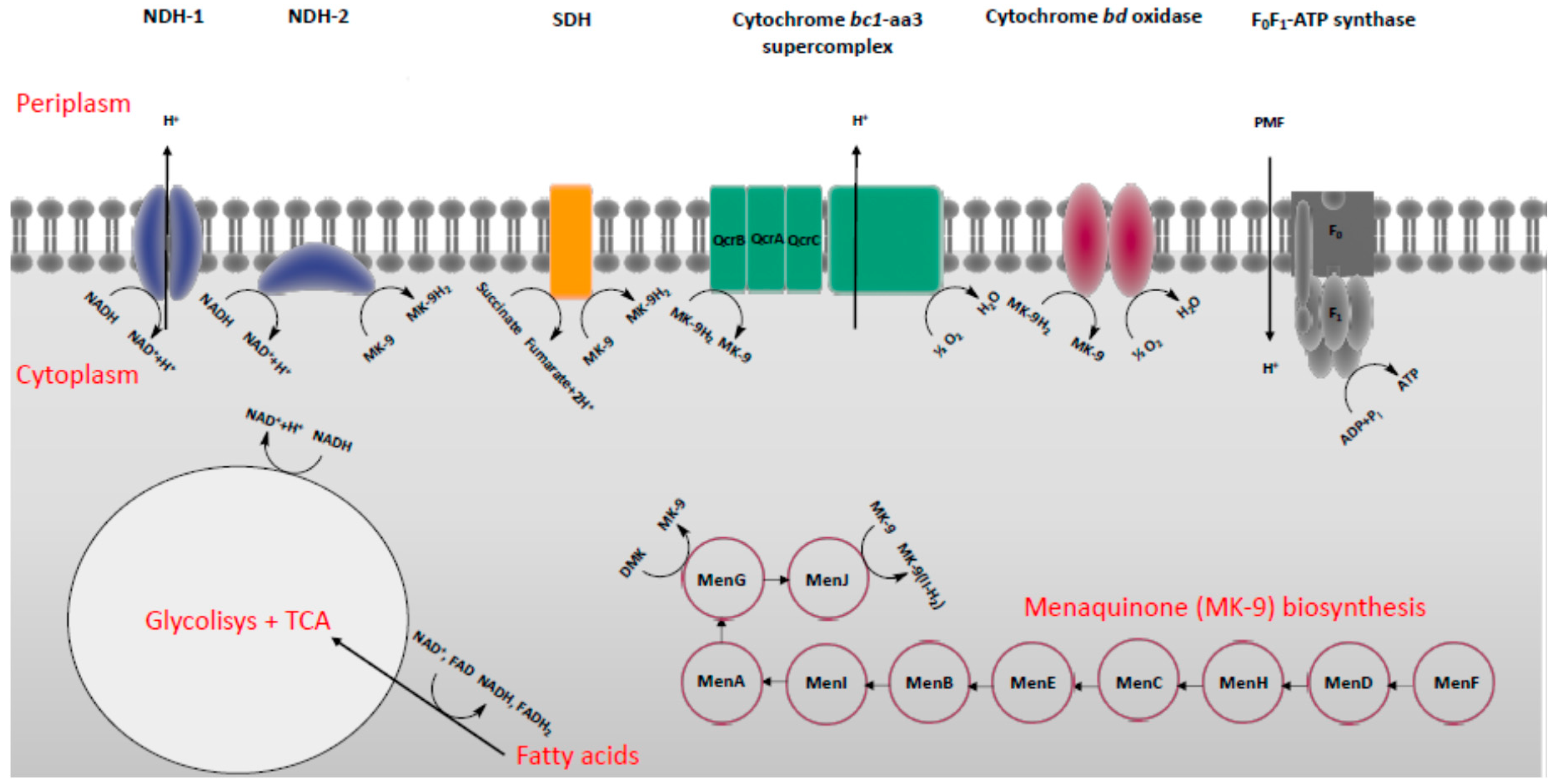
2. Energy-Metabolism in Mycobacterium Tuberculosis
3. Classification of Drugs Targeting Energy-Metabolism in Mtb
3.1. Inhibitors of NDH-2
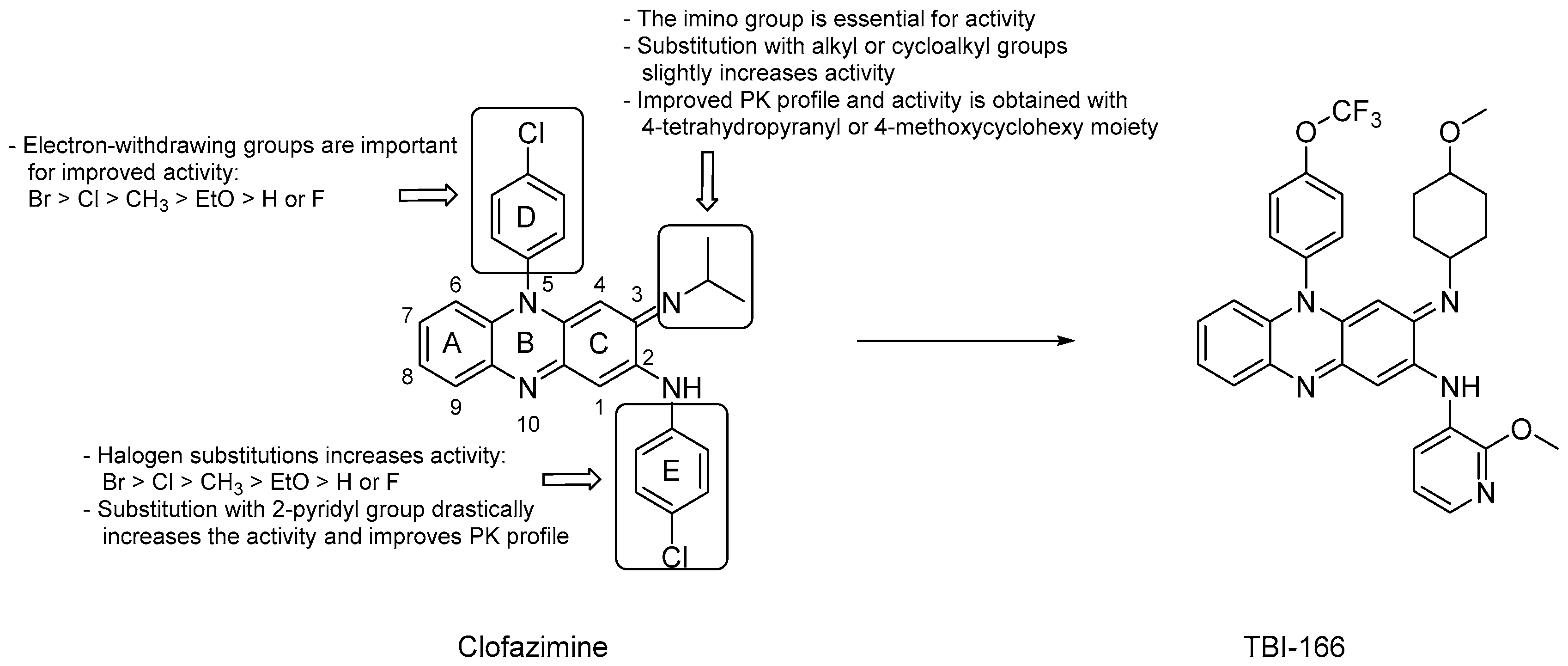
3.1.1. Riminophenazines
- deletion of the A, D or E aromatic rings led to a significant loss of the mycobactericidal activity [29];
- substitution of the iminium group at C3 with an alkyl or cycloalkyl group slightly increased the activity;
- halogen substitution on the phenyl rings was not essential for the anti-TB activity, but when substituted at the para-positions, activity increased according to the following order: Br > Cl > CH3 > EtO > H or F;
- both the phenyl rings at C2 and N5 can be substituted with a pyridyl ring and, notably, the replacement with a 2-pyridyl group at C2 drastically increased the activity and led to favorable PK properties [29];
- replacing the isopropyl group on the imino nitrogen with either a 4-thetrahydopyranyl or a 4-methoxycyclohexyl moiety led to improved activity and more favorable PK [30];
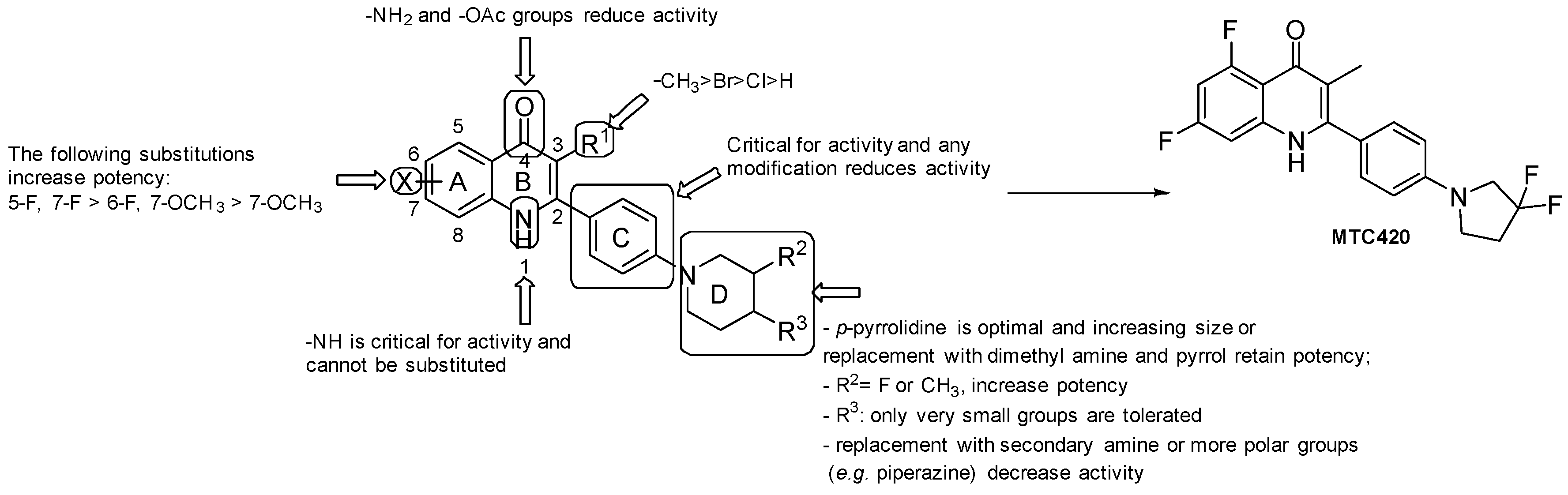
3.1.2. Quinoline/Quinolone Derivatives
- the presence of -NH2 and -OAc at the 4-position led to inactive molecules;
- the replacement of the phenyl ring at position 2 with a pyridyl ring or any other modification resulted in a loss of activity;
- the most favorable X groups on the A ring are: 5-F, 7-F > 6-F, 7-OCH3 > 7-OCH3;
- any substitution of the H on the N1 nitrogen led to a loss of activity;
- substitution at C3 with H and halogenation led to a loss of activity. Compound-specific exception resulted from substitution with Br, but generally, the -CH3 group is considered optimal at this position;
- the piperidine of the side chain has to be located at the para position; small groups at the 4-position are more tolerated than larger ones, such as -CF3 and cyclopropyl; the F and -CH3 groups at the 3-position resulted in improvements in anti-TB activity; when increasing the ring size of the piperidine, the presence of dimethyl amine and substitution with a pyrrole retained potency.
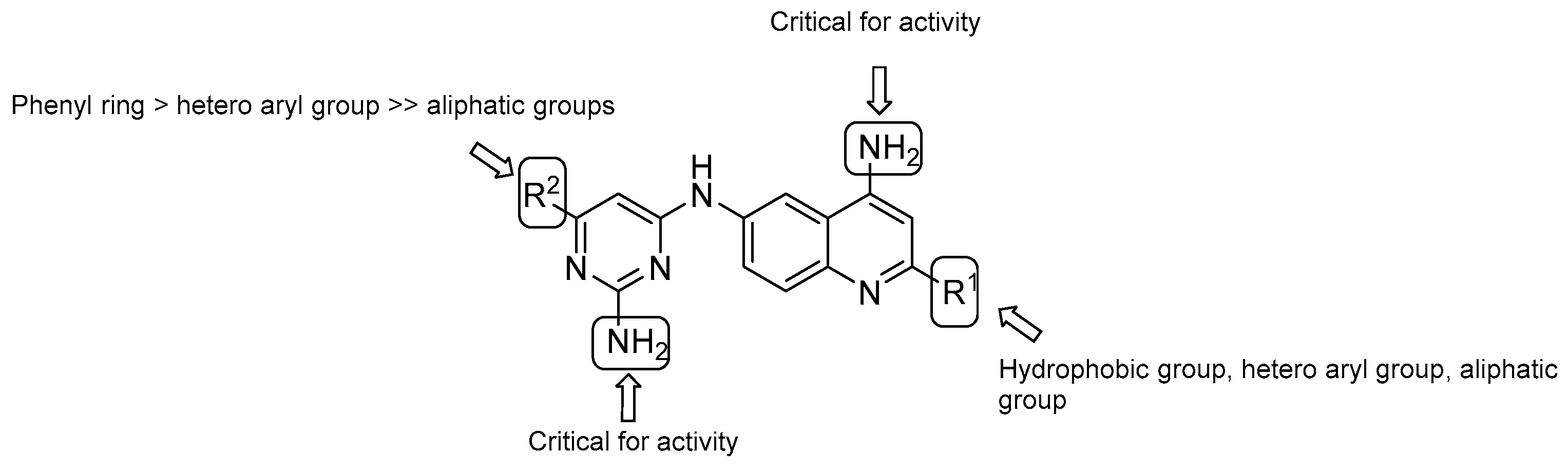
- both –NH2 groups on the quinolone and the pyrimidine rings are critical for activity;
- in vitro evaluation showed an increasing potency regarding the monosubstitution at R2 in the following order: phenyl ring > hetero aryl groups >> aliphatic groups;
- on the quinoline ring, substitution at R1 displays a similar trend as the pyrimidine ring (R2), with the most potent substitution group to be a 4-F or 2-OCH3-phenyl ring.
