New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding


Abstract
1. Introduction
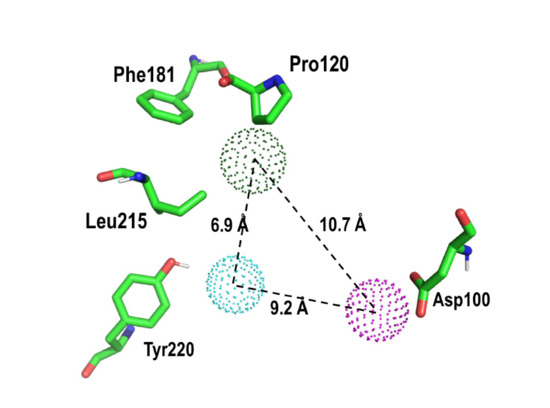
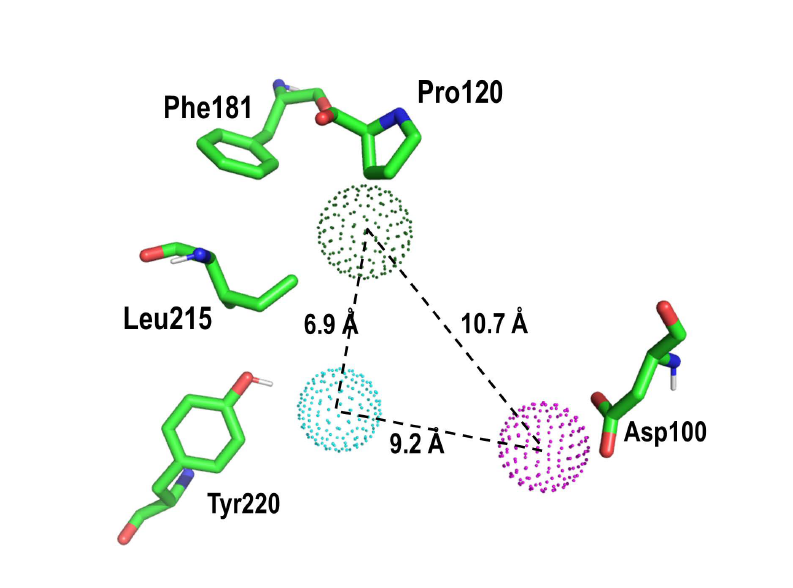
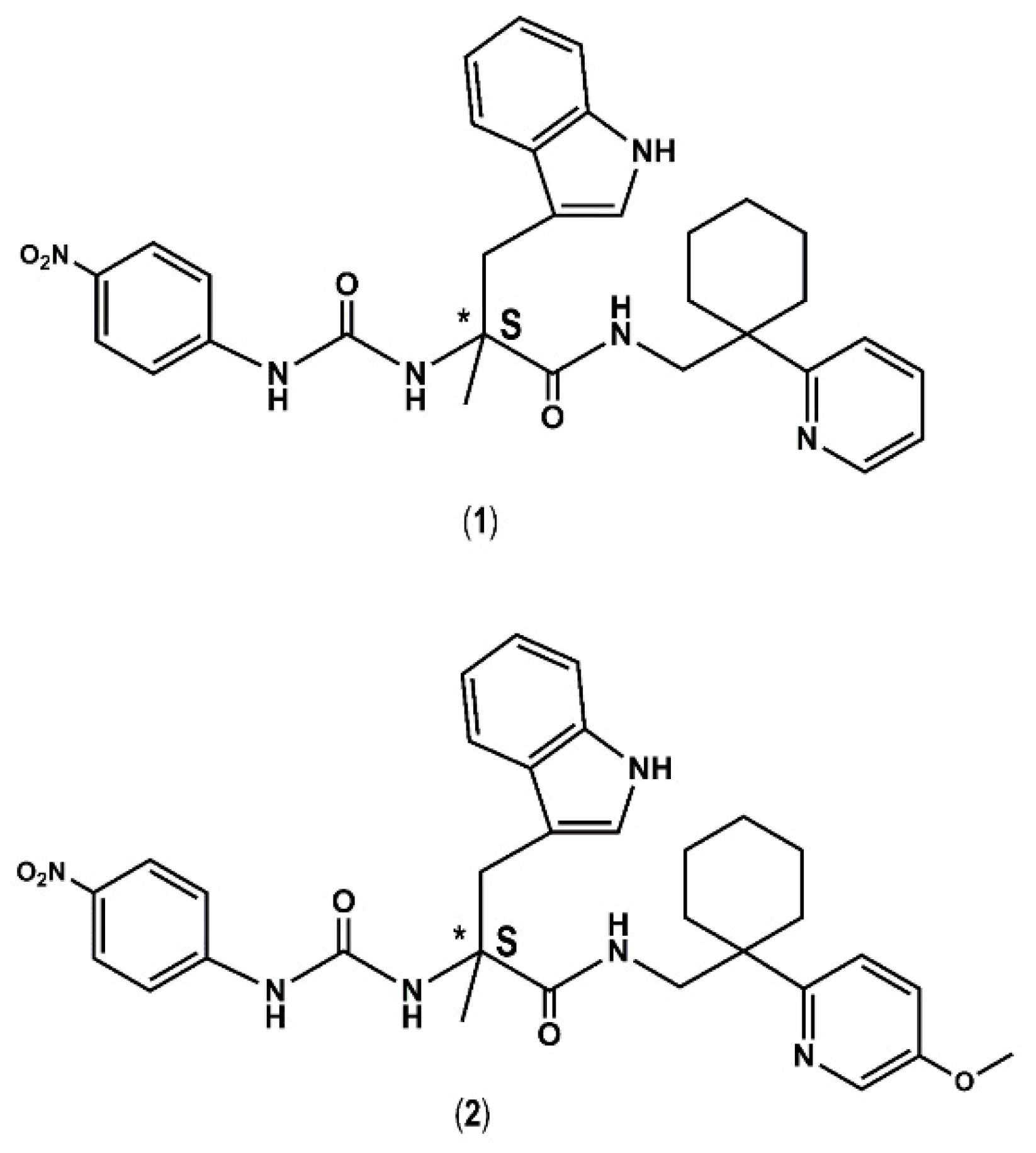
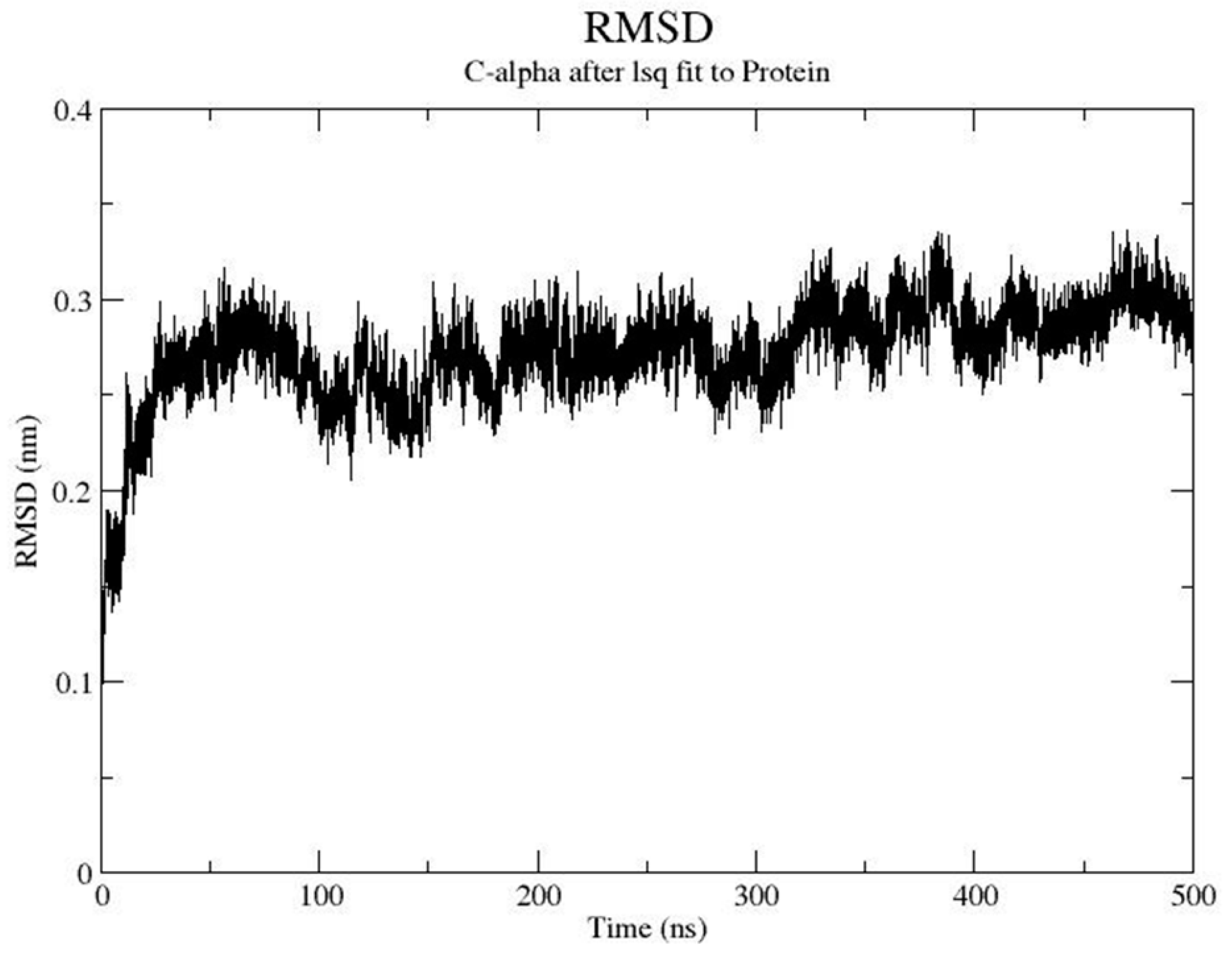
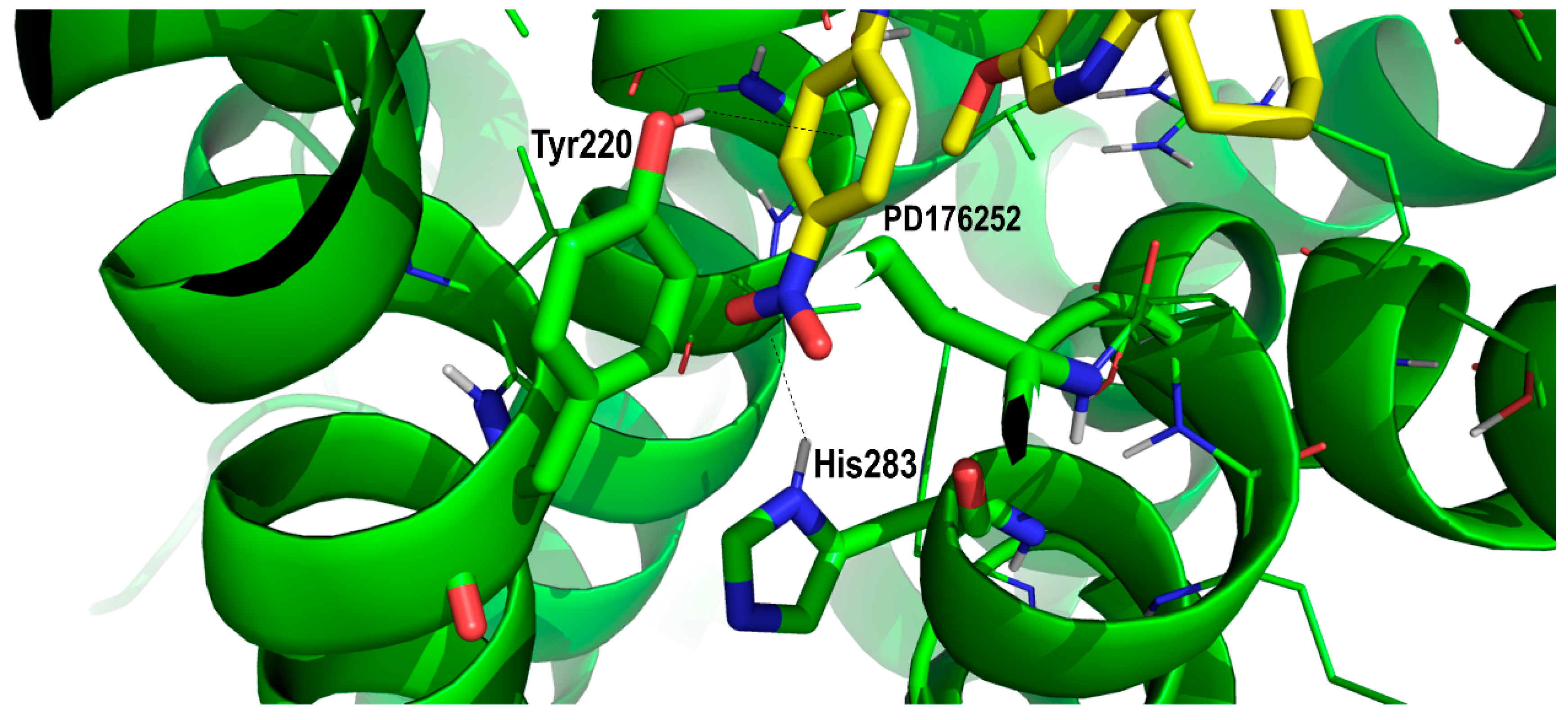
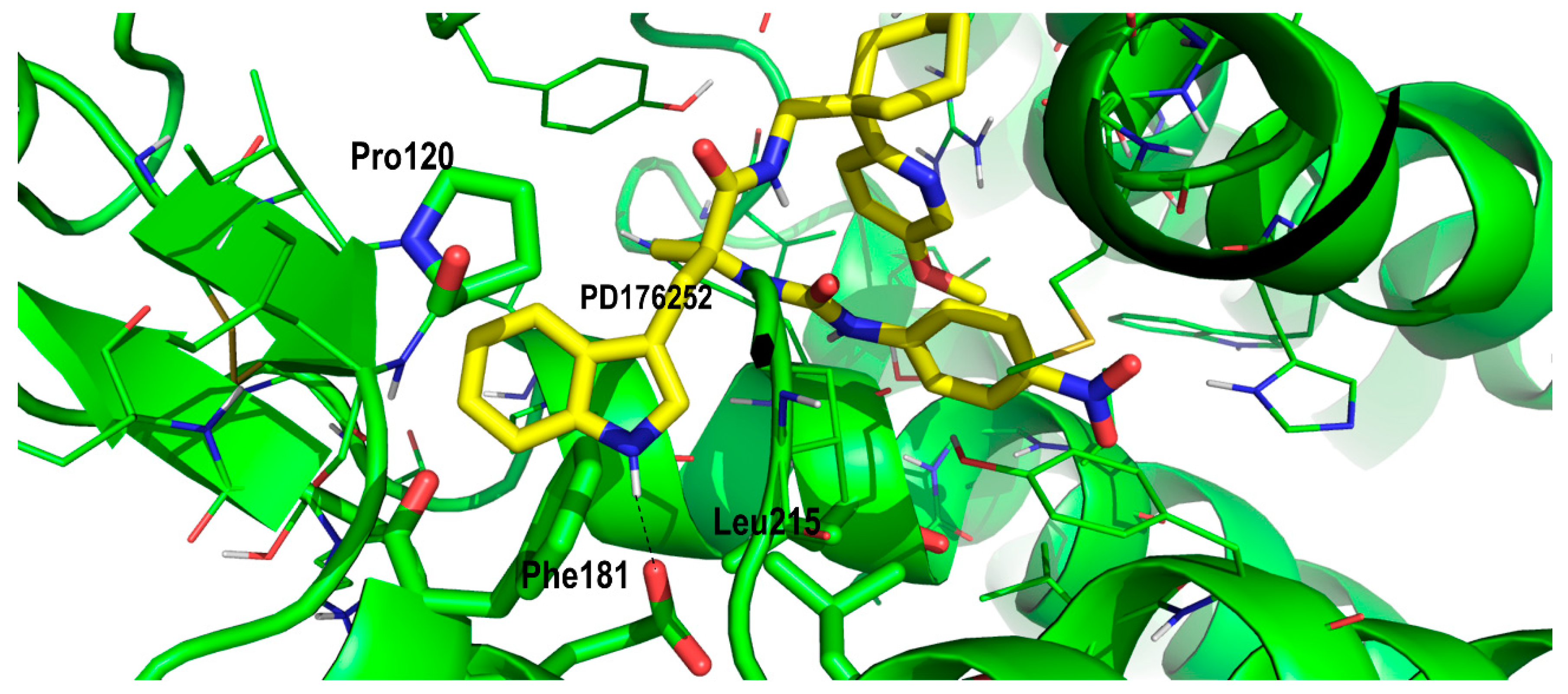
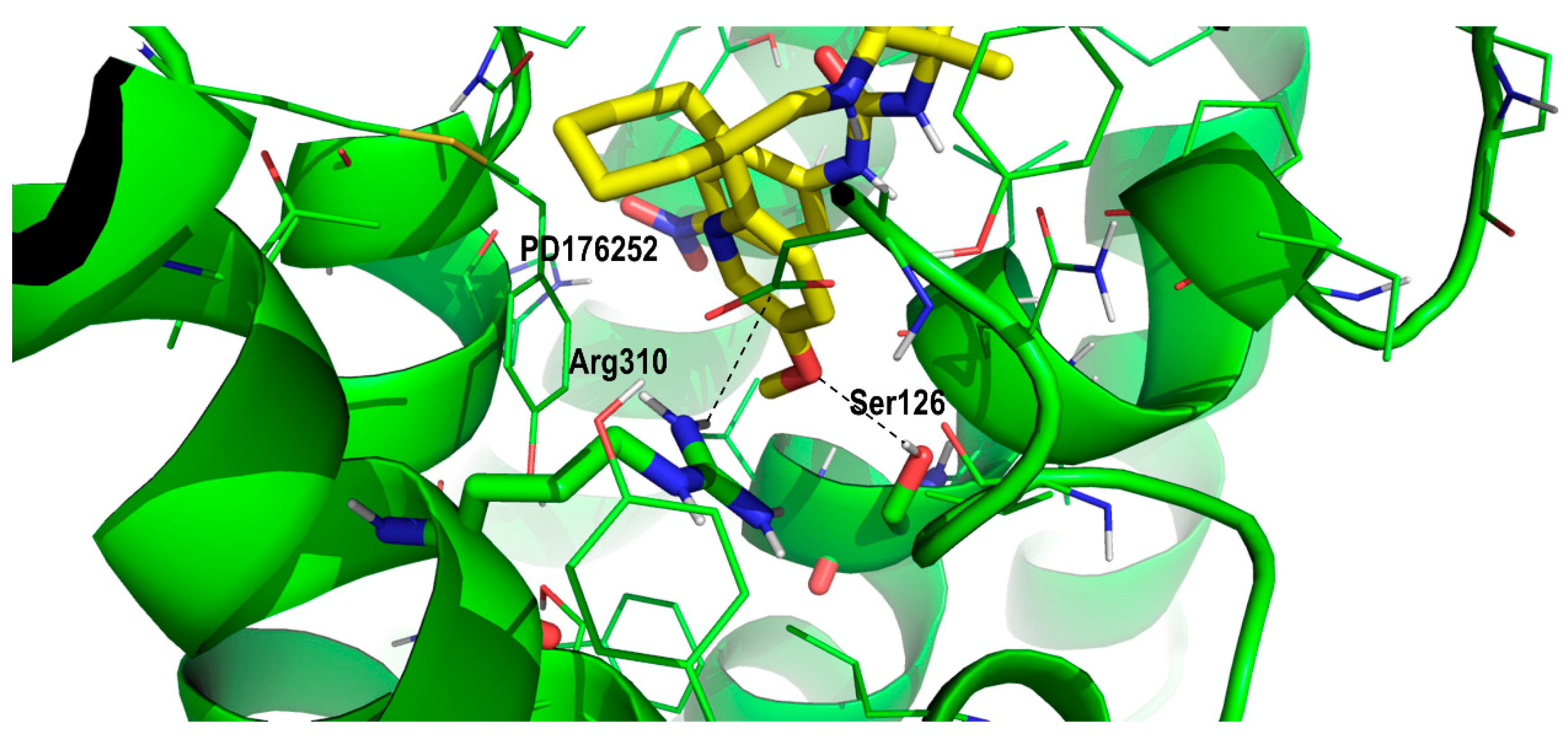
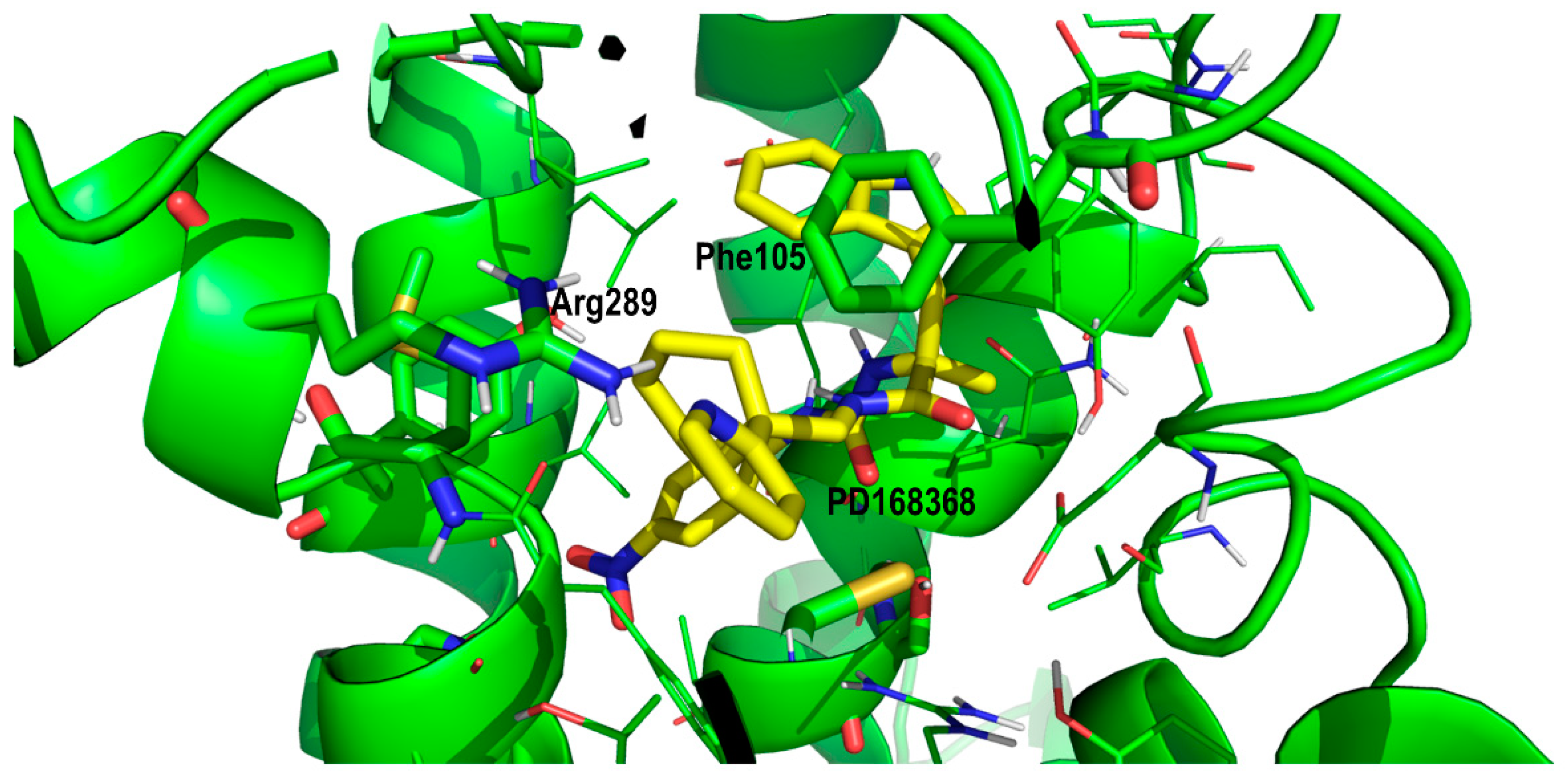
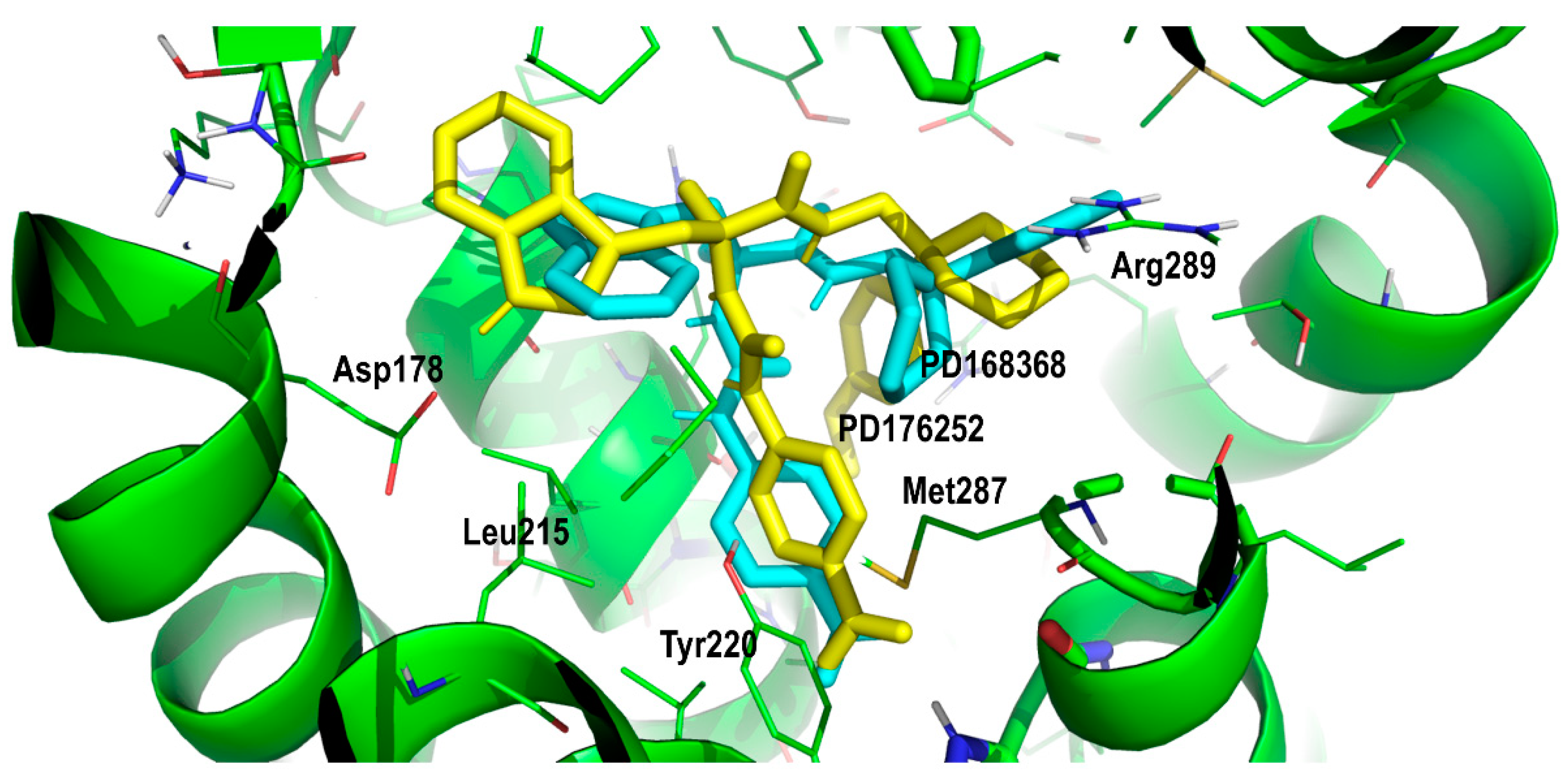
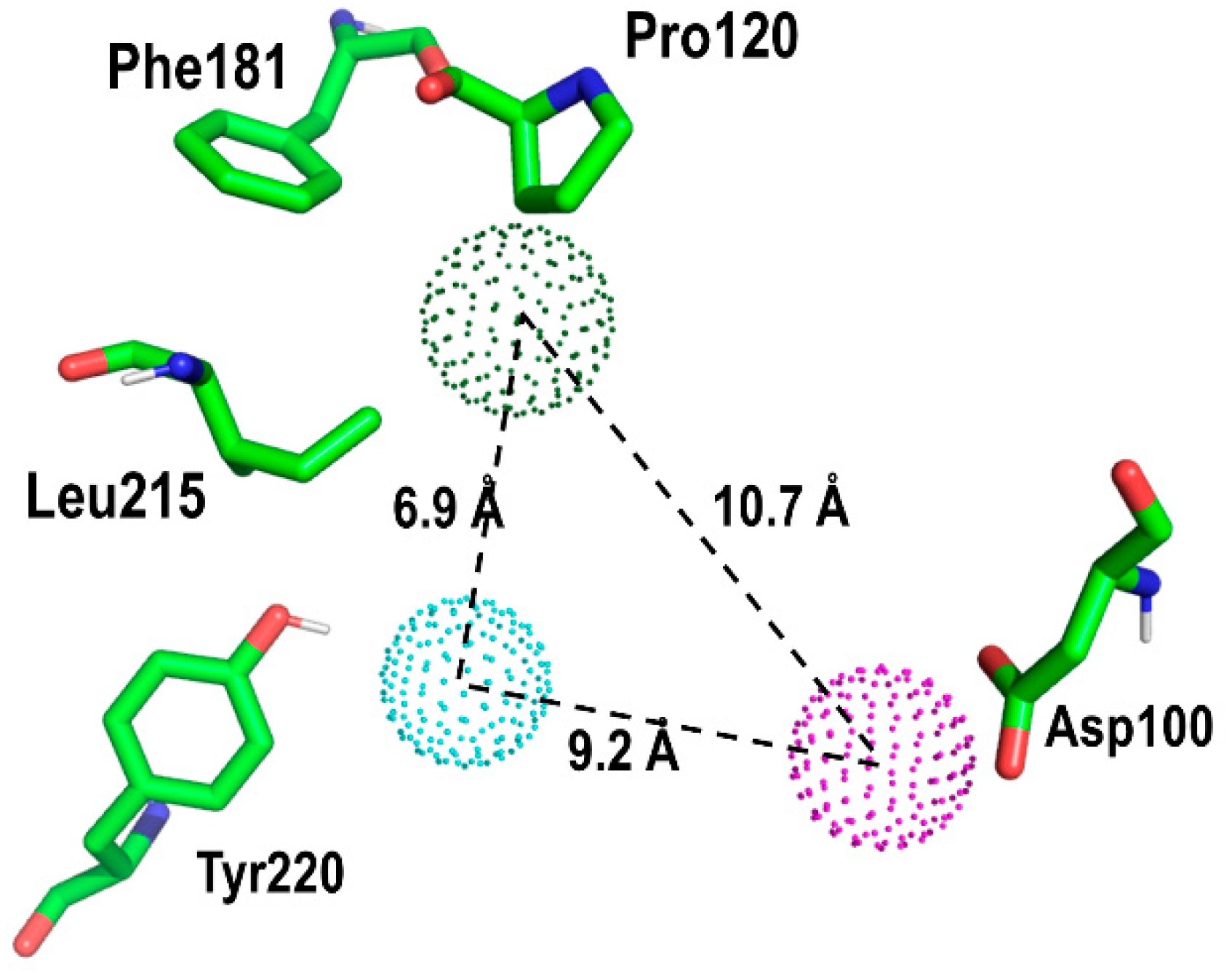
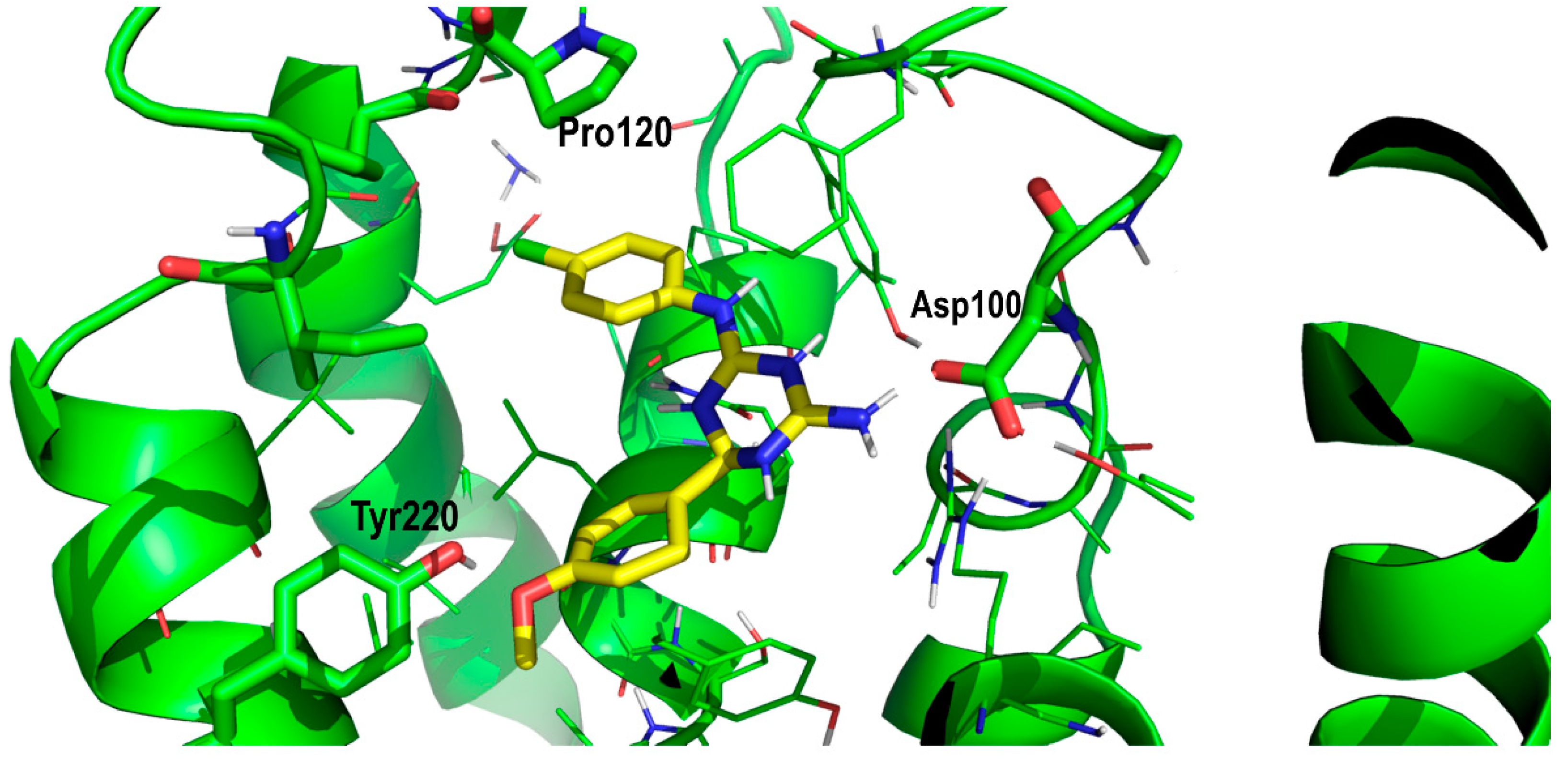
2. Results and Discussion
Proof of Concept
3. Materials and Methods
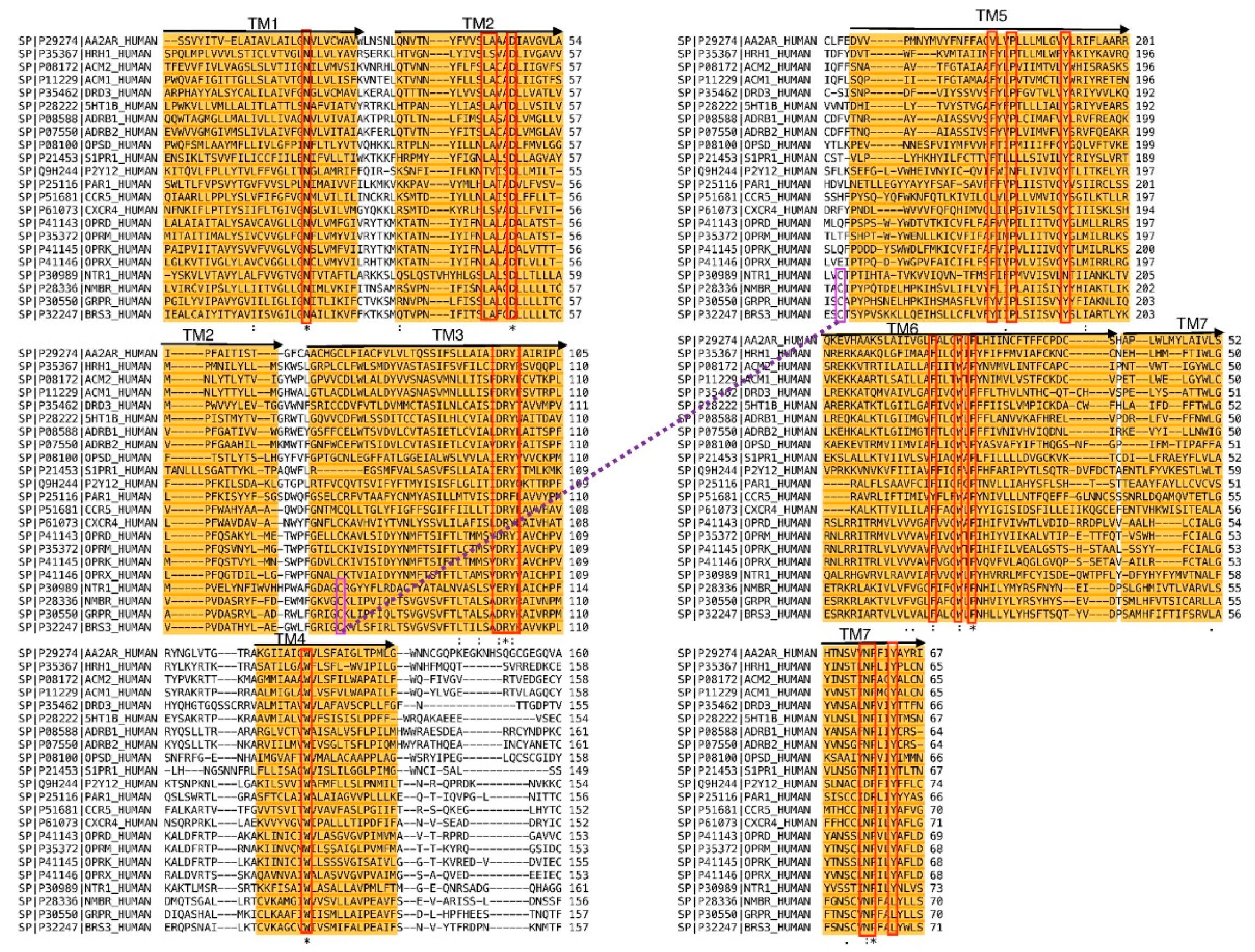
3.1. Molecular Modeling
3.2. Binding Assays
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Jensen, R.T.; Moody, T.W. Bombesin Peptides (Cancer). In Hand-Book of Biologically Active Peptides; Kastin, A.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 506–511. [Google Scholar]
- Ramos-Alvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into bombesin receptors and ligands: Highlighting recent advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef]
- Weber, H.C. Regulation and signaling of human bombesin receptors and their biological effects. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Flores, D.G.; De Farias, C.B.; Leites, J.; De Oliveira, M.S.; Lima, R.C.; Tamajusuku, A.S.; Leone, L.P.D.; Meurer, L.; Brunetto, A.L.; Schwartsmann, G.; et al. Gastrin releasing peptide receptors regulate proliferation of C6 glioma cells through a phosphatidylinositol 3-kinase-dependent mechanism. Curr. Neurovasc. Res. 2008, 5, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Moreno, P.; Jensen, R.T. Neuropeptides as lung cancer growth factors. Peptides 2015, 72, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Benya, R.V.; Kusui, T.; Pradhan, T.K.; Battey, J.F.; Jensen, R.T. Expression and characterization of cloned human bombesin receptors. Mol. Pharmacol. 1995, 47, 10–20. [Google Scholar] [PubMed]
- Fathi, Z.; Corjay, M.H.; Shapira, H.; Wada, E.; Benya, R.; Jensen, R.; Viallet, J.; Sausville, E.A.; Battey, J.F. BRS-3: A novel bombesin receptor subtype selectively expressed in testis and lungcarcinoma cells. J. Biol. Chem. 1993, 268, 5979–8594. [Google Scholar] [PubMed]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Beny, R.V. International Union of Pharmacology. LXVIII. Mammalian Bombesin Receptors: Nomenclature, Distribution, Pharmacology, Signaling, and Functions in Normal and Disease States. Pharm. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging; targeting and treatment. Expert Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, S.R.; Kim, M.K.; Choi, K.S.; Jang, H.O.; Yun, I.; Bae, M.-K. Neuromedin B receptor antagonist suppresses tumor angiogenesis and tumor growth in vitro and in vivo. Cancer Lett. 2011, 312, 117–127. [Google Scholar] [CrossRef]
