Abstract
Polymyxins are vital antibiotics for the treatment of multiresistant Gram-negative ESKAPE pathogen infections. However, their clinical value is limited by their high nephrotoxicity and neurotoxicity, as well as their poor permeability and absorption in the gastrointestinal tract. This review focuses on various polymyxin delivery systems that improve polymyxin bioavailability and reduce drug toxicity through targeted and controlled release. Currently, the most suitable systems for improving oral, inhalation, and parenteral polymyxin delivery are polymer particles, liposomes, and conjugates, while gels, polymer fibers, and membranes are attractive materials for topical administration of polymyxin for the treatment of infected wounds and burns. In general, the application of these systems protects polymyxin molecules from the negative effects of both physiological and pathological factors while achieving higher concentrations at the target site and reducing dosage and toxicity. Improving the properties of polymyxin will be of great interest to researchers who are focused on developing antimicrobial drugs that show increased efficacy and safety.
1. Introduction
The World Health Organization published an R&D list pertaining to antibiotic-resistant bacteria and the measures required to address the global increase in resistance to antimicrobial drugs, including resistance to carbapenems and third-generation cephalosporins. The most critical group of microbes are the multidrug-resistant Gram-negative bacteria, such as Acinetobacter, Pseudomonas, and various Enterobacteriaceae (including Klebsiella, Escherichia coli, and Serratia), as these can cause severe lethal infections (bacteremia and pneumonia) [1].
The need is great for innovation in antibiotic research; however, the development of new antimicrobial agents is complicated due to the regulatory environment and financial risks [2]. One alternative strategy to the synthesis of new drugs is the development of nanomedicine-based forms of existing antimicrobial drugs to control multidrug resistance of microorganisms [3,4,5,6,7,8,9,10,11]. One group of drugs that has seen a revival in medical practice, given the multidrug resistance of Gram-negative bacteria (the so-called ESKAPE pathogens Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter) and the absence of new antibiotics, are the old antimicrobial drugs, the polymyxins. Polymyxins have significant therapeutic potential because they are effective against four of the six ESKAPE pathogens (K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter) [12,13,14,15,16,17,18,19,20,21,22,23,24].
At present, several reviews have addressed delivery systems for polypeptide antibiotics [25,26], and still, others have presented the pharmacological characteristics of polymyxins and the clinical experience of their use for the treatment of multidrug-resistant infections [13,27]. This review focuses on recently developed polymyxin delivery systems and analysis of their effectiveness for different routes of administration.
2. Chemical Structure of Polymyxins
Polymyxins are a group of polypeptide antibiotics that includes several chemically different compounds (polymyxin A–E, M, etc.). Of these, only polymyxin B and polymyxin E have clinical applications [13,28,29].
Polymyxin E (colistin) is a mixture of related decapeptides, polymyxin E1 (colistin A) and polymyxin E2 (colistin B). These have a general structure composed of a cyclic heptapeptide moiety and a side chain acylated at the N-terminus by a fatty acid (Figure 1A). Polymyxins E1 and E2 contain the same amino acids but differ in their fatty acid components (6-methyl-octanoic acid and 6-methyl-heptanoic acid, respectively). Different pharmaceutical formulations can contain different amounts of these two components [13,30,31,32,33,34,35]. For pharmaceutical use, polymyxin E is available in the form of colistin sulfate (topical and oral use) and as sodium colistimethate (injection and inhalation). Sodium colistimethate is a derivative of colistin methanesulfonic acid and is characterized by the presence of methanesulfonic acid in the sodium salt form on each amino group of the five diaminobutyric acid moieties (Figure 1B) [36].
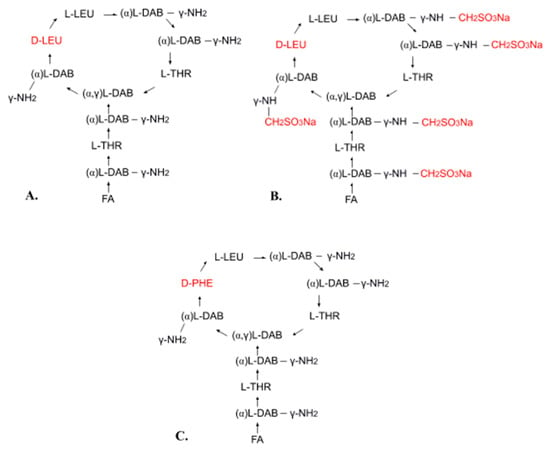
Figure 1.
Chemical structure of polymyxin E (A), sodium colistimethate (B), and polymyxin B (C): FA—fatty acid, THR—threonine, LEU—leucine, DAB—α,γ-diaminobutyric acid, PHE—phenylalanine, α and γ indicate the amino groups forming the peptide linkage.
Polymyxin B is also used in clinical practice. It differs from polymyxin E in its phenylalanine content (Figure 1C). Both polymyxin E and polymyxin B have the same mechanism of action and similar applications, but polymyxin B has less activity and causes more common and severe toxicity [13,37]. Polymyxin B is used for injection in the form of polymyxin B sulfate [13,37].
3. Mechanism of Action and Side Effects
Polymyxins exert their bacteriostatic effect by damaging the membrane of the bacterial cell. Polymyxins are selective for Gram-negative bacteria, since the primary molecular target of these antibacterial agents is the three-domain lipopolysaccharide (lipid A, oligosaccharide, and O-antigen) that is the main component of the outer membrane of Gram-negative bacteria [38,39]. This lipopolysaccharide has several negative charges with which polymyxins can bind by electrostatic interactions. This binding competitively replaces membrane-stabilizing ions, such as Ca2+ and Mg2+, thereby destabilizing the lipopolysaccharide layer and allowing the introduction of the polymyxin hydrophobic chain into the hydrophobic domain (lipid A). As a result, the permeability of the outer membrane increases and the polymyxin penetrates into the periplasmic space (through the process of self-promoted uptake) [40,41]. In the periplasmic space, polymyxin forms contacts between the outer and internal membranes and promotes the fast and selective exchange of anionic phospholipids. This, in turn, causes an osmotic imbalance and ultimately the death of the bacteria. Thus, lipid A is the main target for polymyxins, and the hydrophobic interactions between polymyxin and the membrane lipids are important factors in the mechanism of action of this antibiotic. In fact, one of the mechanisms of bacterial polymyxin resistance is related to lipid A modifications or to the complete loss of lipopolysaccharides, as these changes prevent this key interaction of polymyxins with the outer membrane of Gram-negative bacteria [42,43,44,45].
Importantly, the polymyxin docking with the bacterial membrane is not related to any specific proteins (receptors) but is instead a simple electrostatic interaction of a cationic molecule with a negatively charged membrane. For this reason, bacterial resistance to polymyxins is relatively rare compared to resistance to other antibiotics. Therefore, polymyxins are currently the last-line antibiotic for multidrug-resistant Gram-negative infections [46,47,48,49,50,51,52]. Polymyxin resistance is mainly caused by structural remodeling of lipid A, located on the bacterial cell surface, via biochemical and genetic mechanisms. This remodeling is initiated by the actions of membrane-associated enzymes (ArnT, EptA, and AlmEFG). The phenotypic resistance to polymyxins is divided into two types: chromosomally encoded intrinsic resistance and transposon/plasmid-mediated transferable resistance related to MCR-like enzymes (MCR-1). Different plasmids can mediate the rapid transfer of MCR-1 among various species of bacteria, and this becomes a key factor in the development of global polymyxin resistance [53,54,55,56].
In addition, various pathogens, such as polymyxin-sensitive Salmonella species, are able to penetrate into macrophages, where they persist and multiply within the cell compartments. Several nanomaterials, including nanospheres, liposomes, and mesoporous silica nanoparticles with surfaces modified by specific cell ligands, are able to penetrate into macrophages and kill these pathogens [57]. Metal-based nanoparticles, including gold nanoparticles, are particularly promising platforms for the intracellular delivery of antimicrobial peptides [58].
One strategy for overcoming microbial resistance to polymyxins is the use of adjuvants, such as anthelmintic salicylanilides like niclosamide, oxyclozanide, rafoxanide, and closantel [59,60,61,62]. Another is the use of polymyxin-based combination therapy with carbapenems, rifampicin, β-lactams, vancomycin, fosfomycin, and tigecycline [42,63,64,65,66,67].
Polymyxin is of critical medical importance because it is effective against selected Gram-negative bacteria (including Acinetobacter species, P. aeruginosa, Klebsiella species, and Enterobacter species, E. coli, Salmonella species, Shigella species) that cause severe nosocomial multidrug-resistant infections, including nosocomial pneumonia. Polymyxins are formulated for parenteral use (for treatment of cystic fibrosis, pneumonia, bacteremia, and urinary tract infections), inhalation (cystic fibrosis, pneumonia), and topical use (otic and ophthalmic solutions). Colistin sulfate and colistimethate sodium are not absorbed after oral administration, so they can only be used for the treatment of gastrointestinal tract (GIT) infections [13,68,69,70,71,72,73,74,75]. The most common polymyxin side effects are nephrotoxicity and neurotoxicity, and both renal and neurological toxicity are considered to be dose-dependent [13,76,77,78,79].