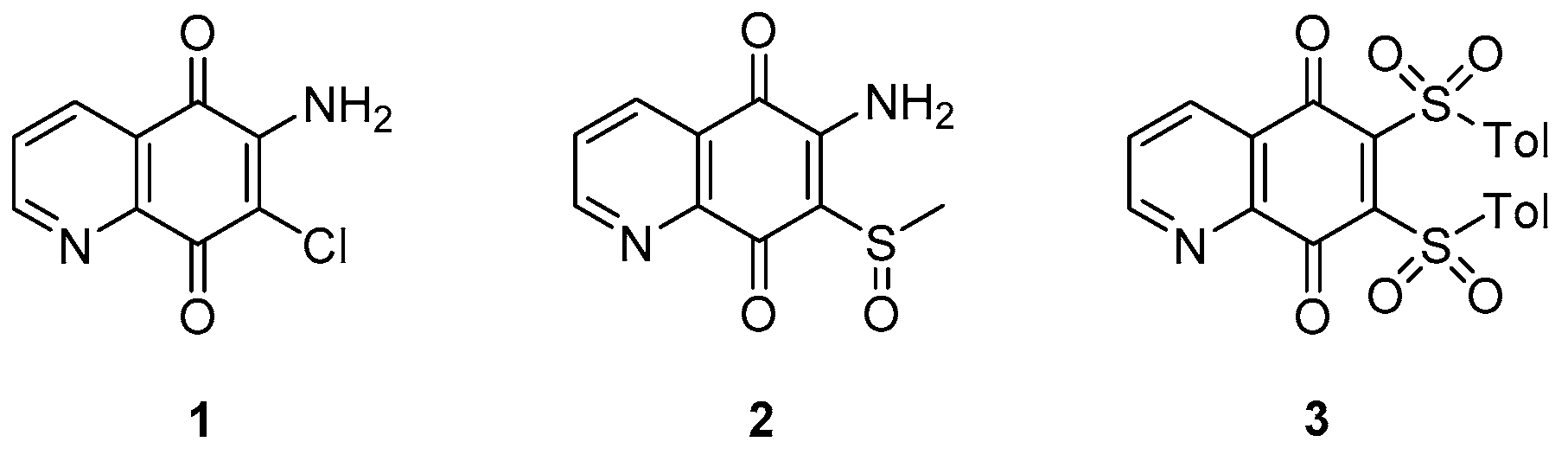
3.1.3. Thioquinazolines and Tetrahydroindazoles
- the pyrimidone core (unit B) cannot be modified;
- generally, increasing/decreasing the saturated ring size brings a reduction of activity, although the solubility slightly increases with the related 5-membered ring;
- replacement of the saturated fused ring with a phenyl (quinazolinone) core increases the anti-TB activity; fluorine analogues were synthesized to increase stability but are less soluble;
- oxidation or replacement of the thioether with O, N, Me and N-methylation of the amide and/or the methylene linker induces to a reduction of activity; thus, the linker modification is generally not tolerated;
- contraction of the cyclohexyl amide at the side chain to cyclopropane or cyclobutane leads to a decrease of activity that is almost retained with cyclopentane and cycloheptane; furthermore, substitution with aromatic rings or disruption of the cycle increases MIC values, suggesting that a bulky hydrophobic group is required to obtain good whole-cell potency;
- -F, -Cl, and -Me substituents are well tolerated on the cyclohexyl ring, with the best activity reached upon substitution with a 4-gem-difluoro;
- solubility significantly increases with -CH2-gem-difluoro cyclohexyl at the end of the side chain.
- resistant MTb strains showed 50-fold higher expression of the gene ndhA;
- neither compound (100 μM) inhibited the growth of M. smegmatis at 48 °C when NDH-2 activity is replaced by the malate/menaquinone oxidoreductase;
- TQZ and THI scaffolds are related to the quinone and adenine molecules;
- TQZ has been previously reported to inhibit the NDH-2 [41].
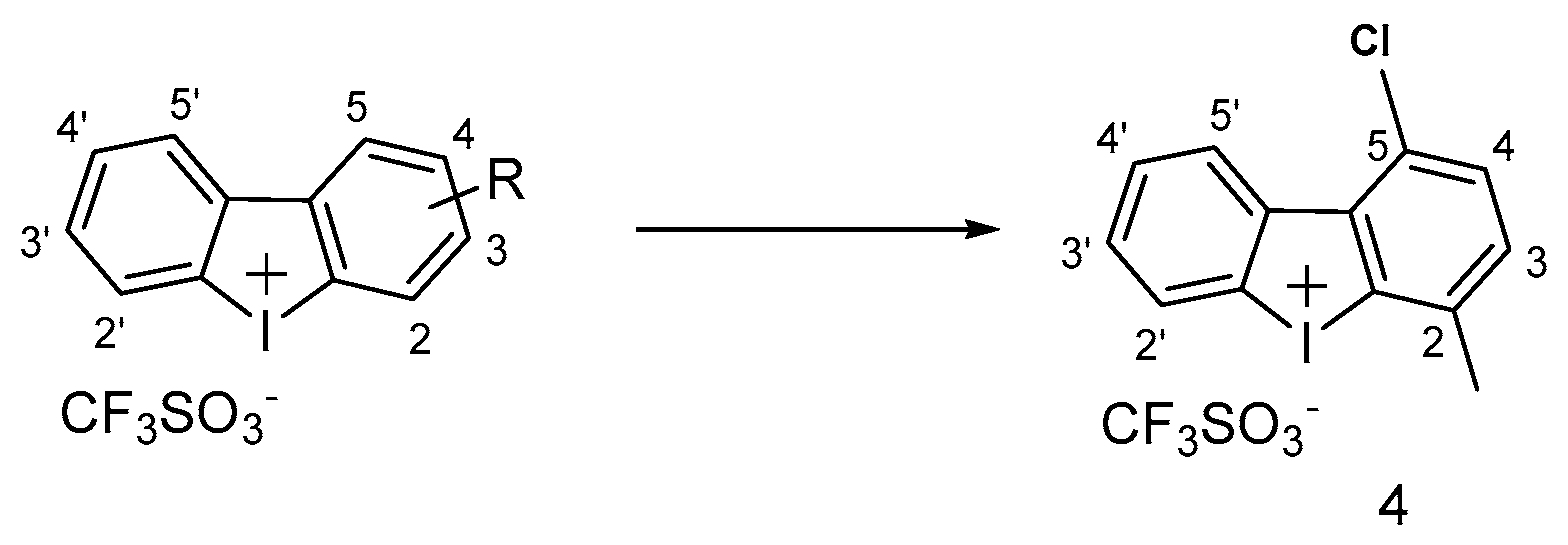
3.1.4. Iodonium Derivatives
3.2. Inhibitors of Cytochrome bc1
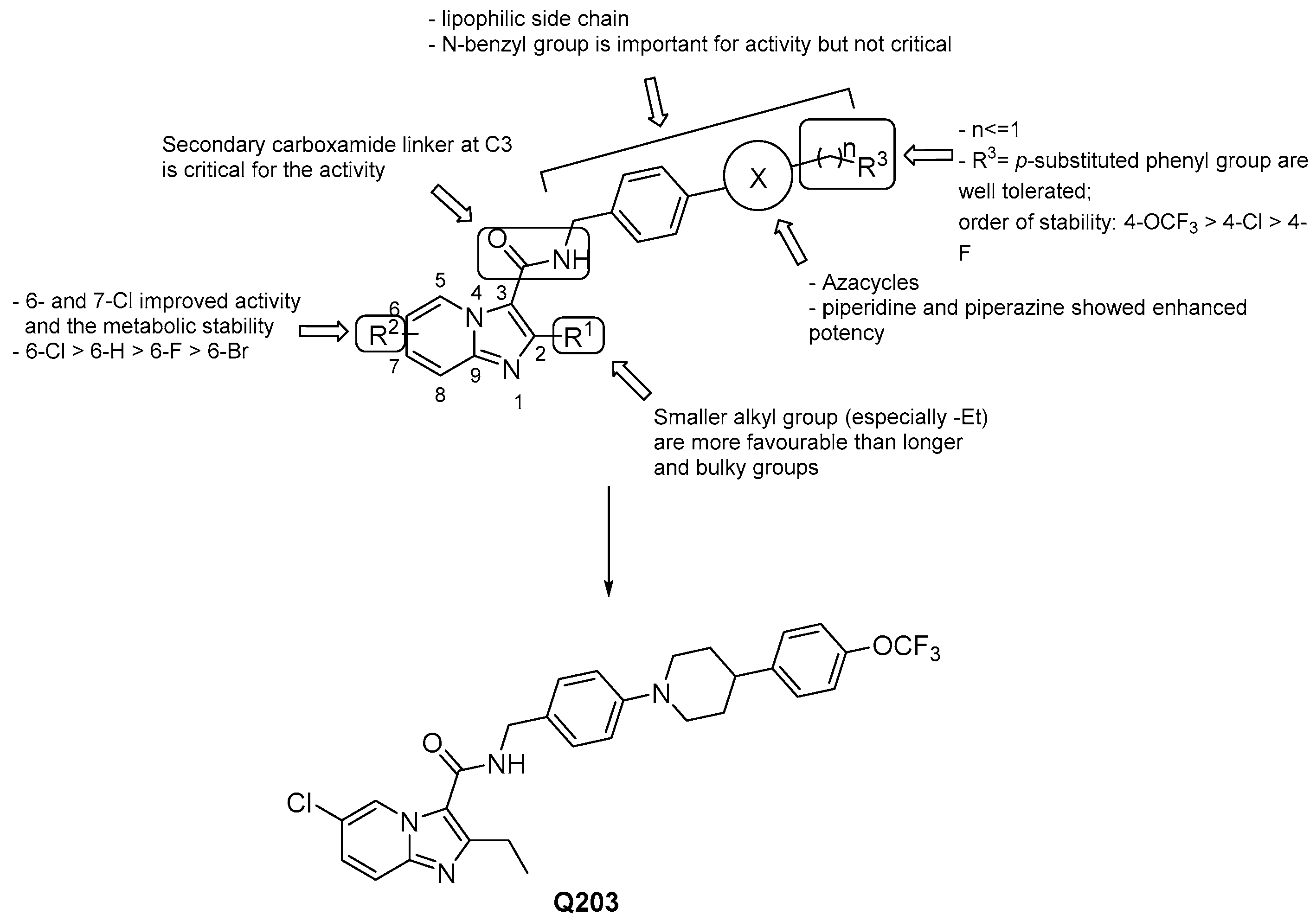
3.2.1. Imidazo [1,2–a] pyridine-3-carboxamides
- a lipophilic side chain is pivotal for the activity, regardless of chain length and linearity [52];
- switching the position of the carboxamide from 3 to 2 results in less effective derivatives.