- Gonzalez, N.; Moreno, P.; Jensen, R.T. Bombesin receptor subtype 3 as a potential target for obesity and diabetes. Expert Opin. Ther. Targets 2015, 19, 1153–1170. [Google Scholar] [CrossRef]
- Zhou, S.; Nissao, E.; Jackson, I.L.; Leong, W.; Dancy, L.; Cuttitta, F.; Vujaskovic, Z.; Sunday, M.E. Radiation-Induced Lung Injury Is Mitigated by Blockade of Gastrin-Releasing Peptide. Am. J. Pathol. 2013, 182, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Ehling, S.; Fukuyama, T.; Ko, M.C.; Olivry, T.; Bäumer, W. Neuromedin B Induces Acute Itch in Mice via the Activation of Peripheral Sensory Neurons. Acta Derm. Venereol. 2019, 99, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Cristau, M.; Devin, C.; Oiry, C.; Chaloin, O.; Amblard, M.; Bernad, N.; Heitz, A.; Fehrentz, J.A.; Martinez, J. Synthesis and Biological Evaluation of Bombesin Constrained Analogues. J. Med. Chem. 2000, 43, 2356–2361. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, N.; Mantey, S.A.; Pradhan, T.K.; Sancho, V.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Characterization of putative GRP- and NMB-receptor antagonist’s interaction with human receptors. Peptides 2009, 30, 1473–1486. [Google Scholar] [CrossRef]