In summary, polymyxins are clinically important antibiotics, but their use is limited to severe site reactions because of their toxicity. The development of targeted and prolonged-release delivery systems that would reduce the dose and frequency of administration, and thereby reduce the polymyxin toxicity, is, therefore, a critical medical need. In addition, polymyxins are essentially not absorbed by the GIT, so their encapsulation into suitable carriers would greatly improve intestinal permeability, thereby imparting a new oral bioavailability for these important drugs.
4. Polymyxin Delivery Systems
The use of suitable nanomedicine-based devices can improve drug delivery and overcome the disadvantages of various routes of administration. Utilization of polymeric particles, or liposomes for intravenous administration, allows control of the residence time of the drug in the systemic circulation due to sustained release, as well as ensuring targeted delivery. The polymeric particles, conjugates, and liposomes in the form of inhalation formulations protect drugs from the aggressive environment of the inflamed lung (high salt concentrations, changes in oxygen levels), improve interaction with mucus, help overcome mucociliary clearance, and contribute to the penetration of antibiotics into the bacterial biofilm. At the same time, a higher concentration of the drug in the lungs is achieved than can be obtained with intravenous administration, leading to a faster pharmacological effect. In addition, solid polymeric particles and liposomes have suitable sizes (1–5 μm, and less than 1 μm) for antibiotics to reach the lower airways and infected foci by inhalation administration. Consequently, these drug carriers improve bioavailability and enhance the drug effect while reducing the required dose and frequency of administration. The end result is a decrease in the toxicity and side effects for both injection and inhalation formulations [80,81,82,83].
Conjugation or encapsulation of antibiotics in polymer and lipid systems for oral administration allows control of the site of release, protects drugs from the aggressive environment of the gastrointestinal tract, and improves intestinal permeability and absorption. These improvements are especially important for polymyxins because they provide control over the release site, rate, and profile, thereby increasing the bioavailability of these drugs [84,85].
Topical administration of polymyxins is limited by their low activity due to their susceptibility to environmental conditions (hydrolysis, oxidation) and the wound environment (pH, proteolysis), in addition to their minimal residence time. Structured gels and polymer nanofibers and membranes can improve the chemical and physical stability of antibiotics, as well as prolong the drug release. Furthermore, these systems are biodegradable and atraumatic, making them suitable topical delivery systems for the treatment of infected skin injuries (acute and chronic wounds, burns, etc.) [86,87,88,89].
Transdermal delivery is also an attractive route for systemic drug administration due to the circumvention of first-pass hepatic metabolism, increased bioavailability, and reduced toxicity, as well as patient convenience. In addition, in the event of side effects, the drug admission into the systemic circulation can be quickly stopped simply by patch removal. The conventional forms for transdermal delivery are patches and films; in addition, the microneedle technology is an innovative approach that overcomes the skin barrier for hydrophilic drugs [90,91,92,93,94].
4.1. Polymeric Nano- and Microparticle Carriers
Several different polymeric nano and microparticles have been successfully used for the delivery of polymyxin. In general, these polymeric materials have unique physicochemical and biological properties, including suitable and controllable size and charge, large specific surface area, functionalizable structures, resistance to chemical and enzymatic destruction, ability to penetrate biological barriers, and affinity for the membranes of bacterial cells. Most importantly, the nano and microparticles have the capacity to target drug delivery to sites of infection, thereby reducing drug doses and eliminating many side effects [4,7,95,96,97,98,99,100].
Polymyxins are positively charged at a neutral pH due to the presence of amine functional groups in their structures; therefore, these molecules are able to interact with anionic and polyanionic molecules to form polyelectrolyte complexes. The polymyxin molecules also contain a lipophilic fatty acyl tail and a hydrophilic head group, so they can form micellar structures with anionic polymers [36,101].
Polysaccharides (such as hyaluronan, sodium alginate, starch) and polyaminoacids are the most commonly used materials for polymyxin delivery systems as they are biocompatible, biodegradable, and non-toxic but still provide suitable drug loading efficiency and controlled drug release. Cationic polymers (e.g., chitosan) are sometimes included to coat these particles to stabilize the resulting complexes, as well as to control the surface charge and colistin release [101,102,103,104]. For example, Yasar et al. [101] developed polycomplexes based on anionic starch (molecular weight (MW) > 100,000) and oligochitosan (MW 5000) for the delivery of colistin and tobramycin. Both tobramycin and colistin were loaded during the formation of the starch/chitosan particles (i.e., the chitosan solution was added to a mixture of starch and antibiotic). The most stable anionic polyplexes were produced using a starch/oligochitosan ratio of 10:1. These polycomplexes had a size of 170–380 nm, a surface charge from −17 mV to −30 mV, an encapsulation efficiency (EE) of 97–99%, and loading efficiencies (LE) of 17% (for colistin) and 3% (for tobramycin). The drug release into phosphate-buffered saline (PBS, pH 7.4) reached 20–40% in 16 h. The antimicrobial activity of antibiotic-loaded polycomplexes against E. coli and P. aeruginosa was similar to that of the corresponding free drugs.
Coppi et al. [105,106,107] designed microparticles (100 nm to 3 μm in size) consisting of alginate (MW 147,000) and chitosan (MW 70,000) for oral administration of polymyxin B. Microparticles that were resistant to gastrointestinal media were prepared from polymyxin–alginate by a spray-drying technique and cross-linking with Ca2+ ions, followed by coating with a chitosan solution at a mass ratio polymyxin–chitosan of 1:1. The EE and LE were about 47% and 12%, respectively. The release of polymyxin in 2 h was 20–25% at pH 3.0 and 50–60% at pH 7.4. The microparticles maintained their antibiotic activity against E. coli (minimal inhibitory concentration (MIC) 0.625–5.00 μg/mL) but showed reduced antibiotic cytotoxicity against Vero cell cultures. A Caco-2 cell monolayer model study further showed that microparticles were endocytosed by the cells by 6 h of incubation. An ex vivo study showed that the fabricated particles could be taken up by M cells.
Liu et al. [108] developed nanoparticles of colistin (8 nm in size) by complexation of colistin with polyglutamic acid and subsequent stabilization with 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] ammonium salt (MW 2700). In vitro and in vivo studies showed that the colistin nanoparticles had equivalent antimicrobial activity against K. pneumoniae and A. baumannii to that of free colistin, but the particles showed improved safety and lower hepatotoxicity than the free drug when administered to mice over a seven-day period (the maximum tolerated dose increased 1.3-fold compared to free colistin).
Zashikhina et al. [109] synthesized amphiphilic charged copolymers of amino acids (hydrophilic lysine and glutamic acid, and hydrophobic phenylalanine) to form interpolyelectrolyte complexes with cationic and anionic peptides, including polymyxin B. The resulting particles were stabilized by coating with cationic or anionic polysaccharides (chitosan, heparin, alginate). The coated particles had a size of 200–2000 nm and a surface charge ranging from −60 mV to 40 mV. The EE was 75–90%, and the LE was 105–630 μg/mg. Drug release in PBS (pH 7.4) over 24 h was 26–60% and 15–30% for noncoated and coated particles, respectively. The coatings also decreased the cytotoxicity and increased the stability of the resulting peptide delivery systems in biological media.
Synthetic polyanionic macromolecules have also been used to design polymyxin delivery systems. For example, Costa et al. [110] fabricated poly-butyl-cyanoacrylate-based nanoparticles of polymyxin B (217 nm in size, with a surface charge of −18 mV), and found them effective in the treatment of leishmaniasis and associated Gram-negative bacterial infections. The release profile of polymyxin from the nanoparticles (into PBS, pH 7.4) was 50% of the loaded drug in 6 h, which was 6–7 times slower than observed with free polymyxin. An in vitro evaluation of the leishmanicidal activity indicated that the number of uninfected macrophages reached approximately 85%, with practically no highly infected macrophages. The MIC against P. aeruginosa for both encapsulated polymyxin and free polymyxin was 2 μg/mL.
Low molecular weight anionic molecules can also be used to produce polyelectrolyte particles of colistin. Abouelmagd et al. [111] studied the formation of pH-sensitive delivery systems for colistin via electrostatic interaction using the anionic polyphenolic compound tannic acid. The resulting particles had a negative surface charge and ranged in size from 1.5 to 5 μm, depending on the ratio of tannic acid/antibiotic. The EE was 50–60%, and the LE was 20–40%. The colistin release was 90–100% in 2 h at pH 4.5 and from 30% to 40% in 2 h at pH 7.4. The fabricated complexes had a MIC approximately two-fold higher than that of free colistin (0.6 and 8 μg/mL against E. coli and S. aureus, respectively) at pH 7.4, and the complexes showed MIC levels comparable to those of free drugs (0.3 and 4 μg/mL against E. coli and S. aureus, respectively) at pH 4.5. Overall, the developed systems appeared suitable for the targeted delivery of drugs to inflammatory tissues.