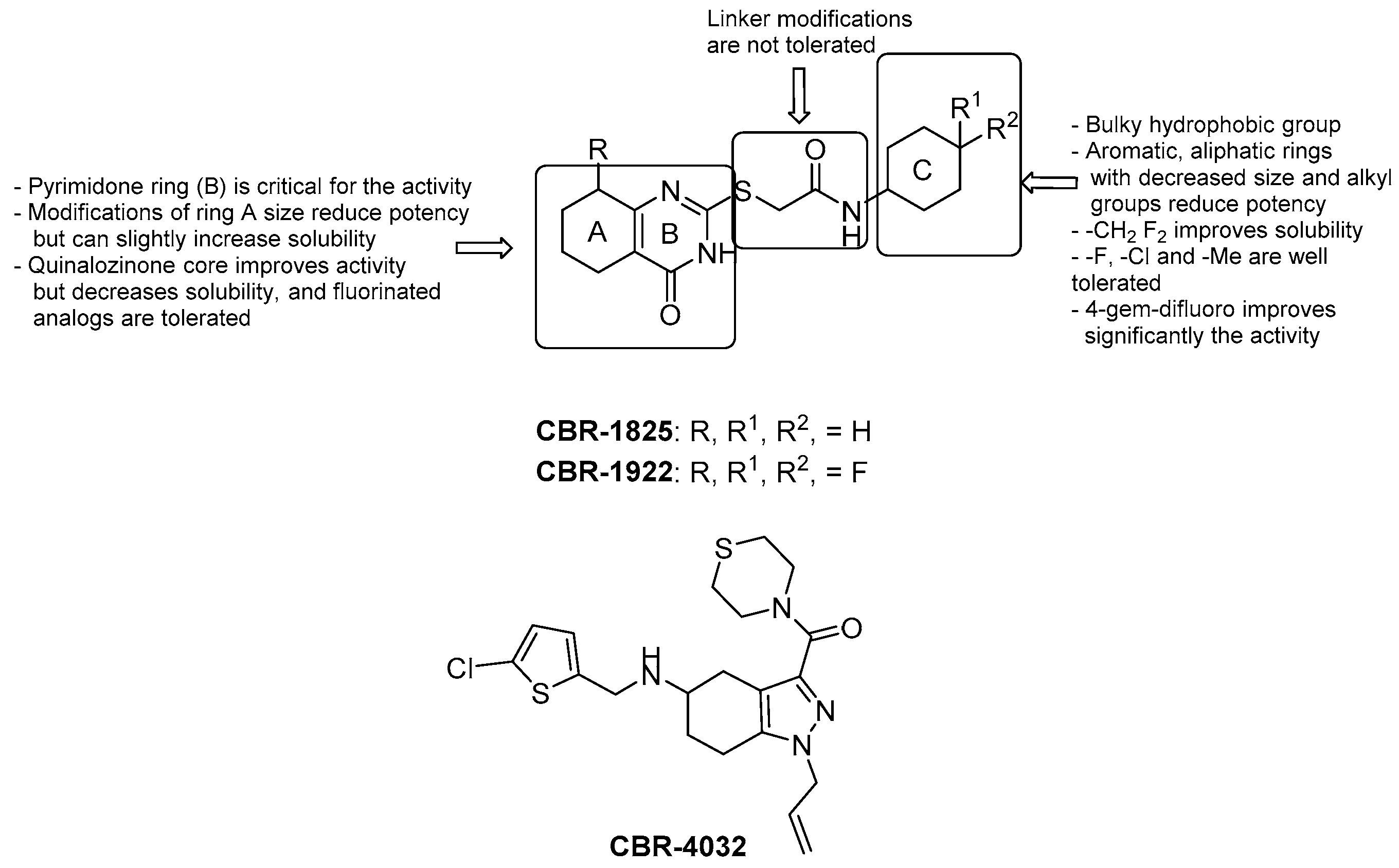
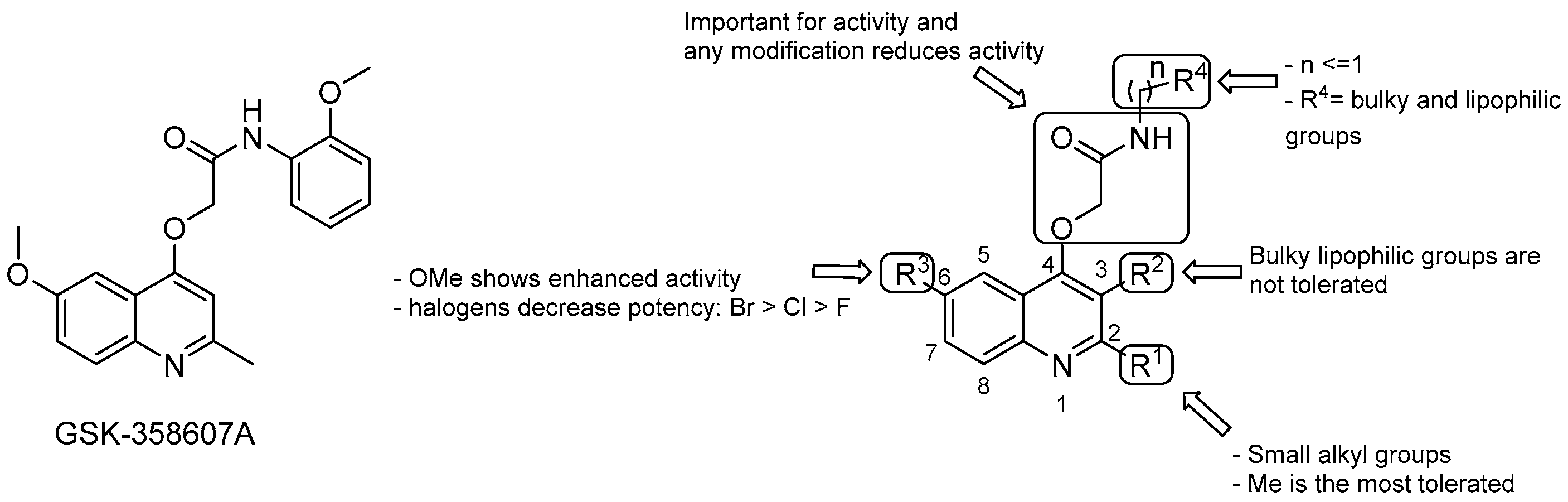
3.2.2. 2-(Quinolin-4-yloxy) Acetamides
- the Me group at R1 seems to be the most suitable one, and as alkyl chain length increases, the potency diminishes;
- the presence of a bulky lipophilic benzyl group at R2 results in a decrease of potency for the respective compound;
- the best group at R3 is the methoxyl one; replacement with halogens decreases the potency (Br > Cl > F);
- bulky and lipophilic substituents of limited conformational flexibility (n ≤ 1) at R4 improve anti-TB activity, regardless of the aromaticity and planarity of the eventual ring;
- the oxygen at position 4 plays an essential role for the activity; thus, substitution with NH to increase the solubility is not tolerated;
- removal or switching to a secondary group or even replacement with bioisosters [69] of the primary amide at the side chain decreases efficacy.
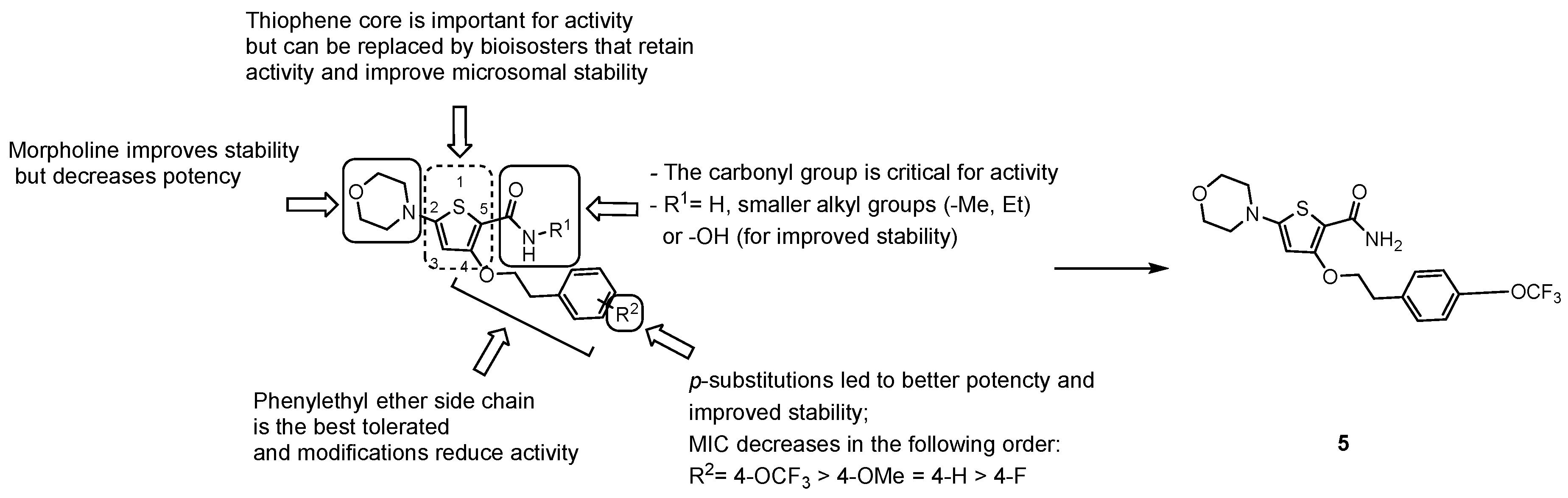
3.2.3. New Chemical Entities
- the carbonyl group of the primary amide is fundamental for the activity and only substitution of the primary amide, to retain activity (e.g., N-methyl-amide) or to increase metabolic stability (e.g., hydroxamic acid), are tolerated;
- the morpholine ring cannot be replaced. Only a few modifications to improve metabolic stability are allowed (e.g., morpholine-3-one), but decrease potency;
- only an ethyl ether linker attached to a p-substituted phenyl ring is tolerated at position 4; moreover, constraining the ethyl linker led to a loss of activity even if it improved stability, and saturation or heterocyclic substitution of the phenyl ring increases polarity but induces a loss of potency;
- the thiophene core may be replaced by bioisosters to improve microsomal stability while maintaining whole-cell activity.
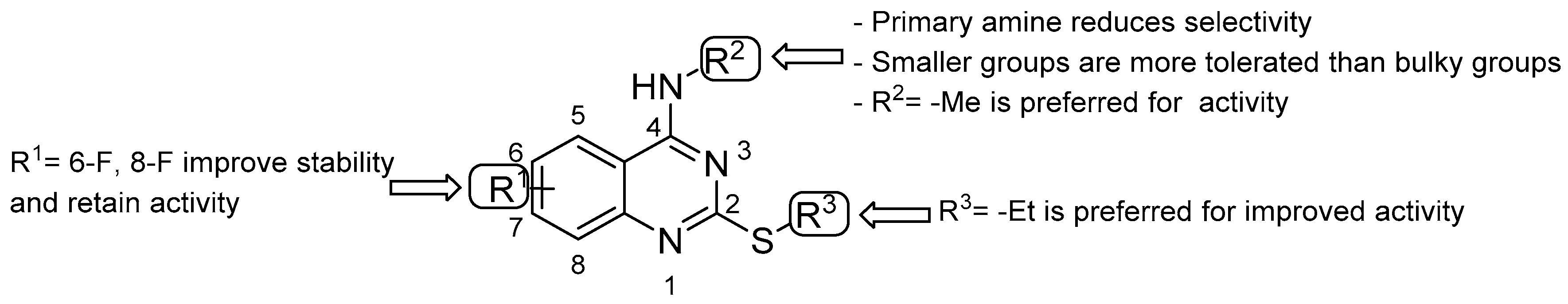
- double substitution with two F atoms at both positions 6 and 8 improves the stability of the metabolism;
- the ethyl group at the thioalkyl side chain (position 2) is the most suitable;
- the secondary amine at position 4 with the smallest substitution (methylamine) is more effective for anti-TB activity; the primary amine or bulky substituents have a detrimental effect on the activity.
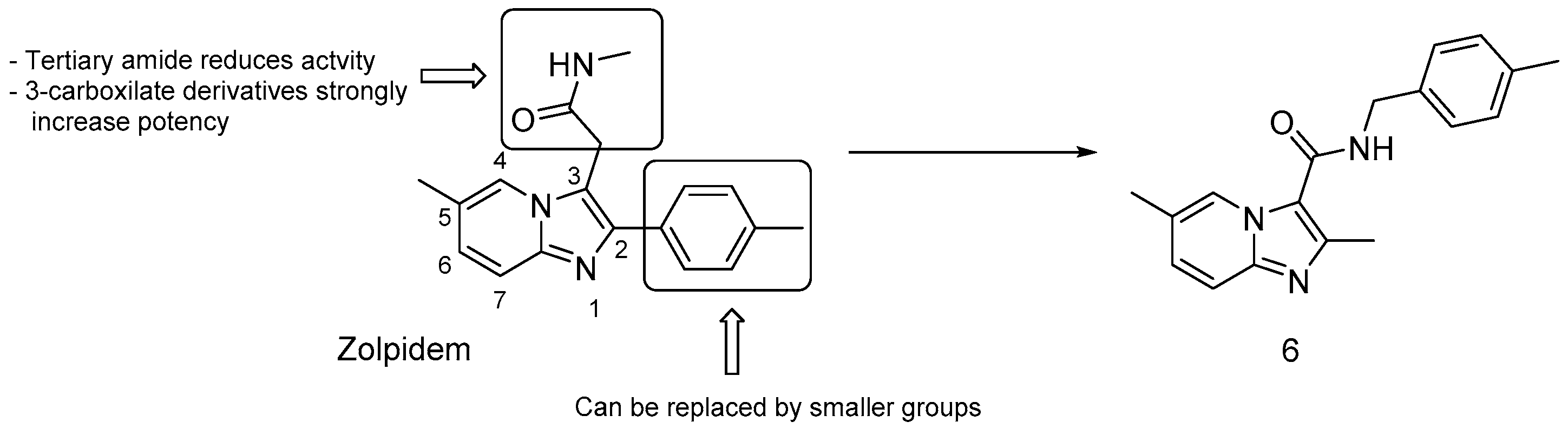
3.2.4. Repurposed Drugs: Zolpidem and Lansoprazole
- bulky groups at position 2 are not essential; thus, the 2-tolyl moiety in Zolpidem can be replaced by a methyl group;
- 3-carboxilate derivatives are much more potent than 3-oxoacetamide and 3-acetamide derivatives;
- secondary amides are more effective than tertiary amides that lack a hydrogen bond donor.