- Perez, J.J.; Corcho, F.; Llorens, O. Molecular modeling in the design of peptidomimetics and peptide surrogates. Curr. Med. Chem. 2002, 9, 2209–2229. [Google Scholar] [CrossRef]
- Perez, J.J. Designing Peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Ashwood, V.; Brownhill, V.; Higginbottom, M.; Horwell, D.C.; Hughes, J.; Lewthwaite, R.A.; McKnight, A.T.; Pinnock, R.D.; Pritchard, M.C.; Suman-Chauhan, N.; et al. PD 176252-The First High Affinity Non-peptide Gastrin-Releasing Peptide (BB2) Receptor Antagonist. Bioorg. Med. Chem. Lett. 1998, 8, 2589–2594. [Google Scholar] [CrossRef]
- Carrieri, A.; Lacivita, E.; Belviso, B.D.; Caliandro, R.; Mastrorilli, P.; Gallo, V.; Niso, M.; Leopoldo, M. Structural determinants in the binding of BB2 receptor ligands: In silico, x-ray and NMR studies in PD176252 analogues. Curr. Top. Med. Chem. 2017, 17, 1599–1610. [Google Scholar] [CrossRef]
- Moody, T.W.; Mantey, S.A.; Moreno, P.; Nakamura, T.; Lacivita, E.; Leopoldo, M.; Jensen, R.T. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides 2015, 64, 55–61. [Google Scholar] [CrossRef]
- Moody, T.W.; Tashakkori, N.; Mantey, S.A.; Moreno, P.; Ramos-Alvarez, I.; Leopoldo, M.; Jensen, R.T. AM-37 and ST-36 are Small Molecule Bombesin Receptor Antagonists. Front. Endocrinol. 2017, 8, 176. [Google Scholar] [CrossRef]