Among inorganic materials, mesoporous silica can be used to produce colistin nanoparticles. The resulting nanoparticles have high porosity for highly effective drug loading. The surface functionalization of mesoporous silica nanoparticles with molecular or polymer moieties provides them with great potency for controlling drug delivery [112,113]. The effectiveness of silica nanoparticles for polymyxin B delivery has been studied by Gounani et al. [114,115], who loaded mesoporous silica nanoparticles and carboxyl-modified mesoporous silica nanoparticles with polymyxin B as well as polymyxin B and vancomycin for the treatment of polymicrobial infections. The fabricated systems had a size of 70–130 nm, and they released 50% of both antibiotics in 24 h, with an 80% release in 80 h (into PBS, pH 7.4). The nanoparticles were effective against both Gram-negative and Gram-positive bacteria, and their efficacy superseded the activity of the free drugs, changing the effect from bacteriostatic to bactericidal at 1 × MIC concentration.
In summary, the delivery of polymyxins can be improved by encapsulating them in carriers based on anionic biopolymers or complexes of anionic and cationic polymers. In this case, the modification of antibiotic release is regulated by the MW of the polymers, the component ratios, particle sizes, and preparation methods.
4.2. Lipid-Based Nanostructures (Liposomes and Niosomes)
Liposome-mediated drug delivery is used to overcome the barriers to cellular and tissue uptake of drugs in both parenteral and oral administration. Liposomes are highly effective against microbial biofilms as they easily penetrate and accumulate throughout the entire thickness of the biofilm [116,117]. The potential use of liposomes for encapsulating polycationic polymyxins is hindered by their phospholipid membrane permeability. The research into polymyxin effects on artificial membrane models has shown various effects, ranging from increasing surface roughness to pore formation and leakage of contents [118]. The preparation of stable liposomal formulations with controlled sustained release often involves surface modification of the liposomes with anionic lipids. In addition, the integrity of bilayer membranes is controlled by the amount of loaded polymyxin [118,119,120,121,122].
Li et al. [119] loaded colistin into liposomes modified with sodium cholesteryl sulfate for intravenous administration. The phospholipid membrane permeability to colistin was reduced by modifying the liposomes with sodium cholesteryl sulfate. This improves the colistin loading by increasing the colistin–bilayer electrostatic interaction. The resulting liposomes had a size of 100–200 nm, ζ-potential of −60 to −66 mV, and EE of 50%. Colistin release (into 10 mM CaCl2) was 15–44% in 24 h. The pharmacokinetics results showed an approximately four-fold increase in the plasma AUC (0–8 h) for fabricated liposomes when compared with a free colistin solution.
Wang et al. [120] encapsulated colistin and ciprofloxacin in anionic liposomes. Incorporation of anionic lipids (1,2-dimyristoyl-sn-glycero-3-phosphoglycerol and hydrogenated soybean phosphatidylcholine) into the liposomes increased the EE of colistin compared to its EE in neutral liposomes (67.0% versus 18.4%). At least 70% of the loaded colistin was associated with the lipid bilayer on the outer surface of the liposomes, while about 30% of the loaded colistin was most likely located within the lipid membrane and oriented by its hydrophilic part toward the inner aqueous phase. The liposomal formulation had sizes of approximately 100 nm and EEs of 67.0% (for colistin) and 85.2% (for ciprofloxacin). In vitro release of ciprofloxacin and colistin into bovine serum for 2 h was greater than 70% and 50%, respectively. Liposome-encapsulated drugs showed enhanced in vitro antimicrobial activities against multidrug-resistant P. aeruginosa when compared to the free drugs.
Wallace et al. [121] obtained liposomes of colistin or colistin methanesulfonate to use in pulmonary inhalation. The prepared carriers had a size of 160–190 nm and EE of 50%. Colistin liposomes had a positive charge (+15 mV), and colistin methanesulfonate liposomes were anionic (−20 mV), indicating a direct association of the amphiphilic polypeptide with the liposome membrane. Investigation of the colloidal stability of the colistin-loaded liposomes showed that the particle size and ζ-potential were stable over the seven days. The particle size and ζ-potential of the colistin methanesulfonate-loaded liposomes were stable only for 48 h, and then a substantial growth in particle size occurred, together with a charge reversal from negative to positive. This instability and the subsequent sedimentation of colistin methanesulfonate-loaded liposomes were explained by the conversion of colistin methanesulfonate to colistin. The release of both colistin and colistimethate into PBS (pH 7.4) was rapid (50% of the total content in 10 min) and remained constant throughout the 72 h experiment.
Yu et al. [122] prepared colistin and ciprofloxacin co-loaded liposomes using mannitol, sucrose, and leucine as formulations for a dry powder inhaler. The EE values of colistin and ciprofloxacin were 47% and 45%, respectively. A cytotoxicity study demonstrated that the liposomal formulations were not cytotoxic at the drug concentrations of 5 μg/mL colistin and 20 μg/mL ciprofloxacin. The liposomal formulation showed superior antibacterial activity against P. aeruginosa when compared to each antibiotic alone.
Menina et al. [118] developed liposomes for oral delivery of colistin for the treatment of intracellular infections by Salmonella enterica. The stability of the liposomes was improved by the inclusion of cholesterol (30 mol%), which increased the integrity of vesicles by promoting alignment of the phospholipid alkyl chains and by enabling a condensed packing of the lipid bilayer. In addition, gastrointestinal-resistant liposomes were prepared using saturated long-carbon-chain phospholipids (1,2-dipalmitoyl phosphatidylcholine, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(glutaryl), and 1,2-distearoyl-sn-glycero-3-phosphocholine). The size of the liposome formulations ranged from 200 to 700 nm, and the negative surface charge ranged from −10 to −30 mV; the EE was 55–62%, while the LE was about 50%. Notably, the EE was inversely proportional to the colistin concentration. This phenomenon can be explained by the ability of colistin to disturb liposomal membranes. Thus, an increase in colistin concentration may lead to disruption of the liposomal structure and a reduced capacity for drug incorporation. Colistin release into biorelevant media (Fasted State Simulated Intestinal Fluid and Fed State Simulated Intestinal Fluid) was 20–50% in 5 h. Cell experiments (HEp-2 and Caco-2 cells infected with S. enterica) showed a significant reduction of intracellular bacteria after liposome treatment when compared with free colistin.
Niosomes are also used to improve antibiotic delivery. A niosome is a non-ionic vesicle that is formed mostly from a non-ionic surfactant, with cholesterol incorporated as an excipient. Niosomes can incorporate both hydrophilic drugs (in an aqueous layer) and lipophilic drugs (in a vesicular lipid membrane). The niosomes are used as delivery systems for poorly absorbable drugs and they increase drug absorption and bioavailability by penetration of the GIT barrier through transcytosis through M cells [123]. For example, Chauhan and Bhatt [124] developed polymyxin B niosomes using sorbitan monostearate (Span 60) and cholesterol to improve intestinal permeability. The fabricated niosomes had a size of 150–750 nm, their surface charge varied from −14 to −31 mV, and the EE was 45–80%. The niosomes protected polymyxin from the GIT environment and increased its absorption in the intestine through M cells. Analysis of rat creatinine concentration as a test of renal toxicity indicated that the creatinine concentration was within normal limits and nephrotoxicity was comparable to that of polymyxin B sulfate injection (500,000 units). In this case, the use of niosomes as a polymyxin delivery system improved its bioavailability by oral administration.
4.3. Conjugates
Conjugation of active pharmaceutical substances with various compounds can change the physicochemical properties of drug molecules or generate prodrugs with a sustained and controlled release. In addition, this type of modification of antimicrobial agents can reduce microbial resistance [125,126,127,128,129].
A polymyxin-cinnamaldehyde conjugate was obtained by the creation of an imine bond between the primary amino groups of polymyxin and the carbonyl group of cinnamaldehyde. This increased the drug lipophilicity and its subsequent incorporation into lipophilic nanoparticles, such as self-emulsifying drug delivery systems for oral administration. At the same time, the molecule polarity (log P) was increased 69-fold. The release of polymyxin ranged from 45% to 81% within 16 h [130].
Dextrin–colistin conjugates were synthesized using a carbodiimide coupling reaction with a colistin content of 7.6% w/w. The fabricated conjugates exhibited lower antimicrobial activity against E. coli than was observed for unmodified colistin (7–12 log-fold reduction in MIC); however, dextrin conjugation reduced colistin in vitro toxicity toward human kidney proximal tubule cells (HK-2, the IC50 concentration improved by four- to five-fold) [131]. Dextrin–colistin conjugates reduced the toxicity of colistin and improved targeting to sites of bacterial infection due to effective binding with bacterial lipopolysaccharide.