3.3. Inhibitors of Cytochrome bd
3.4. Inhibitors of Menaquinone Biosynthesis
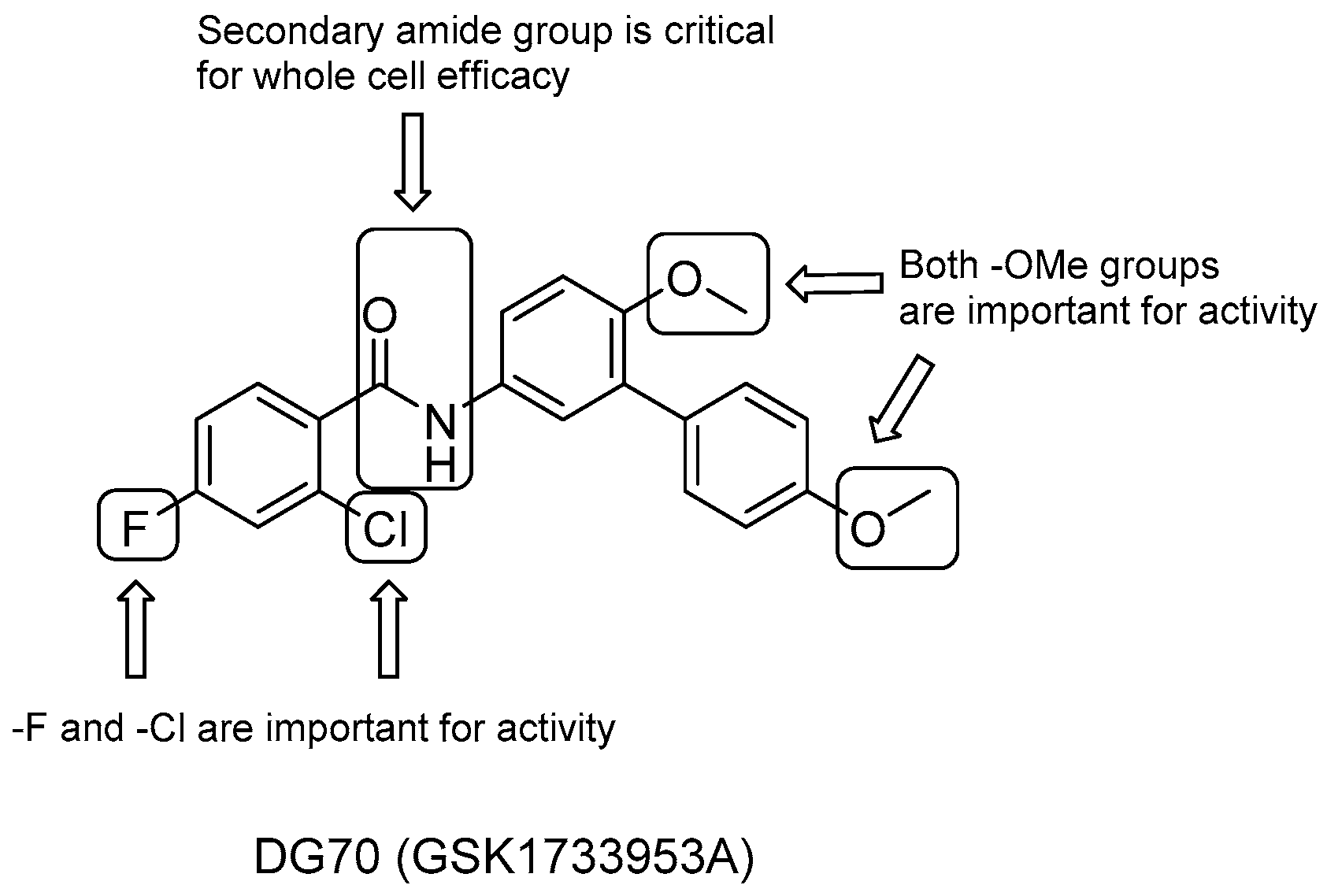
DG70
- the role of the two methoxyl groups appears to be critical for whole-cell efficacy since either removal or substitution with -OH diminishes the activity; a similar loss of anti-TB activity is observed with the eradication of the terminal methoxyphenyl moiety;
- removal of either the F or both F and Cl on the benzoyl moiety causes a small decrease in activity, which is completely lost after the elimination of the solely Cl group;
- the presence of secondary amide is also critical to whole-cell efficacy.
3.5. Inhibitors of FoF1-ATP Synthase (ATPase)
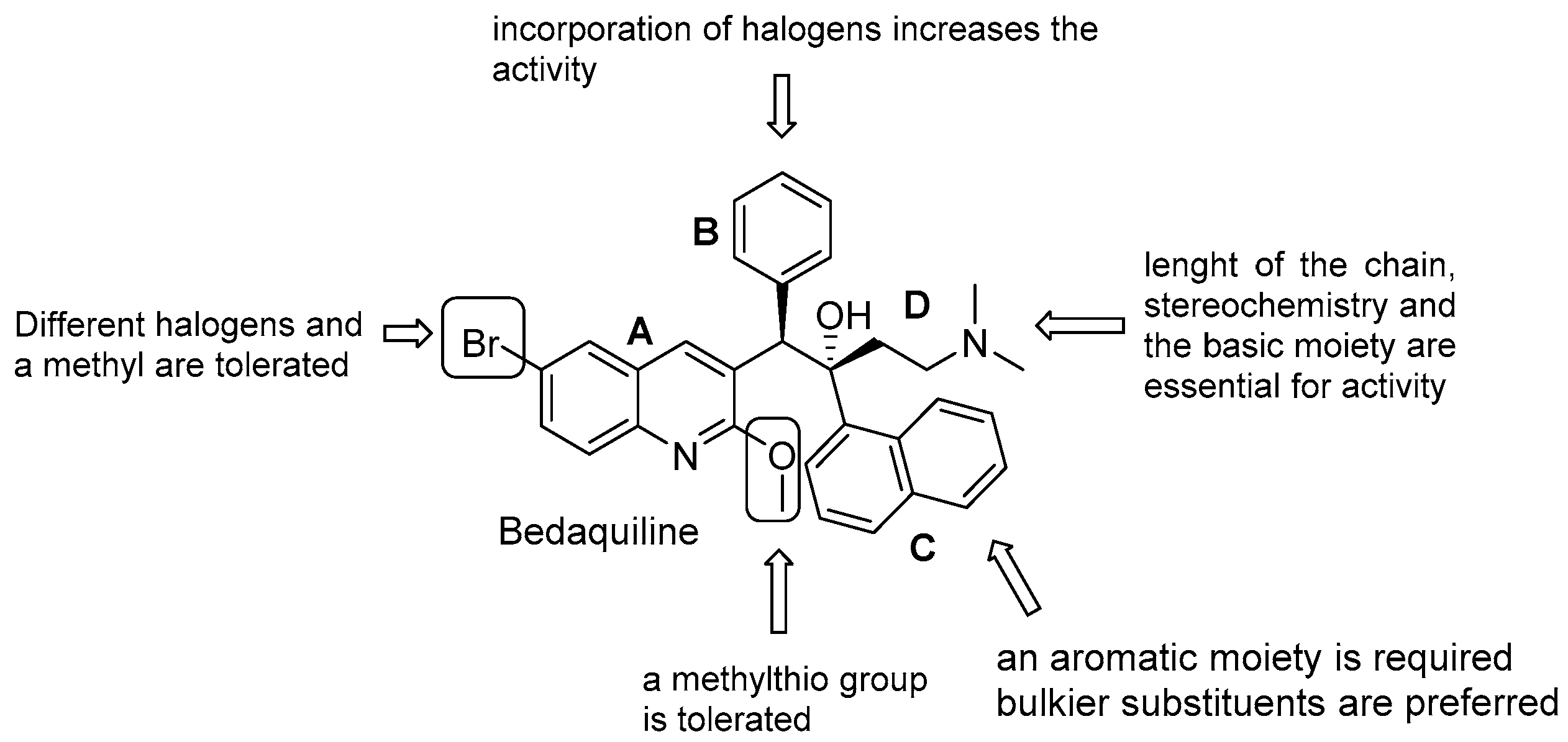
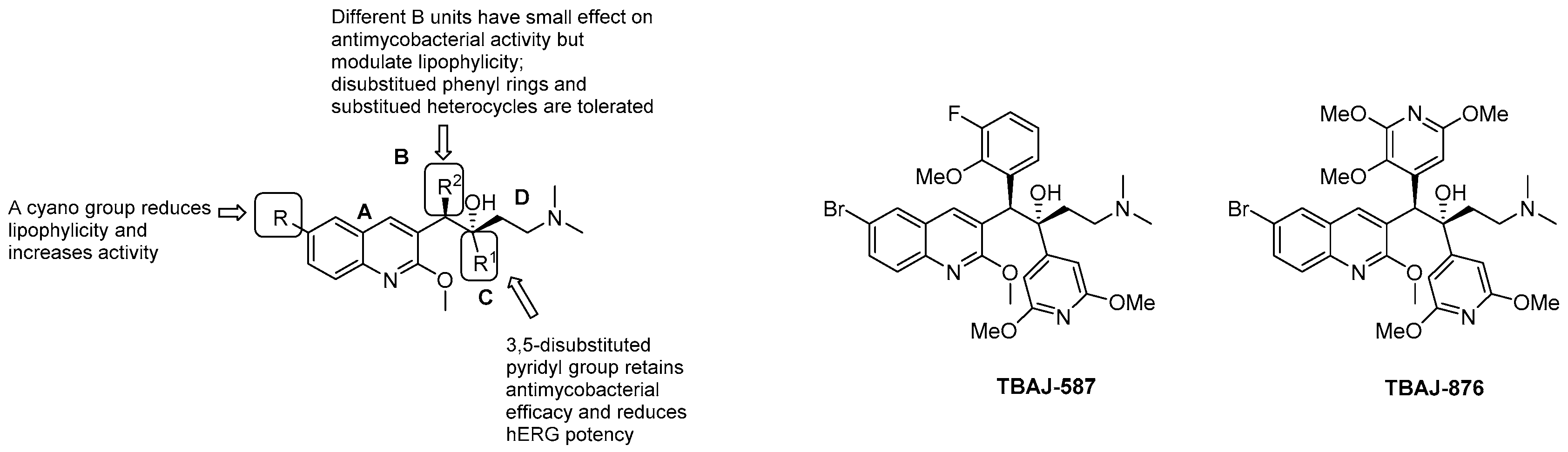
3.5.1. Diarilquinolines (DARQs)
- quinoline core (Unit A): (i) omitting substituents at C2 and C6 is deleterious for the activity; (ii) different halogens or a methyl moiety at position 6 are allowed, but an excess of electron density is not tolerated; (iii) a methylthio moiety can replace the methoxy at C2; (iv) the combination of a halogen at position 6 and methoxy or methylthio at position 2 gives the optimal activity;
- first phenyl ring (Unit B): incorporation of halogens increases the activity;
- second phenyl ring (Unit C): (i) a lipophilic group is essential for activity; (ii) an aromatic ring is required; (iii) an electron-withdrawing substituent and di-substituents on the phenyl ring are well tolerated; (iv) a bulkier substituent is preferred;
- C3 side chain (Unit D): this unit does not tolerate modifications. The two hydrogen bonding acceptors/donors are crucial for activity and any attempt at modifying the length of the chain, the basicity of the amino group, and the configuration of the two stereogenic centers was detrimental.
3.5.2. Squaramides
- a 2-pyridyl moiety is critical for activity, suggesting that the nitrogen is involved in hydrogen bonding with the target;
- replacing the -CF3 group with a more hydrophylic one (such as a cyano) is detrimental for activity, whereas a morpholino ring, having the potential for hydrogen bonding and increased size, has a positive impact.
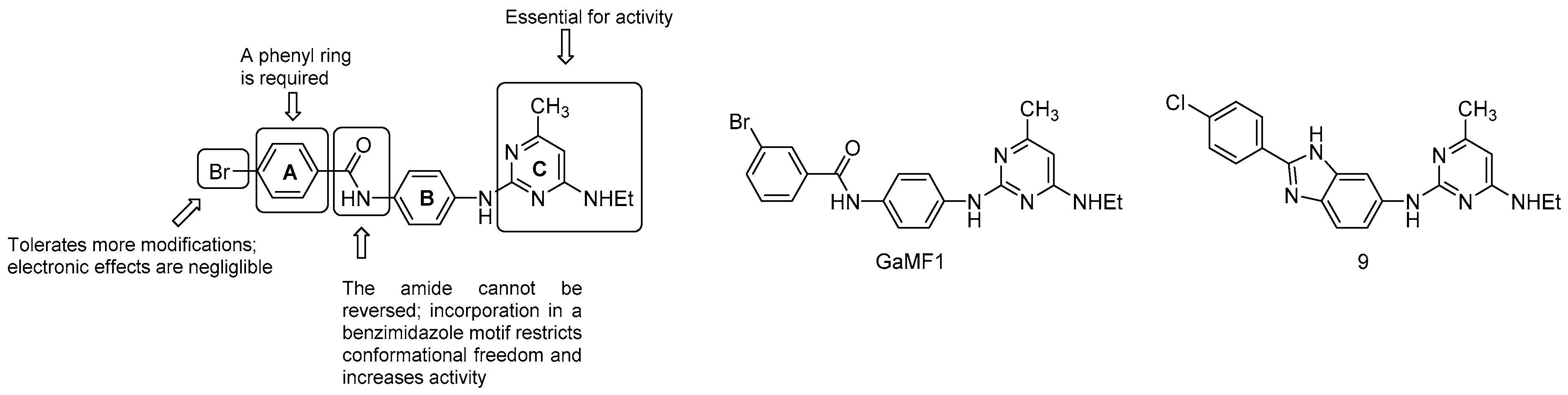
3.5.3. Miscellaneous Compounds
- the C ring is essential;
- the amide link between the A and B ring cannot be reversed; however, the incorporation in a benzimidazole motif to restrict conformational freedom is possible and strongly increases potency (compound 9, MIC50 = 3 μM);
- the phenyl ring A is required but is more amenable to modifications: the electronics of the aromatic ring does not affect the potency.