- Tokita, K.; Hocart, S.J.; Katsuno, T.; Mantey, S.A.; Coy, D.H.; Jensen, R.T. Tyrosine 220 in the 5th Transmembrane Domain of the Neuromedin B Receptor Is Critical for the High Selectivity of the Peptoid Antagonist PD168368. J. Biol. Chem. 2001, 276, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Jutila, M.A.; Quinn, M.T. Gastrin-Releasing Peptide/Neuromedin B Receptor Antagonists PD176252, PD168368, and Related Analogs Are Potent Agonists of Human Formyl-Peptide Receptors. Mol. Pharmacol. 2011, 79, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Shuttleworth, S.J.; Connors, R.V.; Chai, A.; Coward, P. Discovery and optimization of a novel Neuromedin B receptor antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 4264–4267. [Google Scholar] [CrossRef]
- Martinez, A.; Zudaire, E.; Julian, M.; Moody, T.W.; Cuttitta, F. Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene 2005, 24, 4106–4113. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.; Mantey, S.A.; Nuche-Berenguer, B.; Reitman, M.L.; Gonzalez, N.; Coy, D.H.; Jensen, R.T. Comparative pharmacology of bombesin receptor subtype-3 nonpeptide agonist MK-5046, a universal peptide agonist; and peptide antagonist Bantag-1 for human bombesin receptors. J. Pharmacol. Exp. Ther. 2013, 347, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Lupala, C.S.; Rasaifar, B.; Gomez-Gutierrez, P.; Perez, J.J. Using Molecular Dynamics for the refinement of atomistic models of GPCRs by homology modeling. J. Biomol. Struct. Dyn. 2018, 36, 2436–2448. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Venkatakrishnana, A.J.; Maa, A.K.; Fonseca, R.; Latorraca, N.R.; Kelly, B.; Betz, R.M.; Asawa, C.; Kobilka, B.K.; Dror, R.O. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc. Natl. Acad. Sci. USA 2019, 116, 3288–3293. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar]
- Molecular Operating Environment (MOE), Version 2019.01; Chemical Computing Group UCL: Montreal, QC, Canada, 2020.
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J. Managing molecular diversity. Chem. Soc. Rev. 2005, 34, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Chetan, S.; Sukuru, K.; Glick, M.; Davies, J.W. How Similar Are Similarity Searching Methods? A Principal Component Analysis of Molecular Descriptor Space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Lipkus, A.H. A proof of the triangular inequality for the Tanimoto distance. J. Math. Chem. 1999, 26, 263–265. [Google Scholar] [CrossRef]
- Jarvis, R.A.; Patrick, E.A. Clustering Using a Similarity Measure Based on Shared Near Neighbors. IEEE Trans. Comp. 1973, 22, 1025–1034. [Google Scholar] [CrossRef]
- Ohki-Hamazaki, H.; Iwabuchi, M.; Maekawa, F. Development and function of bombesin-like peptides and their receptors. Int. J. Dev. Biol. 2005, 49, 293–300. [Google Scholar] [CrossRef]