In biological studies, both colistin and the dextrin–colistin conjugate effectively inhibited lipopolysaccharide-induced hemolysis and tumor necrosis factor-alpha secretion, but only the dextrin–colistin conjugate showed no additive toxicity at high concentrations [132]. A study of the antibacterial activity of dextrin–colistin conjugate against A. baumannii showed a prolonged antimicrobial effect for up to 48 h, while free colistin was active for only 4 h. An in vivo study showed that colistin was effectively released from the conjugate by endogenous alpha-amylase within a wound environment (up to 86.3% at 48 h) [133]. Colistin–dextrin conjugates exhibited comparable antimicrobial activity to colistimethate sodium against Gram-negative pathogens (A. baumannii, E. coli, K. pneumoniae, and P. aeruginosa), but in vitro toxicity toward kidney cells was significantly reduced (the IC50 increased by four times). In vivo dose-escalation studies in rats demonstrated improved prolonged pharmacokinetics of the conjugates (t1/2 130–1300 min vs. 53 min) and decreased toxicity when compared to colistin sulfate [134].
A poly(ethylene glycol) methyl ether acrylate (PEGA-480) bioconjugate of colistin has been synthesized via copper-mediated, photoinduced, living radical polymerization using the initiator linker 2-(2-bromo-2-methylpropanoyloxy) acetic acid (degrees of polymerization of 5, 10, and 20). The initial colistin release (into PBS, pH 7.4) was 30–40% in 5 h, and the total release occurred by 48 h. In vitro experiments showed that the conjugate’s antimicrobial activity against A. baumannii was similar to or better than that of the clinically relevant colistin prodrug colistimethate sodium [135].
Gold nanoparticles were functionalized with colistin using polyethylene glycol as a linker for later use in binding A. baumannii. The resulting particles had an average size of 20 nm and a surface charge of about −6 mV. In vitro studies showed that particle complexation with A. baumannii occurred rapidly and reached half-maximum saturation in 7 min [136].
4.4. Structured Gels (Hydrogels and Microgels)
Various structured gels based on natural and synthetic polymers are used to develop sustained release antimicrobial dosage forms for the treatment of wounds and burns [137,138,139,140]. Hydrogels and microgels are dispersions of loosely crosslinked polymeric colloids that can be used as delivery systems for polypeptides due to their swelling behavior. They can protect peptides from chemical and enzymatic degradation and increase physical stability by reducing aggregation or conformational changes [141].
For example, phenylboronic acid-functionalized polycarbonate hydrogels loaded with polymyxin B demonstrated controlled in vitro drug release kinetics (75–100% in 50 h) and in vitro antimicrobial activity against P. aeruginosa over 48 h (MIC = 2 μM, determined by the disc diffusion method), as well as in vivo antimicrobial efficacy against P. aeruginosa burn wound infections [142].
A dextran-poly(ethylene glycol) hydrogel covalently conjugated with polymyxin B and vancomycin has been designed as a wound dressing. The releases (into PBS, pH 7.4) of polymyxin B and vancomycin were 9.2% and 19.7%, respectively, on the first day, and the values remained almost unchanged for the first two weeks. The hydrogel exhibits potent antibacterial activities against both Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria. In addition, the hydrogels facilitated the penetration of the drug into the bacterial cell membrane and killed bacteria without a cytotoxic effect on the NIH 3T3 mouse fibroblast cell line [143].
Chitosan hydrogels with zinc phthalocyanine-colistin conjugates demonstrated improved efficiency against P. aeruginosa when compared to free colistin in an in vitro study [144]. Microfluidics-based self-assembled alginate microgels crosslinked with Ca2+ were loaded with polymyxin B to give an EE above 80%. The size of the microgel particles was 100–200 nm and the ζ-potential ranged from −10 to −30 mV. The polymyxin release from the obtained microgels was 20–30% in 10 mM Tris buffer; however, an increase in the ionic strength to 150 mM NaCl resulted in 100% polymyxin release within 1 h [145].
4.5. Polymeric Nanofibers and Membranes
Electrospinning technology is used to produce non-woven fiber mats. The electrospun fibers have a large specific surface, excellent mechanical performance, porous structure, and unique nanometer-scale architecture. Therefore, these materials have great potential for use as antibacterial wound dressings [146,147,148,149,150]. For example, Zhang et al. [151] prepared poly(l-lactide) electrospun mats for loading polymyxin B and dexamethasone. The drug release (into tris-HCl buffer, pH 7.4) from the electrospun mats was 23% (for dexamethasone) and 30% (for polymyxin B) for 35 days. An in vitro study confirmed the antibacterial activity of the developed systems against Gram-positive and Gram-negative bacteria, as the mats completely inhibited the growth of E. coli over a 110 h period, and the S. aureus inhibition efficiencies were 98.8% and 49.5% after 16 and 28 h, respectively. An in vivo study of infected full-thickness burns and infected wounds demonstrated healing on day 14.
Huang et al. [152] prepared a polymyxin B-immobilized nanofiber sponge for endotoxin adsorption by electrospinning and freeze-drying technology. The endotoxin removal rate of a polymyxin-free nanofiber sponge was about 17%, while the polymyxin-immobilized nanofiber sponge adsorbed 99% of the endotoxin. The endotoxin removal rate in human plasma was 90%, and the adsorption reached equilibrium within 60 min, thereby confirming the great potential of polymyxin-immobilized-nanofiber sponges for clinical blood purification.
Polymer membranes can also be used as colistin delivery systems for treating wounds and burns. An elastomer nanocomposite membrane loaded with polymyxin B in halloysite nanotubes with ciprofloxacin has been developed for wound dressing. The membrane released 50% of the polymyxin in 20 h, and 80–100% in 120 h (into PBS, pH 7.4). All the nanocomposites exhibited antimicrobial activity against both Gram-negative (P. aeruginosa) and Gram-positive (S. aureus) bacteria in more than seven days due to sustained drug release. This drug system is highly water-absorbing and shows low cytotoxicity, good biodegradability, and appreciable elasticity [153].
4.6. Microneedles
Microneedle technology is a transdermal, but minimally invasive, delivery system for active pharmaceutical substances, including antimicrobial agents, peptides, and proteins. Microneedles penetrate the stratum corneum and dissolve into the intercellular fluid of the skin to ensure systemic drug delivery [154,155,156,157].
Dillon et al. [156] developed a microneedle system composed of polyvinylpyrrolidone and trehalose and encapsulated polymyxin B for transdermal delivery. The disc diffusion method showed the effectiveness of fabricated microneedles against Salmonella typhimurium. Ex vivo skin diffusion studies showed the system successfully delivered antibiotics through porcine skin during the first 4 h of application at a faster initial rate than a drug-loaded control disc.
5. Future Aspects
The toxicity of polymyxins observed in the clinic is due to their antimicrobial mechanism, which involves interaction with and damage to bilayer membranes. The same mechanism, however, can cause similar and significant damage to the membranes of cells that make up human organs, like the liver and kidney, when polymyxins are administered at high doses. Therefore, dose reduction is a prime concern in the development of safe and effective polymyxin formulations for targeted and controlled drug release. The development of polymyxin dosage forms should be based on the combined achievements of chemistry, nanotechnology, and nanomedicine. Solving polymyxin delivery and toxicity problems will require chemical conjugation of the drug molecules with biocompatible and biodegradable materials, as well as the development of new compositions based on nanoparticles, liposomes, microneedles, and composite nanomaterials.
Currently, polymeric and lipid-based particles with different parameters may be used to improve both oral and parenteral administration of polymyxin via targeted and control release, while also reducing dose and toxicity. These approaches require a search for safe, biocompatible, and biodegradable materials. Undoubtedly, natural polymers and their derivatives will prove to have great medical potential in this respect. Further research is, therefore, needed to develop systems based on natural polymers (e.g., polyelectrolyte complexes, self-assembled particles, electrospray particles, etc.).
Conjugation of polymyxins is a promising method for improving their intestinal permeability and absorption, as well as for preventing microbial resistance to this vital antimicrobial agent. In general, the chemical modification of polymyxin molecules is insufficiently studied; and information is extremely sparse regarding the effect of modification of polymyxin molecules on drug stability, antimicrobial activity, and the ability to overcome microbial resistance. At the moment, the threat of global resistance to polymyxins is a major problem in the treatment of Gram-negative multiresistant infections, since polymyxins are the last-line drugs. Chemical modification of existing drugs may, therefore, prove to be the solution to the drug resistance problem and may represent the future of antimicrobial drug chemistry.
Author Contributions
Conceptualization, N.V.D., Y.A.S.; writing—original draft preparation, N.V.D.; writing—review and editing, Y.A.S.; supervision, Y.A.S.; project administration, Y.A.S.; funding acquisition, Y.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Russian Science Foundation (project 19-73-20157).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- WHO Publishes List of Bacteria for Which New Antibiotics are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 12 April 2020).