3.6. Drugs Dissipating the Proton Motive Force (PMF)
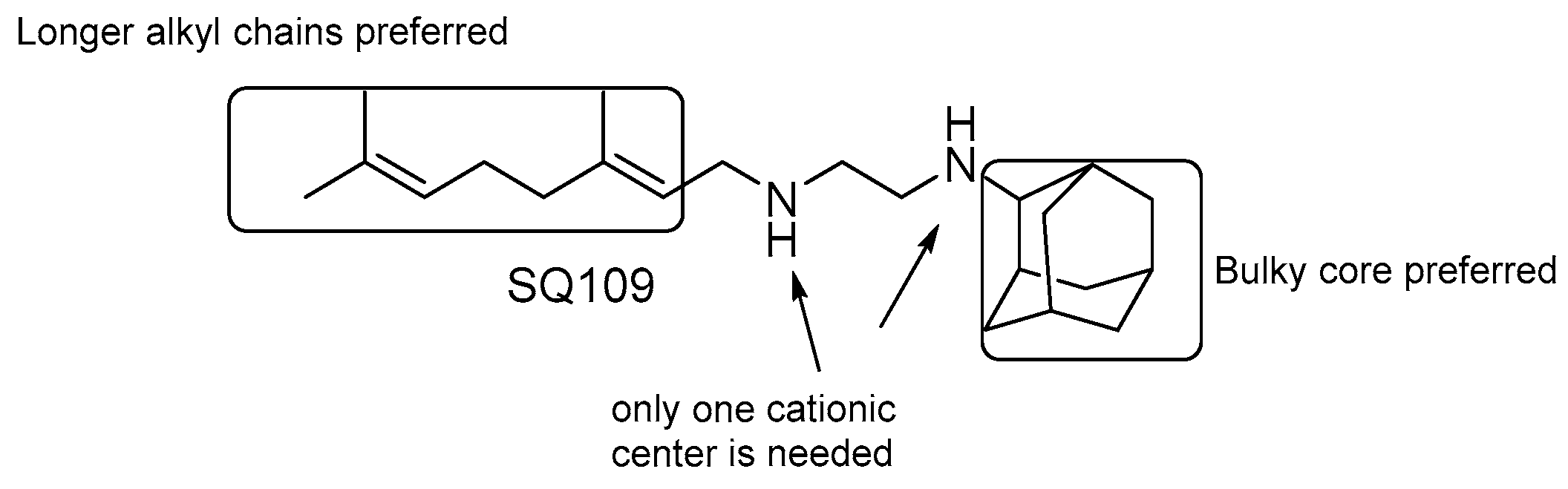
SQ109
- the presence of two nitrogens is not essential for the activity, only one cationic center is needed [124];
- both size and nature of the alkyl substituents scaffold are critical for activity;
- highly α-branched aliphatic moieties are more effective.
3.7. SAR of Small Molecule Inhibitors Targeting CCM
3.7.1. Inhibitors of Isocitrate Lyase
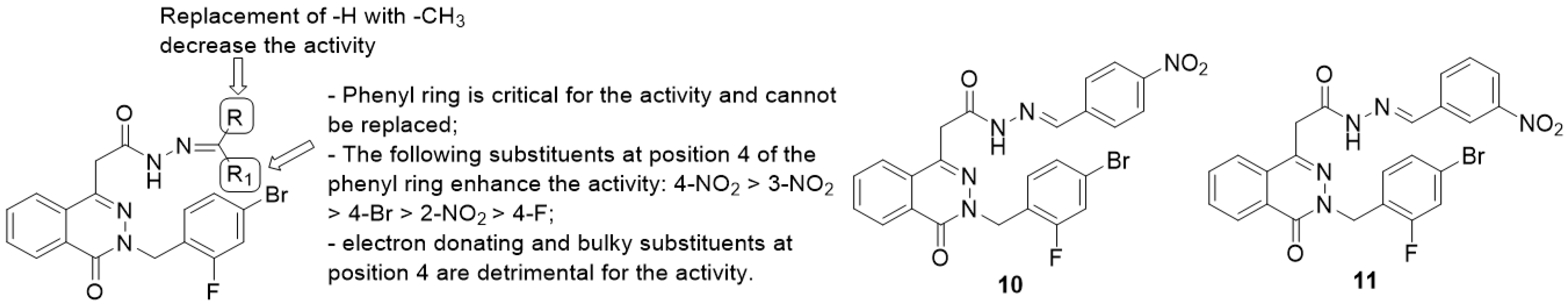
Phthalazinyl Hydrazones and Phthalazin-4-ylacetamide Derivatives
- the phenyl ring is fundamental for the activity while the replacement with heteroaryls, such as the furanyl ring, has a negative impact;
- introduction of electron-withdrawing groups at position 4 of the phenyl ring provided the highest enhancement of the activity (4-NO2 > 3-NO2 > 2-NO2), particularly with the insertion of halogens and nitro groups (4-NO2 > 4-Br > 4-F);
- electron-donating groups (methyl, hydroxyl, methoxyl and dimethylamino substituents) and bulky groups at position 4 of the phenyl ring are detrimental for the activity;
- substitution of -R with methyl group provided less-active compounds.
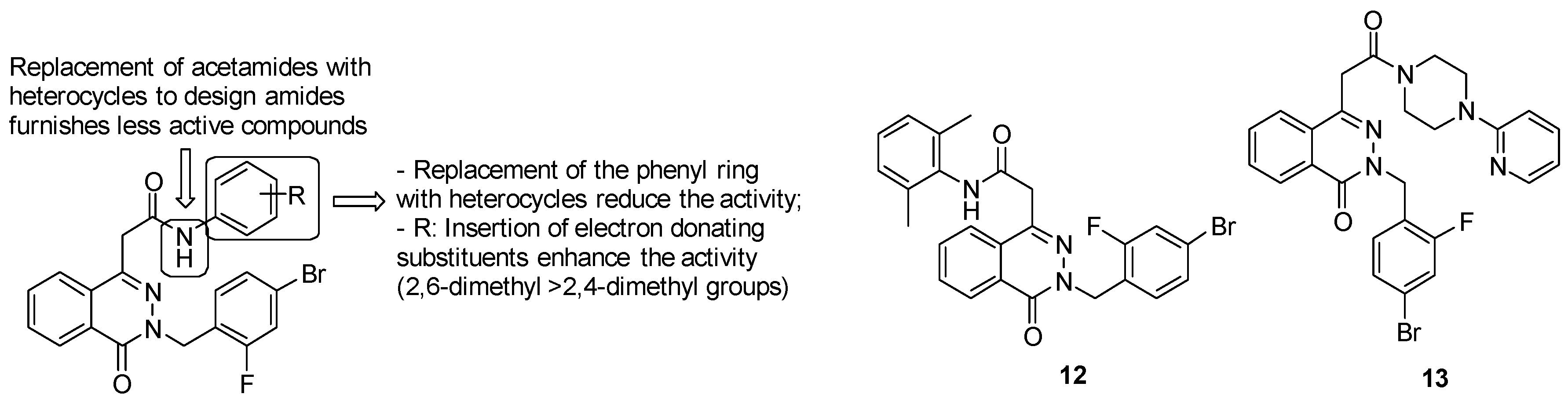
- acetamide derivatives substituted with the phenyl ring provide high activity, while replacement of acetamide moiety with heterocycles led to less active compounds;
- replacement of the phenyl ring with another ring system, such as the pyridyl one, reduces the activity;
- insertion of electron donating groups, such as methyl one, on the ring strongly enhances the antimycobacterial activity.
Salycilanilides
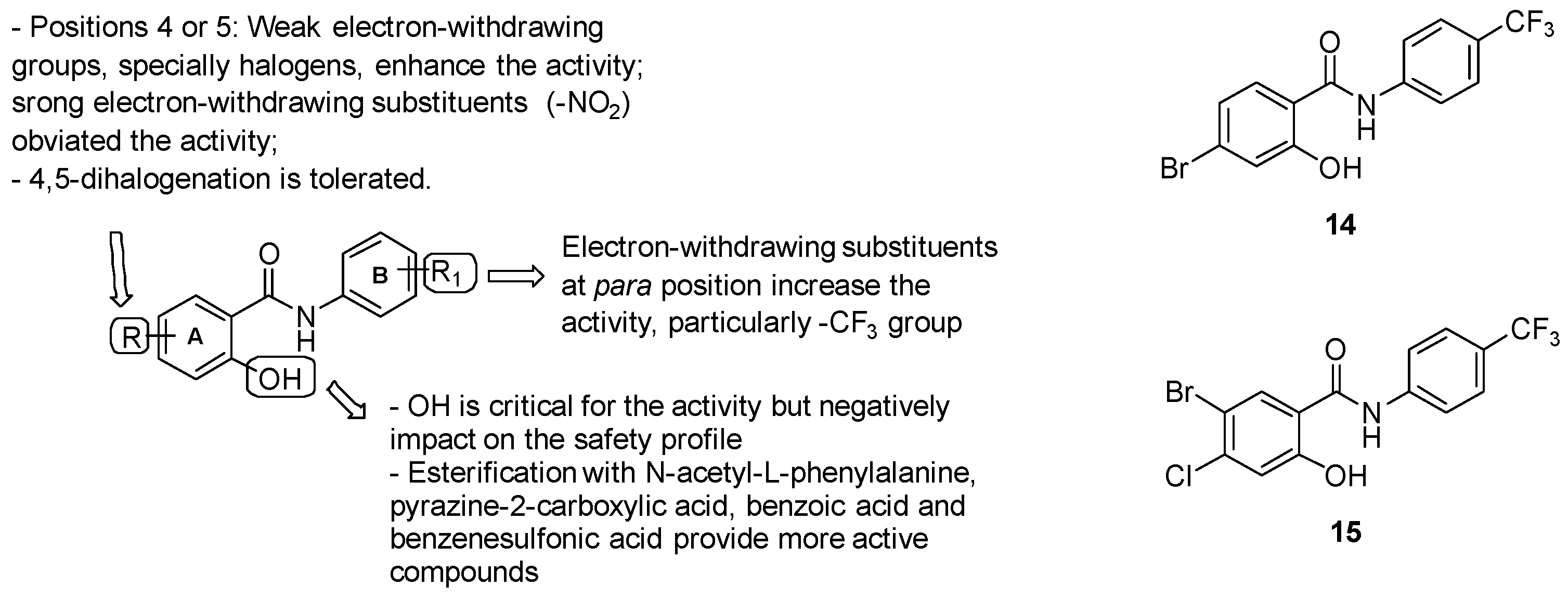
- ring A: substitution with weak electron-withdrawing groups, particularly halogens, is preferred at positions 4 or 5, while strong electron-withdrawing substituents at the same positions (-NO2) affected the activity; notably, bromine derivatives result in being more active than chlorine analogues, likely due to the higher ClogP values of the final molecules bearing a bromine atom. The free phenolic hydroxyl result was essential for the activity but negatively impacted the safety profile;
- Aniline unit (ring B): the introduction of electron-withdrawing and lipophilic substituents enhanced the activity, with the best results provided by the trifluoromethyl group at position 4.
3.7.2. Inhibitors of Malate Synthase
Phenyl-diketo Acids
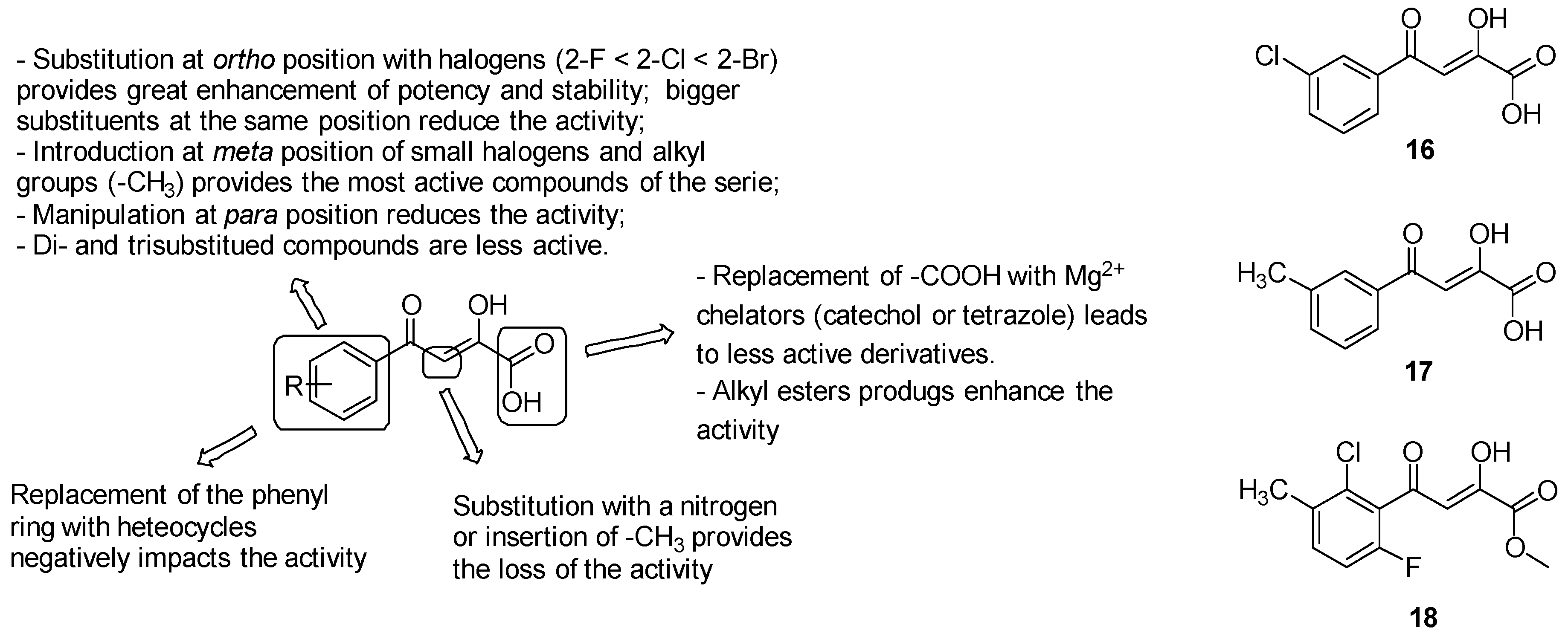
- the oxygens of the diketo acid moiety coordinate Mg2+ and provide hydrogen bonds with Asp462, Leu461, and Asp633, thus removing the diketo acid moiety generated inactive derivatives;
- modifications on the ß-carbon of the diketo acid as substitution with a nitrogen or insertion of a methyl group resulted in the loss of the activity;
- the replacement of the carboxylic acid with well-known Mg2+ chelators, such as catechol or bioisosteres as tetrazole, led to less-active derivatives;
- the aromatic ring is strongly required to establish both van der Waals interactions with Asp633, Met515, Trp541, Met63 and anion-π interactions [143] with the carboxylate of Asp633 side chain that is deprotonated during the catalyst; alternative structures including aliphatic cycles or heterocycles (naphthyl, indole, pyrrole, thiophene, furan, quinoline, benzodioxole, benzothiazole, thiazole, pyridine and pyrimidine) fail to provide a generally favourable combination of inhibitory activity against GlcB and pharmacokinetics;
- o-substituted analogues with small groups, especially halogens (2-F < 2-Cl < 2-Br), provided a great enhancement of potency, while the introduction of bigger or double substituents reduces the activity, probably due to steric interference with the Val118 side chain. Substitutions at the ortho position induced the twist of the phenyl ring out of plane and therefore increased stability by diminishing the degree of conjugation, which is responsible for the retro-Claisen decomposition of unsubstituted PDKAs in in various buffer solutions and cell growth media;
- manipulations at position 4 of the phenyl ring are less effective on the activity due to the undesirable steric clash of the substituents with the Met631 side chain;
- the introduction of substituents at the meta position to extend the parental PDKA structure afforded the best activity and potency by accommodating additional interactions in the acetyl-CoA binding channel, including that of the hydrogen bond with Val119 and van der Waals interactions with Met631, Met515, and Val118-Val119. Notably, the best results have been collected by the introduction of small halogens, such as fluorine, and alkyl groups such as methyl; bigger alkyl and aryl substituents negatively impact the activity, likely due to the steric interference with Met631 and Val118;
- di- and trisubstituted compounds such as 2-Cl-6-F-PDKAs are less active than monosubstituted PDKAs.