- Krishna, S.; Singh, D.K.; Meena, S.; Datta, D.; Siddiqi, M.I.; Banerjee, D. Pharmacophore-Based Screening and Identification of Novel Human Ligase I Inhibitors with Potential Anticancer Activity. J. Chem. Inf. Model. 2014, 54, 781–792. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, P.; Campos, P.M.; Perez, J.J. Identification of a Novel Inhibitory Allosteric Site of MAP Kinases. PLoS ONE 2016, 11, e0167379. [Google Scholar] [CrossRef]
- Noinaj, N.; White, J.F.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. The crystal structure of the neurotensin receptor NTS1 in complex with neurotensin (8–13). Nature 2012, 490, 508–513. [Google Scholar]
- Costanzi, S.; Skorski, M.; Deplano, A.; Habermehl, B.; Mendoza, M.; Wang, K.; Biederman, M.; Dawson, J.; Gao, J. Homology modeling of a Class A GPCR in the inactive conformation: A quantitative analysis of the correlation between model/template sequence identity and model accuracy. J. Mol. Graph. Model. 2016, 70, 140–152. [Google Scholar] [CrossRef]
- Nayeem, A.; Sitkoff, D.; Krystek, S., Jr. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006, 15, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Palomba, D. Expanding the horizons of G protein-coupled receptor structure-based ligand discovery and optimization using homology models. Chem. Commun. 2015, 51, 13576–13594. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Cordomi, A.; Edholm, O.; Perez, J.J. Effect of different treatments of long-range interactions and sampling conditions in molecular dynamic simulations of rhodopsin embedded in a dipalmitoyl phosphatidylcholine bilayer. J. Comput. Chem. 2007, 28, 1017–1030. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast flexible and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kaminski, G.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound# | Chemical Structure | BB1R(Neuromendin B Receptor) Radioligand Displacement (%) | BB2R(Gastrin-Releasing Peptide Receptor) Radioligand Displacement (%) |
|---|---|---|---|
| 1 |  | 19.7 | 0.0 |
| 2 |  | 24.3 | 0.0 |
| 3 |  | 28.1 | 0.0 |
| 4 |  | 30.5 | 0.0 |
| 5 |  | 38.0 | 10.5 |
| 6 |  | 16.1 | 0.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals 2020, 13, 197. https://doi.org/10.3390/ph13080197
Rasaeifar B, Gomez-Gutierrez P, Perez JJ. New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals. 2020; 13(8):197. https://doi.org/10.3390/ph13080197
Chicago/Turabian StyleRasaeifar, Bahareh, Patricia Gomez-Gutierrez, and Juan J. Perez. 2020. "New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding" Pharmaceuticals 13, no. 8: 197. https://doi.org/10.3390/ph13080197
APA StyleRasaeifar, B., Gomez-Gutierrez, P., & Perez, J. J. (2020). New Insights into the Stereochemical Requirements of the Bombesin BB1 Receptor Antagonists Binding. Pharmaceuticals, 13(8), 197. https://doi.org/10.3390/ph13080197