- Gajdacs, M. The concept of an ideal antibiotic: Implications for drug design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bello, C. Antibiotic adjuvants—A strategy to unlock bacterial resistance to antibiotics. Bioorg. Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pornpattananangku, D.; Hu, C.M.; Huang, C.M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef]
- Raza, A.; Sime, F.B.; Cabot, P.J.; Maqbool, F.; Roberts, J.A.; Falconer, J.R. Solid nanoparticles for oral antimicrobial drug delivery: A review. Drug Discov. Today 2019, 24, 858–866. [Google Scholar] [CrossRef]
- Dos Santos Ramos, M.A.; Da Silva, P.B.; Sposito, L.; De Toledo, L.G.; Bonifacio, B.V.; Rodero, C.F.; Dos Santos, K.C.; Chorilli, M.; Bauab, T.M. Nanotechnology-based drug delivery systems for control of microbial biofilms: A review. Int. J. Nanomed. 2018, 13, 1179–1213. [Google Scholar] [CrossRef]
- Martin-Serrano, A.; Gomez, R.; Ortega, P.; de la Mata, F.J. Nanosystems as vehicles for the delivery of antimicrobial peptides (AMPs). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef]
- Kelly, S.A.; Rodgers, A.M.; O’Brien, S.C.; Donnelly, R.F.; Gilmore, B.F. Gut check time: Antibiotic delivery strategies to reduce antimicrobial resistance. Trends Biotechnol. 2020, 38, 447–462. [Google Scholar] [CrossRef]
- Landman, D.; Georgescu, C.; Martin, D.A.; Quale, J. Polymyxins revisited. Clin. Microbiol. Rev. 2008, 21, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.X.; Wang, C.Y.; Li, Y.Y.; Li, J.; Wan, Q.Q.; Chen, J.H.; Tay, F.R.; Niu, L.N. Considerations and caveats in combating eskape pathogens against nosocomial infections. Adv. Sci. 2020, 7, 1901872. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Davin-Regli, A.; Lavigne, J.P.; Pages, J.M. Enterobacter spp.: Update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin. Microbiol. Rev. 2019, 32, e00002–e00019. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-how P. aeruginosa can escape antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef]
- Lupo, A.; Haenni, M.; Madec, J.Y. Antimicrobial resistance in Acinetobacter spp. and Pseudomonas spp. Microbiol. Spectr. 2018, 6, 377–393. [Google Scholar] [CrossRef]
- Felden, B.; Cattoir, V. Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrob. Agents Chemother 2018, 62, e02503–e02517. [Google Scholar] [CrossRef]
- Gaspar, M.C.; Couet, W.; Olivier, J.C.; Pais, A.A.; Sousa, J.J. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: A review. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1231–1252. [Google Scholar] [CrossRef]
- Friebel, C.; Steckel, H. Single-use disposable dry powder inhalers for pulmonary drug delivery. Expert Opin. Drug Deliv. 2010, 7, 1359–1372. [Google Scholar] [CrossRef]
- Safdar, A.; Shelburne, S.A.; Evans, S.E.; Dickey, B.F. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin. Drug Saf. 2009, 8, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.; Mills, N.; Whitaker, P. Nebuliser systems for drug delivery in cystic fibrosis. Cochrane Database Syst. Rev. 2013, 4. [Google Scholar] [CrossRef]
- Banerjee, D.; Stableforth, D. The treatment of respiratory pseudomonas infection in cystic fibrosis: What drug and which way? Drugs 2000, 60, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Novel formulations for antimicrobial peptides. Int. J. Mol. Sci. 2014, 15, 18040–18083. [Google Scholar] [CrossRef]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and their potential next generation as therapeutic antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure—Activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef]
- Martin, N.I.; Hu, H.; Moake, M.M.; Churey, J.J.; Whittal, R.; Worobo, R.W.; Vederas, J.C. Isolation, structural characterization, and properties of mattacin (polymyxin M), a cyclic peptide antibiotic produced by paenibacillus kobensis M. J. Biol. Chem. 2003, 278, 13124–13132. [Google Scholar] [CrossRef]
- Orwa, J.A.; Van Gerven, A.; Roets, E.; Hoogmartens, J. Development and validation of a liquid chromatography method for analysis of colistin sulphate. Chromatographia 2000, 51, 433–436. [Google Scholar] [CrossRef]
- Cao, G.; Ali, F.E.; Chiu, F.; Zavascki, A.P.; Nation, R.L.; Li, J. Development and validation of a reversed-phase high-performance liquid chromatography assay for polymyxin b in human plasma. J. Antimicrob. Chemother 2008, 62, 1009–1014. [Google Scholar] [CrossRef]
- Pfeifer, C.; Fassauer, G.; Gerecke, H.; Jira, T.; Remane, Y.; Frontini, R.; Byrne, J.; Reinhardt, R. Purity determination of amphotericin b, colistin sulfate and tobramycin sulfate in a hydrophilic suspension by HPLC. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 990, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Burkin, M.; Eremin, S.; Dias, A.C.P.; Zhang, X. Development of competitive ELISA and CLEIA for quantitative analysis of polymyxin B. Food Analyt. Methods 2019, 12, 1412–1419. [Google Scholar] [CrossRef]
- Decolin, D.; Leroy, P.; Nicolas, A.; Archimbault, P. Hyphenated liquid chromatographic method for the determination of colistin residues in bovine tissues. J. Chromatogr. Sci. 1997, 35, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Orwa, J.A.; Govaerts, C.; Busson, R.; Roets, E.; Van Schepdael, A.; Hoogmartens, J. Isolation and structural characterization of colistin components. J. Antibiot. 2001, 54, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.J.; Li, J.; Nation, R.L.; Prankerd, R.J.; Velkov, T.; Boyd, B.J. Self-assembly behavior of colistin and its prodrug colistin methanesulfonate: Implications for solution stability and solubilization. J. Phys. Chem. B 2010, 114, 4836–4840. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.D.; Stern, R.C.; O’Riordan, M.A.; Blumer, J.L. The pharmacokinetics of colistin in patients with cystic fibrosis. J. Clin. Pharmacol. 2001, 41, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Lopez, C.A.; Gnanakaran, S. Permeability barrier of gram-negative cell envelopes and approaches to bypass it. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef]
- Nishino, K.; Hsu, F.F.; Turk, J.; Cromie, M.J.; Wosten, M.M.; Groisman, E.A. Identification of the lipopolysaccharide modifications controlled by the Salmonella PmrA/PmrB system mediating resistance to Fe(III) and Al(III). Mol. Microbiol. 2006, 61, 645–654. [Google Scholar] [CrossRef]
- Hancock, R.E. Antibacterial peptides and the outer membranes of gram-negative bacilli. J. Med. Microbiol. 1997, 46, 1–3. [Google Scholar]
- Hancock, R.E.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.E.; Feola, D.J.; Rapp, R.P. Polymyxin B sulfate and colistin: Old antibiotics for emerging multiresistant gram-negative bacteria. Ann. Pharmacother. 1999, 33, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed. Res. Int. 2015, 2015, 679109. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.T.; Cajal, Y.; Skowronska, E.M.; Belkin, S.; Chen, J.; Van Dyk, T.K.; Sasser, M.; Jain, M.K. Cationic peptide antimicrobials induce selective transcription of micF and osmY in Escherichia coli. Biochim. Biophys. Acta 2000, 1463, 43–54. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Khondker, A.; Alsop, R.J.; Dhaliwal, A.; Saem, S.; Moran-Mirabal, J.M.; Rheinstadter, M.C. Membrane cholesterol reduces polymyxin b nephrotoxicity in renal membrane analogs. Biophys. J. 2017, 113, 2016–2028. [Google Scholar] [CrossRef]
- Khondker, A.; Dhaliwal, A.K.; Saem, S.; Mahmood, A.; Fradin, C.; Moran-Mirabal, J.; Rheinstadter, M.C. Membrane charge and lipid packing determine polymyxin-induced membrane damage. Commun. Biol. 2019, 2, 67. [Google Scholar] [CrossRef]
- Khondker, A.; Rheinstadter, M.C. How do bacterial membranes resist polymyxin antibiotics? Commun. Biol. 2020, 3, 77. [Google Scholar] [CrossRef]
- Fu, L.; Wan, M.; Zhang, S.; Gao, L.; Fang, W. Polymyxin B loosens lipopolysaccharide bilayer but stiffens phospholipid bilayer. Biophys. J. 2020, 118, 138–150. [Google Scholar] [CrossRef]
- Dupuy, F.G.; Pagano, I.; Andenoro, K.; Peralta, M.F.; Elhady, Y.; Heinrich, F.; Tristram-Nagle, S. Selective interaction of colistin with lipid model membranes. Biophys. J. 2018, 114, 919–928. [Google Scholar] [CrossRef]