3.7.3. Inhibitors of Cholesterol Catabolism
Azasteroids
- position C17: the introduction of aryl/alkyl amides was tolerated; oxazole-bearing derivatives exhibited highly ameliorated activity, while the incorporation of carboxylic acid and polar heterocycles was detrimental for the activity; compound 19 with the hydrophobic 8-carbon side-chain at the C17 position, which mostly mimics the cholestenone substrate, proved to be the greatest at binding competitive inhibitors;
- substitutions at C1, C2, C7, C4 and N6 on the A-B ring system are poorly effective due to the stringent steric requirements for effective interaction with the enzyme.
3.8. Inhibitors of PrpC (Methyl Citrate Cycle)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global tuberculosis report. 2019. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 8 July 2020).
- Pipeline. Working Group for New TB Drugs. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 9 July 2020).
- Shao, M.; McNeil, M.; Cook, G.M.; Lu, X. MmpL3 inhibitors as antituberculosis drugs. Eur. J. Med. Chem. 2020, 200, 112390. [Google Scholar] [CrossRef]
- Poce, G.; Consalvi, S.; Venditti, G.; Alfonso, S.; Desideri, N.; Fernandez-Menendez, R.; Bates, R.H.; Ballell, L.; Barros Aguirre, D.; Rullas, J.; et al. Novel pyrazole-containing compounds active against Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2019, 10, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Poce, G.; Cocozza, M.; Alfonso, S.; Consalvi, S.; Venditti, G.; Fernandez-Menendez, R.; Bates, R.H.; Barros Aguirre, D.; Ballell, L.; De Logu, A.; et al. In vivo potent BM635 analogue with improved drug-like properties. Eur. J. Med. Chem. 2018, 145, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.-S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef] [PubMed]
- Piton, J.; Foo, C.S.-Y.; Cole, S.T. Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov. Today 2017, 22, 526–533. [Google Scholar] [CrossRef]
- Gawad, J.; Bonde, C. Decaprenyl-phosphoryl-ribose 2′-epimerase (DprE1): Challenging target for antitubercular drug discovery. Chem. Cent. J. 2018, 12, 72. [Google Scholar] [CrossRef]
- Christophe, T.; Jackson, M.; Jeon, H.K.; Fenistein, D.; Contreras-Dominguez, M.; Kim, J.; Genovesio, A.; Carralot, J.-P.; Ewann, F.; Kim, E.H.; et al. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog. 2009, 5, e1000645. [Google Scholar] [CrossRef]
- Stanley, S.A.; Kawate, T.; Iwase, N.; Shimizu, M.; Clatworthy, A.E.; Kazyanskaya, E.; Sacchettini, J.C.; Ioerger, T.R.; Siddiqi, N.A.; Minami, S.; et al. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD. Proc. Natl. Acad. Sci. USA 2013, 110, 11565–11570. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinšová, J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012, 19, 6126–6137. [Google Scholar] [CrossRef]
- Li, Y.; Sun, F.; Zhang, W. Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis: Promising but challenging. Drug Dev. Res. 2019, 80, 98–105. [Google Scholar] [CrossRef]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a New paradigm in antimycobacterial drug discovery. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative phosphorylation as a target space for tuberculosis: Success, caution, and future directions. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Hards, K.; Vilchèze, C.; Hartman, T.; Berney, M. Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Kurosu, M.; Begari, E. Vitamin K2 in electron transport system: Are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules 2010, 15, 1531–1553. [Google Scholar] [CrossRef]
- Boersch, M.; Rudrawar, S.; Grant, G.; Zunk, M. Menaquinone biosynthesis inhibition: A review of advancements toward a new antibiotic mechanism. RSC Adv. 2018, 8, 5099–5105. [Google Scholar] [CrossRef]
- Upadhyay, A.; Kumar, S.; Rooker, S.A.; Koehn, J.T.; Crans, D.C.; McNeil, M.R.; Lott, J.S.; Crick, D.C. Mycobacterial MenJ: An oxidoreductase involved in menaquinone biosynthesis. ACS Chem. Biol. 2018, 13, 2498–2507. [Google Scholar] [CrossRef]
- Harikishore, A.; Chong, S.S.M.; Ragunathan, P.; Bates, R.W.; Grüber, G. Targeting the menaquinol binding loop of mycobacterial cytochrome bd oxidase. Mol. Divers. 2020. [Google Scholar] [CrossRef]
- Thompson, A.M.; Denny, W.A. Chapter Four—Inhibitors of enzymes in the electron transport chain of Mycobacterium tuberculosis. In Annual Reports in Medicinal Chemistry; Chibale, K., Ed.; Medicinal Chemistry Approaches to Tuberculosis and Trypanosomiasis; Academic Press: Cambridge, MA, USA, 2019; Volume 52, pp. 97–130. [Google Scholar]
- Weinstein, E.A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A.A.; Duncan, K.; Lonsdale, J.T.; Rubin, H. Inhibitors of type II NADH: Menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. USA 2005, 102, 4548–4553. [Google Scholar] [CrossRef]
- Blaza, J.N.; Bridges, H.R.; Aragão, D.; Dunn, E.A.; Heikal, A.; Cook, G.M.; Nakatani, Y.; Hirst, J. The mechanism of catalysis by type-II NADH:quinone oxidoreductases. Sci Rep. 2017, 7, 40165. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.; Hu, M.; Huo, F.; Guo, S.; Jing, W.; Nuermberger, E.; Lu, Y. Primary Clofazimine and Bedaquiline Resistance among isolates from patients with multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Banerjee, D.K.; Ellard, G.A.; Gammon, P.T.; Waters, M.F. Some observations on the pharmacology of clofazimine (B663). Am. J. Trop. Med. Hyg. 1974, 23, 1110–1115. [Google Scholar] [CrossRef]
- Swanson, R.V.; Adamson, J.; Moodley, C.; Ngcobo, B.; Ammerman, N.C.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Tapley, A.; Bester, L.A.; et al. Pharmacokinetics and pharmacodynamics of clofazimine in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 3042–3051. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef]
- Liu, B.; Liu, K.; Lu, Y.; Zhang, D.; Yang, T.; Li, X.; Ma, C.; Zheng, M.; Wang, B.; Zhang, G.; et al. Systematic evaluation of structure-activity relationships of the riminophenazine class and discovery of a C2 pyridylamino series for the treatment of multidrug-resistant tuberculosis. Molecules 2012, 17, 4545–4559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lu, Y.; Liu, K.; Liu, B.; Wang, J.; Zhang, G.; Zhang, H.; Liu, Y.; Wang, B.; Zheng, M.; et al. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J. Med. Chem. 2012, 55, 8409–8417. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zhang, C.; Zhang, H.; Wang, B.; Xu, J.; Fu, L.; Yin, D.; Cooper, C.B.; Ma, Z.; et al. Synthesis and biological evaluation of novel 2-methoxypyridylamino-substituted riminophenazine derivatives as antituberculosis agents. Molecules 2014, 19, 4380–4394. [Google Scholar] [CrossRef]
- Lu, Y.; Zheng, M.; Wang, B.; Fu, L.; Zhao, W.; Li, P.; Xu, J.; Zhu, H.; Jin, H.; Yin, D.; et al. Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob. Agents Chemother. 2011, 55, 5185–5193. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.; Fu, L.; Zhu, H.; Guo, S.; Huang, H.; Yin, D.; Zhang, Y.; Lu, Y. In vitro and in vivo activities of the riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Biagini, G.A.; Fisher, N.; Shone, A.E.; Mubaraki, M.A.; Srivastava, A.; Hill, A.; Antoine, T.; Warman, A.J.; Davies, J.; Pidathala, C.; et al. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc. Natl. Acad. Sci. USA 2012, 109, 8298–8303. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Patil, S.A. Quinoline: A promising antitubercular target. Biomed. Pharmacother. 2014, 68, 1161–1175. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.D.; Gibbons, P.D.; Leung, S.C.; Amewu, R.; Stocks, P.A.; Stachulski, A.; Horta, P.; Cristiano, M.L.S.; Shone, A.E.; Moss, D.; et al. Rational design, synthesis, and biological evaluation of heterocyclic quinolones targeting the respiratory chain of Mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 3703–3726. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Paul, B.; Roy Choudhury, N.; Kedari, C.; Bandodkar, B.; Ugarkar, B.G. Quinolinyl Pyrimidines: Potent inhibitors of NDH-2 as a novel class of anti-TB agents. ACS Med. Chem. Lett. 2012, 3, 736–740. [Google Scholar] [CrossRef]
- Mulchin, B.J.; Newton, C.G.; Baty, J.W.; Grasso, C.H.; Martin, W.J.; Walton, M.C.; Dangerfield, E.M.; Plunkett, C.H.; Berridge, M.V.; Harper, J.L.; et al. The anti-cancer, anti-inflammatory and tuberculostatic activities of a series of 6,7-substituted-5,8-quinolinequinones. Bioorganic Med. Chem. 2010, 18, 3238–3251. [Google Scholar] [CrossRef]
- Harbut, M.B.; Yang, B.; Liu, R.; Yano, T.; Vilchèze, C.; Cheng, B.; Lockner, J.; Guo, H.; Yu, C.; Franzblau, S.G.; et al. small molecules targeting Mycobacterium tuberculosis Type II NADH dehydrogenase exhibit antimycobacterial activity. Angew. Chem. Int. Ed. Engl. 2018, 57, 3478–3482. [Google Scholar] [CrossRef]
- Murugesan, D.; Ray, P.C.; Bayliss, T.; Prosser, G.A.; Harrison, J.R.; Green, K.; Soares de Melo, C.; Feng, T.-S.; Street, L.J.; Chibale, K.; et al. 2-Mercapto-quinazolinones as inhibitors of type II NADH dehydrogenase and Mycobacterium tuberculosis: Structure-activity relationships, mechanism of action and absorption, distribution, metabolism, and excretion characterization. ACS Infect. Dis. 2018, 4, 954–969. [Google Scholar] [CrossRef]
- Ioerger, T.R.; O’Malley, T.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Mohaideen, N.; Murphy, K.C.; Boshoff, H.I.M.; Mizrahi, V.; Rubin, E.J.; et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e75245. [Google Scholar] [CrossRef]
- Shiemke, A.K.; Arp, D.J.; Sayavedra-Soto, L.A. Inhibition of membrane-bound methane monooxygenase and ammonia monooxygenase by diphenyliodonium: Implications for electron transfer. J. Bacteriol. 2004, 186, 928–937. [Google Scholar] [CrossRef]
- Deris, Z.Z.; Akter, J.; Sivanesan, S.; Roberts, K.D.; Thompson, P.E.; Nation, R.L.; Li, J.; Velkov, T. A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. 2014, 67, 147–151. [Google Scholar] [CrossRef]
- Pandey, M.; Singh, A.K.; Thakare, R.; Talwar, S.; Karaulia, P.; Dasgupta, A.; Chopra, S.; Pandey, A.K. Diphenyleneiodonium chloride (DPIC) displays broad-spectrum bactericidal activity. Sci. Rep. 2017, 7, 11521. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Wilson, D.W.; Nagalingam, G.; Triccas, J.A.; Schneider, E.K.; Li, J.; Velkov, T.; Baell, J. Broad activity of diphenyleneiodonium analogues against Mycobacterium tuberculosis, malaria parasites and bacterial pathogens. Eur. J. Med. Chem. 2018, 148, 507–518. [Google Scholar] [CrossRef]
- Gong, H.; Li, J.; Xu, A.; Tang, Y.; Ji, W.; Gao, R.; Wang, S.; Yu, L.; Tian, C.; Li, J.; et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; O’Brien, K.; Tiwari, D.; Engelhart, C.A.; Walters, S.; Andrews, J.; Yang, H.-J.; Sutphen, M.L.; Weiner, D.M.; Dayao, E.K.; et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat. Commun. 2019, 10, 4970. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Cox, J.A.G.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinidou, C.; Fernández, R.; Alemparte, C.; Remuiñán, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef] [PubMed]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Ochoa-Montaño, B.; Tsang, P.S.; Blundell, T.L.; Dawes, S.S.; Mizrahi, V.; Bayliss, T.; Mackenzie, C.J.; Cleghorn, L.A.T.; Ray, P.C.; et al. Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6962–6965. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Sellamuthu, S.; Bhat, M.F.; Kumar, A.; Singh, S.K. Phenothiazine: A better scaffold against tuberculosis. Mini Rev. Med. Chem. 2018, 18, 1442–1451. [Google Scholar] [CrossRef]
- Kang, S.; Kim, Y.M.; Jeon, H.; Park, S.; Seo, M.J.; Lee, S.; Park, D.; Nam, J.; Lee, S.; Nam, K.; et al. Synthesis and structure-activity relationships of novel fused ring analogues of Q203 as antitubercular agents. Eur. J. Med. Chem. 2017, 136, 420–427. [Google Scholar] [CrossRef]