- Zhang, H.; Srinivas, S.; Xu, Y.; Wei, W.; Feng, Y. Genetic and biochemical mechanisms for bacterial lipid a modifiers associated with polymyxin resistance. Trends Biochem. Sci. 2019, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wei, W.; Lei, S.; Lin, J.; Srinivas, S.; Feng, Y. An evolutionarily conserved mechanism for intrinsic and transferable polymyxin resistance. mBio 2018, 9, e02317. [Google Scholar] [CrossRef] [PubMed]
- Nang, S.C.; Li, J.; Velkov, T. The rise and spread of mcr plasmid-mediated polymyxin resistance. Crit. Rev. Microbiol. 2019, 45, 131–161. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, Z.; Gholizadeh, P.; Ganbarov, K.; Bialvaei, A.Z.; Mahmood, S.S.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Molecular mechanisms related to colistin resistance in enterobacteriaceae. Infect. Drug Resist. 2019, 12, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Kiani, M.H.; Imran, M.; Raza, A.; Shahnaz, G. Multi-functionalized nanocarriers targeting bacterial reservoirs to overcome challenges of multi drug-resistance. Daru 2020. [Google Scholar] [CrossRef] [PubMed]
- Yeom, J.H.; Lee, B.; Kim, D.; Lee, J.K.; Kim, S.; Bae, J.; Park, Y.; Lee, K. Gold nanoparticle-DNA aptamer conjugate-assisted delivery of antimicrobial peptide effectively eliminates intracellular Salmonella enterica serovar Typhimurium. Biomaterials 2016, 104, 43–51. [Google Scholar] [CrossRef]
- Domalaon, R.; De Silva, P.M.; Kumar, A.; Zhanel, G.G.; Schweizer, F. The anthelmintic drug niclosamide synergizes with colistin and reverses colistin resistance in gram-negative bacilli. Antimicrob. Agents Chemother 2019, 63. [Google Scholar] [CrossRef]
- Domalaon, R.; Okunnu, O.; Zhanel, G.G.; Schweizer, F. Synergistic combinations of anthelmintic salicylanilides oxyclozanide, rafoxanide, and closantel with colistin eradicates multidrug-resistant colistin-resistant gram-negative bacilli. J. Antibiot. 2019, 72, 605–616. [Google Scholar] [CrossRef]
- Ayerbe-Algaba, R.; Gil-Marques, M.L.; Miro-Canturri, A.; Parra-Millan, R.; Pachon-Ibanez, M.E.; Jimenez-Mejias, M.E.; Pachon, J.; Smani, Y. The anthelmintic oxyclozanide restores the activity of colistin against colistin-resistant gram-negative bacilli. Int. J. Antimicrob. Agents 2019, 54, 507–512. [Google Scholar] [CrossRef]
- Zimmerman, S.M.; Lafontaine, A.J.; Herrera, C.M.; McLean, A.B.; Trent, M.S. A whole-cell screen identifies small bioactives that synergize with polymyxin and exhibit antimicrobial activities against multidrug-resistant bacteria. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Niu, H.; Wang, R.; Cai, Y. Safety and efficacy of colistin alone or in combination in adults with Acinetobacter baumannii infection: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2019, 53, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Ioannidou, E.; Falagas, M.E. Colistin monotherapy vs. combination therapy: Evidence from microbiological, animal and clinical studies. Clin. Microbiol. Infect. 2008, 14, 816–827. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I.; Kasiakou, S.K.; Hatzopoulou, P.; Michalopoulos, A. Effectiveness and nephrotoxicity of colistin monotherapy vs. colistin-meropenem combination therapy for multidrug-resistant gram-negative bacterial infections. Clin. Microbiol. Infect. 2006, 12, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Ribera, A.; Benavent, E.; Lora-Tamayo, J.; Tubau, F.; Pedrero, S.; Cabo, X.; Ariza, J.; Murillo, O. Osteoarticular infection caused by MDR Pseudomonas aeruginosa: The benefits of combination therapy with colistin plus β-lactams. J. Antimicrob. Chemother 2015, 70, 3357–3365. [Google Scholar] [PubMed]
- Zhang, X.; Guo, F.; Shao, H.; Zheng, X. Clinical translation of polymyxin-based combination therapy: Facts, challenges and future opportunities. J. Infect. 2017, 74, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Falagas, M.E. Colistin versus polymyxin b for the treatment of patients with multidrug-resistant gram-negative infections: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2017, 49, 233–238. [Google Scholar] [CrossRef]
- Liu, Q.; Li, W.; Feng, Y.; Tao, C. Efficacy and safety of polymyxins for the treatment of Acinectobacter baumannii infection: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98091. [Google Scholar] [CrossRef]
- Zusman, O.; Altunin, S.; Koppel, F.; Dishon Benattar, Y.; Gedik, H.; Paul, M. Polymyxin monotherapy or in combination against carbapenem-resistant bacteria: Systematic review and meta-analysis. J. Antimicrob. Chemother 2017, 72, 29–39. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Liu, H.X.; Zhu, Y.G.; Qu, J.M. Intravenous combined with aerosolised polymyxin versus intravenous polymyxin alone in the treatment of pneumonia caused by multidrug-resistant pathogens: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2015, 46, 603–609. [Google Scholar] [CrossRef]
- Terayama, T.; Yamakawa, K.; Umemura, Y.; Aihara, M.; Fujimi, S. Polymyxin B hemoperfusion for sepsis and septic shock: A systematic review and meta-analysis. Surg. Infect. 2017, 18, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, S.C.; Wong, H.; Hop, C.E.C.A. Oral absorption. In Drug Metabolism and Pharmacokinetics Quick Guide; Springer: New York, NY, USA, 2011; pp. 47–56. [Google Scholar]
- Sorli, L.; Luque, S.; Li, J.; Campillo, N.; Danes, M.; Montero, M.; Segura, C.; Grau, S.; Horcajada, J.P. Colistin for the treatment of urinary tract infections caused by extremely drug-resistant Pseudomonas aeruginosa: Dose is critical. J. Infect. 2019, 79, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.K.; Nichols, B.L.B.; Edgar, K.J.; Murgia, X.; Loretz, B.; Lehr, C.M. Challenges and strategies in drug delivery systems for treatment of pulmonary infections. Eur. J. Pharm. Biopharm. 2019, 144, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef] [PubMed]
- Oliota, A.F.; Penteado, S.T.; Tonin, F.S.; Fernandez-Llimos, F.; Sanches, A.C. Nephrotoxicity prevalence in patients treated with polymyxins: A systematic review with meta-analysis of observational studies. Diagn. Microbiol. Infect. Dis. 2019, 94, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, F.; Dashti-Khavidaki, S. Colistin: Efficacy and safety in different populations. Expert Rev. Clin. Pharmacol. 2015, 8, 423–448. [Google Scholar] [CrossRef]
- Dai, C.; Xiao, X.; Li, J.; Ciccotosto, G.D.; Cappai, R.; Tang, S.; Schneider-Futschik, E.K.; Hoyer, D.; Velkov, T.; Shen, J. Molecular mechanisms of neurotoxicity induced by polymyxins and chemoprevention. ACS Chem. Neurosci. 2019, 10, 120–131. [Google Scholar] [CrossRef]
- Pinto-Alphandary, H.; Andremont, A.; Couvreur, P. Targeted delivery of antibiotics using liposomes and nanoparticles: Research and applications. Int. J. Antimicrob. Agents 2000, 13, 155–168. [Google Scholar] [CrossRef]
- Falciani, C.; Zevolini, F.; Brunetti, J.; Riolo, G.; Gracia, R.; Marradi, M.; Loinaz, I.; Ziemann, C.; Cossio, U.; Llop, J.; et al. Antimicrobial peptide-loaded nanoparticles as inhalation therapy for Pseudomonas aeruginosa infections. Int. J. Nanomed. 2020, 15, 1117–1128. [Google Scholar] [CrossRef]
- Weers, J.G.; Miller, D.P.; Tarara, T.E. Spray-dried pulmosphere formulations for inhalation comprising crystalline drug particles. AAPS Pharm. Sci. Tech. 2019, 20, 103. [Google Scholar] [CrossRef]
- Woods, A.; Rahman, K.M. Antimicrobial molecules in the lung: Formulation challenges and future directions for innovation. Future Med. Chem. 2018, 10, 575–604. [Google Scholar] [CrossRef] [PubMed]
- Faustino, C.; Pinheiro, L. Lipid systems for the delivery of amphotericin b in antifungal therapy. Pharmaceutics 2020, 12, 29. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Diep, D.B.; Tonnesen, H.H. Topical antimicrobial peptide formulations for wound healing: Current developments and future prospects. Acta Biomater. 2020, 103, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Ohnstedt, E.; Lofton Tomenius, H.; Vagesjo, E.; Phillipson, M. The discovery and development of topical medicines for wound healing. Expert Opin. Drug Discov. 2019, 14, 485–497. [Google Scholar] [CrossRef]
- Ambekar, R.S.; Kandasubramanian, B. Advancements in nanofibers for wound dressing: A review. Eur. Polym. J. 2019, 117, 304–336. [Google Scholar] [CrossRef]
- Mofazzal Jahromi, M.A.; Sahandi Zangabad, P.; Moosavi Basri, S.M.; Sahandi Zangabad, K.; Ghamarypour, A.; Aref, A.R.; Karimi, M.; Hamblin, M.R. Nanomedicine and advanced technologies for burns: Preventing infection and facilitating wound healing. Adv. Drug Deliv. Rev. 2018, 123, 33–64. [Google Scholar] [CrossRef]
- Kurmi, B.D.; Tekchandani, P.; Paliwal, R.; Paliwal, S.R. Transdermal drug delivery: Opportunities and challenges for controlled delivery of therapeutic agents using nanocarriers. Curr. Drug Metab. 2017, 18, 481–495. [Google Scholar] [CrossRef]
- Nematpour, N.; Farhadian, N.; Ebrahimi, K.S.; Arkan, E.; Seyedi, F.; Khaledian, S.; Shahlaei, M.; Moradi, S. Sustained release nanofibrous composite patch for transdermal antibiotic delivery. Colloids Surfaces A Physicochem. Eng. Aspects 2020, 586, 124267. [Google Scholar] [CrossRef]
- Kodoth, A.K.; Ghate, V.M.; Lewis, S.A.; Prakash, B.; Badalamoole, V. Pectin-based silver nanocomposite film for transdermal delivery of donepezil. Int. J. Biol. Macromol. 2019, 134, 269–279. [Google Scholar] [CrossRef]
- Gonzalez-Vazquez, P.; Larraneta, E.; McCrudden, M.T.C.; Jarrahian, C.; Rein-Weston, A.; Quintanar-Solares, M.; Zehrung, D.; McCarthy, H.; Courtenay, A.J.; Donnelly, R.F. Transdermal delivery of gentamicin using dissolving microneedle arrays for potential treatment of neonatal sepsis. J. Control Release 2017, 265, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Tsioris, K.; Raja, W.K.; Pritchard, E.M.; Panilaitis, B.; Kaplan, D.L.; Omenetto, F.G. Fabrication of silk microneedles for controlled-release drug delivery. Adv. Funct. Mater. 2012, 22, 330–335. [Google Scholar] [CrossRef]
- Lee, N.Y.; Ko, W.C.; Hsueh, P.R. Nanoparticles in the treatment of infections caused by multidrug-resistant organisms. Front. Pharmacol. 2019, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Giri, T.K. (Eds.) Recent advances of chitosan nanoparticles as a carrier for delivery of antimicrobial drugs. In Polysaccharide Based Nano-Biocarrier in Drug Delivery; CRC Press: Boca Raton, FL, USA, 2018; pp. 637–679. [Google Scholar]
- Parisi, O.I.; Scrivano, L.; Sinicropi, M.S.; Puoci, F. Polymeric nanoparticle constructs as devices for antibacterial therapy. Curr. Opin. Pharmacol. 2017, 36, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Opatz, T.; Landfester, K.; Wurm, F.R. Carbohydrate nanocarriers in biomedical applications: Functionalization and construction. Chem. Soc. Rev. 2015, 44, 8301–8325. [Google Scholar] [CrossRef]
- D’ANGELO, I.; Conte, C.; Miro, A.; Quaglia, F.; Ungaro, F. Pulmonary drug delivery: A role for polymeric nanoparticles? Curr. Top. Med. Chem. 2015, 15, 386–400. [Google Scholar] [CrossRef][Green Version]
- Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-based local antimicrobial drug delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57. [Google Scholar] [CrossRef]
- Yasar, H.; Ho, D.K.; De Rossi, C.; Herrmann, J.; Gordon, S.; Loretz, B.; Lehr, C.M. Starch-chitosan polyplexes: A versatile carrier system for anti-infectives and gene delivery. Polymers 2018, 10, 252. [Google Scholar] [CrossRef]
- Raik, S.V.; Gasilova, E.R.; Dubashynskaya, N.V.; Dobrodumov, A.V.; Skorik, Y.A. Diethylaminoethyl chitosan-hyaluronic acid polyelectrolyte complexes. Int. J. Biol. Macromol. 2020, 146, 1161–1168. [Google Scholar] [CrossRef]
- Deacon, J.; Abdelghany, S.M.; Quinn, D.J.; Schmid, D.; Megaw, J.; Donnelly, R.F.; Jones, D.S.; Kissenpfennig, A.; Elborn, J.S.; Gilmore, B.F.; et al. Antimicrobial efficacy of tobramycin polymeric nanoparticles for pseudomonas aeruginosa infections in cystic fibrosis: Formulation, characterisation and functionalisation with dornase alfa (dnase). J. Control Release 2015, 198, 55–61. [Google Scholar] [CrossRef]
- Balmayor, E.R.; Baran, E.T.; Azevedo, H.S.; Reis, R.L. Injectable biodegradable starch/chitosan delivery system for the sustained release of gentamicin to treat bone infections. Carbohydr. Polym. 2012, 87, 32–39. [Google Scholar] [CrossRef]
- Coppi, G.; Iannuccelli, V.; Sala, N.; Bondi, M. Alginate microparticles for polymyxin B peyer’s patches uptake: Microparticles for antibiotic oral administration. J. Microencapsul. 2004, 21, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Coppi, G.; Sala, N.; Bondi, M.; Sergi, S.; Iannuccelli, V. Ex-vivo evaluation of alginate microparticles for polymyxin B oral administration. J. Drug Target. 2006, 14, 599–606. [Google Scholar] [CrossRef]
- Coppi, G.; Montanari, M.; Rossi, T.; Bondi, M.; Iannuccelli, V. Cellular uptake and toxicity of microparticles in a perspective of polymyxin b oral administration. Int. J. Pharm. 2010, 385, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Kuo, S.C.; Yao, B.Y.; Fang, Z.S.; Lee, Y.T.; Chang, Y.C.; Chen, T.L.; Hu, C.J. Colistin nanoparticle assembly by coacervate complexation with polyanionic peptides for treating drug-resistant gram-negative bacteria. Acta Biomater. 2018, 82, 133–142. [Google Scholar] [CrossRef]
- Zashikhina, N.N.; Yudin, D.V.; Tarasenko, I.I.; Osipova, O.M.; Korzhikova-Vlakh, E.G. Multilayered particles based on biopolyelectrolytes as potential peptide delivery systems. Polym. Sci. Ser. A 2020, 62, 43–53. [Google Scholar] [CrossRef]
- Souza Ribeiro Costa, J.; Medeiros, M.; Yamashiro-Kanashiro, E.H.; Rocha, M.C.; Cotrim, P.C.; Stephano, M.A.; Lancellotti, M.; Tavares, G.D.; Oliveira-Nascimento, L. Biodegradable nanocarriers coated with polymyxin B: Evaluation of leishmanicidal and antibacterial potential. PLoS Negl. Trop. Dis. 2019, 13, e0007388. [Google Scholar] [CrossRef] [PubMed]
- Abouelmagd, S.A.; Abd Ellah, N.H.; Amen, O.; Abdelmoez, A.; Mohamed, N.G. Self-assembled tannic acid complexes for ph-responsive delivery of antibiotics: Role of drug-carrier interactions. Int. J. Pharm. 2019, 562, 76–85. [Google Scholar] [CrossRef]
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I. Mesoporous silica nanoparticles in biomedical applications. Chem. Soc. Rev. 2012, 41, 2590–2605. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.Y. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Gounani, Z.; Asadollahi, M.A.; Pedersen, J.N.; Lyngso, J.; Skov Pedersen, J.; Arpanaei, A.; Meyer, R.L. Mesoporous silica nanoparticles carrying multiple antibiotics provide enhanced synergistic effect and improved biocompatibility. Colloids Surf. B Biointerfaces 2019, 175, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Gounani, Z.; Asadollahi, M.A.; Meyer, R.L.; Arpanaei, A. Loading of polymyxin B onto anionic Mesoporous silica nanoparticles retains antibacterial activity and enhances biocompatibility. Int. J. Pharm. 2018, 537, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Naeem, S.; Viswanathan, G.; Misran, M.B. Liposomes as colloidal nanovehicles: On the road to success in intravenous drug delivery. Rev. Chem. Eng. 2018, 34, 365–383. [Google Scholar] [CrossRef]
- Wang, D.Y.; van der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-based antimicrobial delivery-systems for the treatment of bacterial infections. Front. Chem. 2019, 7, 872. [Google Scholar] [CrossRef] [PubMed]
- Menina, S.; Eisenbeis, J.; Kamal, M.A.M.; Koch, M.; Bischoff, M.; Gordon, S.; Loretz, B.; Lehr, C.M. Bioinspired liposomes for oral delivery of colistin to combat intracellular infections by Salmonella enterica. Adv. Healthc. Mater. 2019, 8, e1900564. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, L.; Tang, C.; Zhang, E.; Ding, L.; Yang, L. Preparation and characterisation of the colistin-entrapped liposome driven by electrostatic interaction for intravenous administration. J. Microencapsul. 2016, 33, 427–437. [Google Scholar] [CrossRef]
- Wang, S.; Yu, S.; Lin, Y.; Zou, P.; Chai, G.; Yu, H.H.; Wickremasinghe, H.; Shetty, N.; Ling, J.; Li, J.; et al. Co-delivery of ciprofloxacin and colistin in liposomal formulations with enhanced in vitro antimicrobial activities against multidrug resistant Pseudomonas aeruginosa. Pharm. Res. 2018, 35, 187. [Google Scholar] [CrossRef]