- Kang, S.; Kim, Y.M.; Kim, R.Y.; Seo, M.J.; No, Z.; Nam, K.; Kim, S.; Kim, J. Synthesis and structure-activity studies of side chain analogues of the anti-tubercular agent, Q203. Eur. J. Med. Chem. 2017, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Lv, K.; Li, L.; Liu, H.; Tao, Z.; Wang, B.; Liu, M.; Ma, C.; Ma, X.; Han, B.; et al. Design, synthesis and biological activity of N-(2-phenoxy)ethyl imidazo[1,2-a]pyridine-3-carboxamides as new antitubercular agents. Eur. J. Med. Chem. 2019, 178, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wang, H.; Geng, Y.; Fu, L.; Gu, J.; Wang, B.; Lv, K.; Liu, M.; Tao, Z.; Ma, C.; et al. Design, synthesis and antimycobacterial activity of less lipophilic Q203 derivatives containing alkaline fused ring moieties. Bioorganic Med. Chem. 2019, 27, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Z.; Liu, M.; Shen, W.; Wang, B.; Guo, H.; Lu, Y. Design, synthesis and antimycobacterial activity of novel imidazo[1,2-a]pyridine amide-cinnamamide hybrids. Molecules 2015, 21, 49. [Google Scholar] [CrossRef]
- Wang, H.; Wang, A.; Gu, J.; Fu, L.; Lv, K.; Ma, C.; Tao, Z.; Wang, B.; Liu, M.; Guo, H.; et al. Synthesis and antitubercular evaluation of reduced lipophilic imidazo[1,2-a]pyridine-3-carboxamide derivatives. Eur. J. Med. Chem. 2019, 165, 11–17. [Google Scholar] [CrossRef]
- Moraski, G.C.; Miller, P.A.; Bailey, M.A.; Ollinger, J.; Parish, T.; Boshoff, H.I.; Cho, S.; Anderson, J.R.; Mulugeta, S.; Franzblau, S.G.; et al. Putting tuberculosis (TB) to rest: Transformation of the sleep aid, ambien, and “anagrams” generated potent antituberculosis agents. ACS Infect. Dis. 2015, 1, 85–90. [Google Scholar] [CrossRef]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, M.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, synthesis, and biological evaluation of pyrazolo[1,5-a]pyridine-3-carboxamides as novel antitubercular agents. ACS Med. Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef]
- Moraski, G.C.; Seeger, N.; Miller, P.A.; Oliver, A.G.; Boshoff, H.I.; Cho, S.; Mulugeta, S.; Anderson, J.R.; Franzblau, S.G.; Miller, M.J. Arrival of imidazo[2,1-b]thiazole-5-carboxamides: Potent anti-tuberculosis agents that target QcrB. ACS Infect. Dis. 2016, 2, 393–398. [Google Scholar] [CrossRef]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo[2,1-b]thiazole-5-carboxamide anti-tuberculosis compounds. PLoS ONE 2020, 15, e0227224. [Google Scholar] [CrossRef]
- Bouvier, G.; Simenel, C.; Jang, J.; Kalia, N.P.; Choi, I.; Nilges, M.; Pethe, K.; Izadi-Pruneyre, N. Target engagement and binding mode of an antituberculosis drug to its bacterial target deciphered in whole living cells by NMR. Biochemistry 2019, 58, 526–533. [Google Scholar] [CrossRef]
- de Jager, V.R.; Dawson, R.; van Niekerk, C.; Hutchings, J.; Kim, J.; Vanker, N.; van der Merwe, L.; Choi, J.; Nam, K.; Diacon, A.H. Telacebec (Q203), a new antituberculosis agent. N. Engl. J. Med. 2020, 382, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef]
- Phummarin, N.; Boshoff, H.I.; Tsang, P.S.; Dalton, J.; Wiles, S.; Barry Rd, C.E.; Copp, B.R. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc1 inhibitors. Medchemcomm 2016, 7, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Pissinate, K.; Villela, A.D.; Rodrigues-Junior, V.; Giacobbo, B.C.; Grams, E.S.; Abbadi, B.L.; Trindade, R.V.; Roesler Nery, L.; Bonan, C.D.; Back, D.F.; et al. 2-(Quinolin-4-yloxy)acetamides are active against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. ACS Med. Chem. Lett 2016, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Giacobbo, B.C.; Pissinate, K.; Rodrigues-Junior, V.; Villela, A.D.; Grams, E.S.; Abbadi, B.L.; Subtil, F.T.; Sperotto, N.; Trindade, R.V.; Back, D.F.; et al. New insights into the SAR and drug combination synergy of 2-(quinolin-4-yloxy)acetamides against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2017, 126, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Pitta, E.; Rogacki, M.K.; Balabon, O.; Huss, S.; Cunningham, F.; Lopez-Roman, E.M.; Joossens, J.; Augustyns, K.; Ballell, L.; Bates, R.H.; et al. Searching for new leads for tuberculosis: Design, synthesis, and biological evaluation of novel 2-quinolin-4-yloxyacetamides. J. Med. Chem. 2016, 59, 6709–6728. [Google Scholar] [CrossRef] [PubMed]
- Borsoi, A.F.; Paz, J.D.; Abbadi, B.L.; Macchi, F.S.; Sperotto, N.; Pissinate, K.; Rambo, R.S.; Ramos, A.S.; Machado, D.; Viveiros, M.; et al. Design, synthesis, and evaluation of new 2-(quinoline-4-yloxy)acetamide-based antituberculosis agents. Eur. J. Med. Chem. 2020, 192, 112179. [Google Scholar] [CrossRef]
- Subtil, F.T.; Villela, A.D.; Abbadi, B.L.; Rodrigues-Junior, V.S.; Bizarro, C.V.; Timmers, L.F.S.M.; de Souza, O.N.; Pissinate, K.; Machado, P.; López-Gavín, A.; et al. Activity of 2-(quinolin-4-yloxy)acetamides in Mycobacterium tuberculosis clinical isolates and identification of their molecular target by whole-genome sequencing. Int. J. Antimicrob. Agents 2018, 51, 378–384. [Google Scholar] [CrossRef]
- Cleghorn, L.A.T.; Ray, P.C.; Odingo, J.; Kumar, A.; Wescott, H.; Korkegian, A.; Masquelin, T.; Lopez Moure, A.; Wilson, C.; Davis, S.; et al. Identification of morpholino thiophenes as novel Mycobacterium tuberculosis inhibitors, targeting QcrB. J. Med. Chem. 2018, 61, 6592–6608. [Google Scholar] [CrossRef]
- Lu, X.; Tang, J.; Cui, S.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Zhang, X.; Ding, K. Pyrazolo[1,5-a]pyridine-3-carboxamide hybrids: Design, synthesis and evaluation of anti-tubercular activity. Eur. J. Med. Chem. 2017, 125, 41–48. [Google Scholar] [CrossRef]
- Foo, C.S.; Lupien, A.; Kienle, M.; Vocat, A.; Benjak, A.; Sommer, R.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; Piton, J.; et al. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.A.; Mayer Bridwell, A.E.; Singh, M.; Jayaraman, K.; Weiss, L.A.; Kinsella, R.L.; Aneke, J.S.; Flentie, K.; Schene, M.E.; Gaggioli, M.; et al. Identification of 4-amino-thieno[2,3-d]pyrimidines as QcrB inhibitors in Mycobacterium tuberculosis. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Lupien, A.; Foo, C.S.-Y.; Savina, S.; Vocat, A.; Piton, J.; Monakhova, N.; Benjak, A.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; et al. New 2-Ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008270. [Google Scholar] [CrossRef] [PubMed]
- Reddyrajula, R.; Dalimba, U.K. Structural modification of zolpidem led to potent antimicrobial activity in imidazo[1,2-a]pyridine/pyrimidine-1,2,3-triazoles. New J. Chem. 2019, 43, 16281–16299. [Google Scholar] [CrossRef]
- Rybniker, J.; Vocat, A.; Sala, C.; Busso, P.; Pojer, F.; Benjak, A.; Cole, S.T. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 2015, 6, 7659. [Google Scholar] [CrossRef]
- Mdanda, S.; Baijnath, S.; Shobo, A.; Singh, S.D.; Maguire, G.E.M.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Govender, T. Lansoprazole-sulfide, pharmacokinetics of this promising anti-tuberculous agent. Biomed. Chromatogr. 2017, 31, e4035. [Google Scholar] [CrossRef]
- Kalia, N.P.; Hasenoehrl, E.J.; Ab Rahman, N.B.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Grüber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426–7431. [Google Scholar] [CrossRef]
- Kunze, B.; Höfle, G.; Reichenbach, H. The aurachins, new quinoline antibiotics from myxobacteria: Production, physico-chemical and biological properties. J. Antibiot. 1987, 40, 258–265. [Google Scholar] [CrossRef]
- Li, X.-W.; Herrmann, J.; Zang, Y.; Grellier, P.; Prado, S.; Müller, R.; Nay, B. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J. Org. Chem. 2013, 9, 1551–1558. [Google Scholar] [CrossRef]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef]
- Lu, P.; Asseri, A.H.; Kremer, M.; Maaskant, J.; Ummels, R.; Lill, H.; Bald, D. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci. Rep. 2018, 8, 2625. [Google Scholar] [CrossRef]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.-G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A novel small-molecule inhibitor of the Mycobacterium tuberculosis demethylmenaquinone methyltransferase MenG is bactericidal to both growing and nutritionally deprived persister cells. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biuković, G.; Subramanian Manimekalai, M.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit α of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef]
- Zhang, A.T.; Montgomery, M.G.; Leslie, A.G.W.; Cook, G.M.; Walker, J.E. The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs. Proc. Natl. Acad. Sci. USA 2019, 116, 4206–4211. [Google Scholar] [CrossRef] [PubMed]
- Preiss, L.; Langer, J.D.; Yildiz, Ö.; Eckhardt-Strelau, L.; Guillemont, J.E.G.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Ragunathan, P.; Shin, J.; Saw, W.-G.; Wong, C.-F.; Dick, T.; Grüber, G. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: Implications for drug design. Prog. Biophys. Mol. Biol. 2020, 152, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Gruber, G.; Dick, T. Re-understanding the mechanisms of action of the anti-mycobacterial drug bedaquiline. Antibiotics 2019, 8, 261. [Google Scholar] [CrossRef]
- Hards, K.; McMillan, D.G.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef]
- Guillemont, J.; Meyer, C.; Poncelet, A.; Bourdrez, X.; Andries, K. Diarylquinolines, synthesis pathways and quantitative structure--activity relationship studies leading to the discovery of TMC. Future Med. Chem. 2011, 3, 1345–1360. [Google Scholar] [CrossRef]
- Diacon, A.H.; Donald, P.R. The early bactericidal activity of antituberculosis drugs. Expert Rev. Anti Infect. Ther. 2014, 12, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Schnippel, K.; Ndjeka, N.; Maartens, G.; Meintjes, G.; Master, I.; Ismail, N.; Hughes, J.; Ferreira, H.; Padanilam, X.; Romero, R.; et al. Effect of bedaquiline on mortality in South African patients with drug-resistant tuberculosis: A retrospective cohort study. Lancet Respir. Med. 2018, 6, 699–706. [Google Scholar] [CrossRef]
- Mohr, E.; Ferlazzo, G.; Hewison, C.; De Azevedo, V.; Isaakidis, P. Bedaquiline and delamanid in combination for treatment of drug-resistant tuberculosis. Lancet Infect. Dis. 2019, 19, 470. [Google Scholar] [CrossRef]
- Jones, J.; Mudaly, V.; Voget, J.; Naledi, T.; Maartens, G.; Cohen, K. Adverse drug reactions in South African patients receiving bedaquiline-containing tuberculosis treatment: An evaluation of spontaneously reported cases. BMC Infect. Dis. 2019, 19, 544. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Pawara, R.; Pawara, K.; Ahmed, F.; Shirkhedkar, A.; Surana, S. A structural insight of bedaquiline for the cardiotoxicity and hepatotoxicity. Tuberculosis 2019, 117, 79–84. [Google Scholar] [CrossRef]
- Singh, S.; Roy, K.K.; Khan, S.R.; Kashyap, V.K.; Sharma, A.; Jaiswal, S.; Sharma, S.K.; Krishnan, M.Y.; Chaturvedi, V.; Lal, J.; et al. Novel, potent, orally bioavailable and selective mycobacterial ATP synthase inhibitors that demonstrated activity against both replicating and non-replicating M. tuberculosis. Bioorganic Med. Chem. 2015, 23, 742–752. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Vandavasi, J.K.; Vasireddy, N.R.; Sharma, V.; Dixit, S.S.; Chattopadhyaya, J. Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorganic Med. Chem. 2009, 17, 2830–2841. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorganic Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Lahore, S.V.; Sayyed, A.Y.; Dixit, S.S.; Shinde, P.D.; Chattopadhyaya, J. Conformationally-constrained indeno[2,1-c]quinolines—A new class of anti-mycobacterial agents. Org. Biomol. Chem. 2010, 8, 2180–2197. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Shinde, P.D.; Sayyed, A.Y.; Kadam, S.A.; Bawane, A.N.; Poddar, A.; Plashkevych, O.; Földesi, A.; Chattopadhyaya, J. Synthesis and structure of azole-fused indeno[2,1-c]quinolines and their anti-mycobacterial properties. Org. Biomol. Chem. 2010, 8, 5661–5673. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Vandavasi, J.K.; Kardile, R.A.; Lahore, S.V.; Dixit, S.S.; Deokar, H.S.; Shinde, P.D.; Sarmah, M.P.; Chattopadhyaya, J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Preiss, L.; Wang, B.; Fu, L.; Wen, H.; Zhang, X.; Cui, H.; Meier, T.; Yin, D. Structural simplification of bedaquiline: The discovery of 3-(4-(N,N-dimethylaminomethyl)phenyl)quinoline-derived antitubercular lead compounds. ChemMedChem 2017, 12, 106–119. [Google Scholar] [CrossRef]