- Wallace, S.J.; Li, J.; Nation, R.L.; Prankerd, R.J.; Boyd, B.J. Interaction of colistin and colistin methanesulfonate with liposomes: Colloidal aspects and implications for formulation. J. Pharm. Sci. 2012, 101, 3347–3359. [Google Scholar] [CrossRef]
- Yu, S.; Wang, S.; Zou, P.; Chai, G.; Lin, Y.W.; Velkov, T.; Li, J.; Pan, W.; Zhou, Q.T. Inhalable liposomal powder formulations for co-delivery of synergistic ciprofloxacin and colistin against multi-drug resistant gram-negative lung infections. Int. J. Pharm. 2020, 575, 118915. [Google Scholar] [CrossRef]
- Jadon, P.S.; Gajbhiye, V.; Jadon, R.S.; Gajbhiye, K.R.; Ganesh, N. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS Pharm. Sci. Tech. 2009, 10, 1186–1192. [Google Scholar] [CrossRef]
- Chauhan, M.K.; Bhatt, N. Bioavailability enhancement of polymyxin b with novel drug delivery: Development and optimization using quality-by-design approach. J. Pharm. Sci. 2019, 108, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Laing, B. Bioconjugates for targeted delivery of therapeutic oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Turos, E.; Shim, J.Y.; Wang, Y.; Greenhalgh, K.; Reddy, G.S.; Dickey, S.; Lim, D.V. Antibiotic-conjugated polyacrylate nanoparticles: New opportunities for development of anti-MRSA agents. Bioorg. Med. Chem. Lett. 2007, 17, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Page, M.G. Siderophore conjugates. Ann N. Y. Acad. Sci. 2013, 1277, 115–126. [Google Scholar] [CrossRef]
- Berezin, A.S.; Lomkova, E.A.; Skorik, Y.A. Chitosan conjugates with biologically active compounds: Design strategies, properties, and targeted drug delivery. Rus. Chem. Bull. 2012, 61, 781–795. [Google Scholar] [CrossRef]
- Berezin, A.S.; Skorik, Y.A. Chitosan-isoniazid conjugates: Synthesis, evaluation of tuberculostatic activity, biodegradability and toxicity. Carbohydr. Polym. 2015, 127, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Dizdarevic, A.; Efiana, N.A.; Phan, T.N.Q.; Matuszczak, B.; Bernkop-Schnurch, A. Imine bond formation: A novel concept to incorporate peptide drugs in self-emulsifying drug delivery systems (SEDDS). Eur. J. Pharm. Biopharm. 2019, 142, 92–100. [Google Scholar] [CrossRef]
- Varache, M.; Powell, L.C.; Aarstad, O.A.; Williams, T.L.; Wenzel, M.N.; Thomas, D.W.; Ferguson, E.L. Polymer masked-unmasked protein therapy: Identification of the active species after amylase activation of dextrin-colistin conjugates. Mol. Pharm. 2019, 16, 3199–3207. [Google Scholar] [CrossRef]
- Roberts, J.L.; Cattoz, B.; Schweins, R.; Beck, K.; Thomas, D.W.; Griffiths, P.C.; Ferguson, E.L. In vitro evaluation of the interaction of dextrin-colistin conjugates with bacterial lipopolysaccharide. J. Med. Chem. 2016, 59, 647–654. [Google Scholar] [CrossRef]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. Development and validation of an in vitro pharmacokinetic/pharmacodynamic model to test the antibacterial efficacy of antibiotic polymer conjugates. Antimicrob. Agents Chemother 2015, 59, 1837–1843. [Google Scholar] [CrossRef]
- Ferguson, E.L.; Azzopardi, E.; Roberts, J.L.; Walsh, T.R.; Thomas, D.W. Dextrin-colistin conjugates as a model bioresponsive treatment for multidrug resistant bacterial infections. Mol. Pharm. 2014, 11, 4437–4447. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Schneider, E.K.; Nikolaou, V.; Klein, T.; Li, J.; Davis, T.P.; Whittaker, M.R.; Wilson, P.; Kempe, K.; Velkov, T.; et al. Hydrolyzable poly[poly(ethylene glycol) methyl ether acrylate]-colistin prodrugs through copper-mediated photoinduced living radical polymerization. Bioconjug. Chem. 2017, 28, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.E.; Bell, C.S.; Mejias, R.; McClain, M.S.; Cover, T.L.; Giorgio, T.D. Colistin-functionalized nanoparticles for the rapid capture of Acinetobacter baumannii. J. Biomed. Nanotechnol. 2016, 12, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control Release 2007, 119, 5–24. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Petrova, V.A.; Elokhovskiy, V.Y.; Raik, S.V.; Poshina, D.N.; Romanov, D.P.; Skorik, Y.A. Alginate gel reinforcement with chitin nanowhiskers modulates rheological properties and drug release profile. Biomolecules 2019, 9, 291. [Google Scholar] [CrossRef]
- Malmsten, M.; Bysell, H.; Hansson, P. Biomacromolecules in microgels—Opportunities and challenges for drug delivery. Curr. Opin. Colloid Interface Sci. 2010, 15, 435–444. [Google Scholar] [CrossRef]
- Obuobi, S.; Voo, Z.X.; Low, M.W.; Czarny, B.; Selvarajan, V.; Ibrahim, N.L.; Yang, Y.Y.; Ee, P.L.R. Phenylboronic acid functionalized polycarbonate hydrogels for controlled release of polymyxin B in pseudomonas aeruginosa infected burn wounds. Adv. Healthc. Mater. 2018, 7, e1701388. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Sun, T.; Tsou, Y.H.; Chen, H.; Xu, X. Dual-functional dextran-peg hydrogel as an antimicrobial biomedical material. Macromol. Biosci. 2018, 18, 1700325. [Google Scholar] [CrossRef]
- Bayat, F.; Karimi, A.R. Design of photodynamic chitosan hydrogels bearing phthalocyanine-colistin conjugate as an antibacterial agent. Int. J. Biol. Macromol. 2019, 129, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Borro, B.C.; Bohr, A.; Bucciarelli, S.; Boetker, J.P.; Foged, C.; Rantanen, J.; Malmsten, M. Microfluidics-based self-assembly of peptide-loaded microgels: Effect of three dimensional (3D) printed micromixer design. J. Colloid Interface Sci. 2019, 538, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Wendorff, J.H.; Greiner, A. Use of electrospinning technique for biomedical applications. Polymer 2008, 49, 5603–5621. [Google Scholar] [CrossRef]
- Kim, K.; Luu, Y.K.; Chang, C.; Fang, D.; Hsiao, B.S.; Chu, B.; Hadjiargyrou, M. Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based electrospun nanofibrous scaffolds. J. Control Release 2004, 98, 47–56. [Google Scholar] [CrossRef]
- Liang, D.; Hsiao, B.S.; Chu, B. Functional electrospun nanofibrous scaffolds for biomedical applications. Adv. Drug Deliv. Rev. 2007, 59, 1392–1412. [Google Scholar] [CrossRef]
- Garg, T.; Singh, O.; Arora, S.; Murthy, R. Scaffold: A novel carrier for cell and drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 1–63. [Google Scholar] [CrossRef]
- Zienkiewicz-Strzalka, M.; Derylo-Marczewska, A.; Skorik, Y.A.; Petrova, V.A.; Choma, A.; Komaniecka, I. Silver nanoparticles on chitosan/silica nanofibers: Characterization and antibacterial activity. Int. J. Mol. Sci. 2020, 21, 166. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, R.; Xu, J.; Lan, Y.; Jiao, Y.; Zhou, C.; Zhao, Y. Poly(l-lactide)/halloysite nanotube electrospun mats as dual-drug delivery systems and their therapeutic efficacy in infected full-thickness burns. J. Biomater. Appl. 2015, 30, 512–525. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, Z.; Zhao, D.; Wang, F.; Zhang, K.; Li, Y.; Wen, Y.; Wang, C. Polymyxin b immobilized nanofiber sponge for endotoxin adsorption. Eur. Polym. J. 2019, 110, 69–75. [Google Scholar] [CrossRef]
- Shi, R.; Niu, Y.; Gong, M.; Ye, J.; Tian, W.; Zhang, L. Antimicrobial gelatin-based elastomer nanocomposite membrane loaded with ciprofloxacin and polymyxin b sulfate in halloysite nanotubes for wound dressing. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 87, 128–138. [Google Scholar] [CrossRef]
- Ita, K. Dissolving microneedles for transdermal drug delivery: Advances and challenges. Biomed. Pharmacother. 2017, 93, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Hughes, H.; O’Reilly, N.J.; Allender, C.J.; Barrow, D.A.; McLoughlin, P. Dissolving microneedle based transdermal delivery of therapeutic peptide analogues. Int. J. Pharm. 2019, 565, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Hughes, H.; O’Reilly, N.J.; McLoughlin, P. Formulation and characterisation of dissolving microneedles for the transdermal delivery of therapeutic peptides. Int. J. Pharm. 2017, 526, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Pamornpathomkul, B.; Ngawhirunpat, T.; Tekko, I.A.; Vora, L.; McCarthy, H.O.; Donnelly, R.F. Dissolving polymeric microneedle arrays for enhanced site-specific acyclovir delivery. Eur. J. Pharm. Sci. 2018, 121, 200–209. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).