- Tong, A.S.T.; Choi, P.J.; Blaser, A.; Sutherland, H.S.; Tsang, S.K.Y.; Guillemont, J.; Motte, M.; Cooper, C.B.; Andries, K.; Van den Broeck, W.; et al. 6-Cyano analogues of bedaquiline as less lipophilic and potentially safer diarylquinolines for tuberculosis. ACS Med. Chem. Lett. 2017, 8, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.J.; Sutherland, H.S.; Tong, A.S.T.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Lotlikar, M.U.; Upton, A.M.; Guillemont, J.; Motte, M.; et al. Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorganic Med. Chem. Lett. 2017, 27, 5190–5196. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.U.; Denny, W.A.; et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorganic Med. Chem. 2018, 26, 1797–1809. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorganic Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.; Denny, W.A.; Palmer, B.D. Variations in the C-unit of bedaquiline provides analogues with improved biology and pharmacology. Bioorganic Med. Chem. 2020, 28, 115213. [Google Scholar] [CrossRef]
- Blaser, A.; Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.; Denny, W.A.; et al. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorganic Med. Chem. 2019, 27, 1283–1291. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Ragunathan, P.; Cooper, C.B.; Upton, A.M.; Grüber, G.; Dick, T. TBAJ-876 Displays bedaquiline-like mycobactericidal potency without retaining the parental drug’s uncoupler activity. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Saw, W.-G.; Wu, M.-L.; Ragunathan, P.; Biuković, G.; Lau, A.-M.; Shin, J.; Harikishore, A.; Cheung, C.-Y.; Hards, K.; Sarathy, J.P.; et al. Disrupting coupling within mycobacterial F-ATP synthases subunit ε causes dysregulated energy production and cell wall biosynthesis. Sci. Rep. 2019, 9, 16759. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehra, R.; Sharma, S.; Bokolia, N.P.; Raina, D.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of antitubercular compound library identifies novel ATP synthase inhibitors of Mycobacterium tuberculosis. Tuberculosis 2018, 108, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Tantry, S.J.; Shinde, V.; Balakrishnan, G.; Markad, S.D.; Gupta, A.K.; Bhat, J.; Narayan, A.; Raichurkar, A.; Jena, L.K.; Sharma, S.; et al. Scaffold morphing leading to evolution of 2,4-diaminoquinolines and aminopyrazolopyrimidines as inhibitors of the ATP synthesis pathway. Med. Chem. Commun. 2016, 7, 1022–1032. [Google Scholar] [CrossRef]
- Hotra, A.; Ragunathan, P.; Ng, P.S.; Seankongsuk, P.; Harikishore, A.; Sarathy, J.P.; Saw, W.-G.; Lakshmanan, U.; Sae-Lao, P.; Kalia, N.P.; et al. Discovery of a novel mycobacterial F-ATP synthase inhibitor and its potency in combination with diarylquinolines. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef]
- Hotra, A.; Suter, M.; Biuković, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Grüber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase γ subunit sheds light on its inhibitory role in ATP hydrolysis-driven H(+) pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef]
- Kushner, S.; Dalalian, H.; Sanjurjo, J.L.; Bach, F.L.; Safir, S.R.; Smith, V.K.; Williams, J.H. Experimental chemotherapy of tuberculosis. II. The synthesis of pyrazinamides and related compounds. J. Am. Chem. Soc. 1952, 74, 3617–3621. [Google Scholar] [CrossRef]
- Shi, W.; Cui, P.; Niu, H.; Zhang, S.; Tønjum, T.; Zhu, B.; Zhang, Y. Introducing RpsA point mutations Δ438A and D123A into the chromosome of Mycobacterium tuberculosis confirms their role in causing resistance to pyrazinamide. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Sun, Q.; Li, X.; Perez, L.M.; Shi, W.; Zhang, Y.; Sacchettini, J.C. The molecular basis of pyrazinamide activity on Mycobacterium tuberculosis PanD. Nat. Commun. 2020, 11, 339. [Google Scholar] [CrossRef]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef]
- Li, W.; Upadhyay, A.; Fontes, F.L.; North, E.J.; Wang, Y.; Crans, D.C.; Grzegorzewicz, A.E.; Jones, V.; Franzblau, S.G.; Lee, R.E.; et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. [Google Scholar] [CrossRef] [PubMed]
- Poce, G.; Cocozza, M.; Consalvi, S.; Biava, M. SAR analysis of new anti-TB drugs currently in pre-clinical and clinical development. Eur. J. Med. Chem. 2014, 86, 335–351. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Makobongo, M.O.; Einck, L.; Peek, R.M.; Merrell, D.S. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS ONE 2013, 8, e68917. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Santos, P.; Li, K.; Lameira, L.; de Carvalho, T.M.U.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a new drug lead for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 1950–1961. [Google Scholar] [CrossRef]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef]
- Rizvi, A.; Shankar, A.; Chatterjee, A.; More, T.H.; Bose, T.; Dutta, A.; Balakrishnan, K.; Madugulla, L.; Rapole, S.; Mande, S.S.; et al. Rewiring of metabolic network in Mycobacterium tuberculosis During adaptation to different stresses. Front. Microbiol. 2019, 10, 2417. [Google Scholar] [CrossRef]
- Baughn, A.D.; Rhee, K.Y. Metabolomics of central carbon metabolism in Mycobacterium tuberculosis. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Rhee, K.Y.; de Carvalho, L.P.S.; Bryk, R.; Ehrt, S.; Marrero, J.; Park, S.W.; Schnappinger, D.; Venugopal, A.; Nathan, C. Central carbon metabolism in Mycobacterium tuberculosis: An unexpected frontier. Trends Microbiol. 2011, 19, 307–314. [Google Scholar] [CrossRef]
- Fieweger, R.A.; Wilburn, K.M.; VanderVen, B.C. Comparing the metabolic capabilities of bacteria in the Mycobacterium tuberculosis Complex. Microorganisms 2019, 7, 177. [Google Scholar] [CrossRef]
- Marrero, J.; Rhee, K.Y.; Schnappinger, D.; Pethe, K.; Ehrt, S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. USA 2010, 107, 9819–9824. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.A.; van de Langemheen, H.; Muñoz-Elías, E.J.; McKinney, J.D.; Sacchettini, J.C. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol. Microbiol. 2006, 61, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Sriram, D.; Yogeeswari, P.; Senthilkumar, P.; Dewakar, S.; Rohit, N.; Debjani, B.; Bhat, P.; Veugopal, B.; Pavan, V.V.S.; Thimmappa, H.M. Novel phthalazinyl derivatives: Synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis isocitrate lyase enzyme. Med. Chem. 2009, 5, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Waisser, K.; Bures, O.; Holý, P.; Kunes, J.; Oswald, R.; Jirásková, L.; Pour, M.; Klimesová, V.; Kubicová, L.; Kaustová, J. Relationship between the structure and antimycobacterial activity of substituted salicylanilides. Arch. Pharm. (Weinh.) 2003, 336, 53–71. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Wsól, V.; Trejtnar, F.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; et al. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis 2012, 92, 434–439. [Google Scholar] [CrossRef]
- Paraskevopoulos, G.; Monteiro, S.; Vosátka, R.; Krátký, M.; Navrátilová, L.; Trejtnar, F.; Stolaříková, J.; Vinšová, J. Novel salicylanilides from 4,5-dihalogenated salicylic acids: Synthesis, antimicrobial activity and cytotoxicity. Bioorganic Med. Chem. 2017, 25, 1524–1532. [Google Scholar] [CrossRef]
- Anstrom, D.M.; Remington, S.J. The product complex of M. tuberculosis malate synthase revisited. Protein Sci. 2006, 15, 2002–2007. [Google Scholar] [CrossRef]
- Quartararo, C.E.; Blanchard, J.S. Kinetic and chemical mechanism of malate synthase from Mycobacterium tuberculosis. Biochemistry 2011, 50, 6879–6887. [Google Scholar] [CrossRef]
- Krieger, I.V.; Freundlich, J.S.; Gawandi, V.B.; Roberts, J.P.; Gawandi, V.B.; Sun, Q.; Owen, J.L.; Fraile, M.T.; Huss, S.I.; Lavandera, J.-L.; et al. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 2012, 19, 1556–1567. [Google Scholar] [CrossRef]
- Berryman, O.B.; Bryantsev, V.S.; Stay, D.P.; Johnson, D.W.; Hay, B.P. Structural criteria for the design of anion receptors: The interaction of halides with electron-deficient arenes. J. Am. Chem. Soc. 2007, 129, 48–58. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- Yam, K.C.; D’Angelo, I.; Kalscheuer, R.; Zhu, H.; Wang, J.-X.; Snieckus, V.; Ly, L.H.; Converse, P.J.; Jacobs, W.R.; Strynadka, N.; et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000344. [Google Scholar] [CrossRef] [PubMed]
- Frye, S.V.; Haffner, C.D.; Maloney, P.R.; Mook, R.A.; Dorsey, G.F.; Hiner, R.N.; Cribbs, C.M.; Wheeler, T.N.; Ray, J.A.; Andrews, R.C. 6-Azasteroids: Structure-activity relationships for inhibition of type 1 and 2 human 5 alpha-reductase and human adrenal 3 beta-hydroxy-delta 5-steroid dehydrogenase/3-keto-delta 5-steroid isomerase. J. Med. Chem. 1994, 37, 2352–2360. [Google Scholar] [CrossRef]
- Thomas, S.T.; Yang, X.; Sampson, N.S. Inhibition of the M. tuberculosis 3β-hydroxysteroid dehydrogenase by azasteroids. Bioorganic Med. Chem. Lett. 2011, 21, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Dolan, S.K.; Wijaya, A.; Geddis, S.M.; Spring, D.R.; Silva-Rocha, R.; Welch, M. Loving the poison: The methylcitrate cycle and bacterial pathogenesis. Microbiology 2018, 164, 251–259. [Google Scholar] [CrossRef]
- VanderVen, B.C.; Fahey, R.J.; Lee, W.; Liu, Y.; Abramovitch, R.B.; Memmott, C.; Crowe, A.M.; Eltis, L.D.; Perola, E.; Deininger, D.D.; et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 2015, 11, e1004679. [Google Scholar] [CrossRef]
- Black, P.A.; Warren, R.M.; Louw, G.E.; van Helden, P.D.; Victor, T.C.; Kana, B.D. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2491–2503. [Google Scholar] [CrossRef]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J.C. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | clogP | hERG IC50 (μM) | Mtb MIC90 (μM) | Log10 CFUreduction | IV cl (mL/min/kg) |
|---|---|---|---|---|---|
| Bedaquiline | 7.25 | 1.6 | 0.03 | 4.5–6.1 | 7 |
| TBAJ-876 | 5.15 | >30 | 0.004 | >5.5 | 13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Appetecchia, F.; Consalvi, S.; Scarpecci, C.; Biava, M.; Poce, G. SAR Analysis of Small Molecules Interfering with Energy-Metabolism in Mycobacterium tuberculosis. Pharmaceuticals 2020, 13, 227. https://doi.org/10.3390/ph13090227
Appetecchia F, Consalvi S, Scarpecci C, Biava M, Poce G. SAR Analysis of Small Molecules Interfering with Energy-Metabolism in Mycobacterium tuberculosis. Pharmaceuticals. 2020; 13(9):227. https://doi.org/10.3390/ph13090227
Chicago/Turabian StyleAppetecchia, Federico, Sara Consalvi, Cristina Scarpecci, Mariangela Biava, and Giovanna Poce. 2020. "SAR Analysis of Small Molecules Interfering with Energy-Metabolism in Mycobacterium tuberculosis" Pharmaceuticals 13, no. 9: 227. https://doi.org/10.3390/ph13090227
APA StyleAppetecchia, F., Consalvi, S., Scarpecci, C., Biava, M., & Poce, G. (2020). SAR Analysis of Small Molecules Interfering with Energy-Metabolism in Mycobacterium tuberculosis. Pharmaceuticals, 13(9), 227. https://doi.org/10.3390/ph13090227