Juvenile Hemochromatosis: A Case Report and Review of the Literature

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
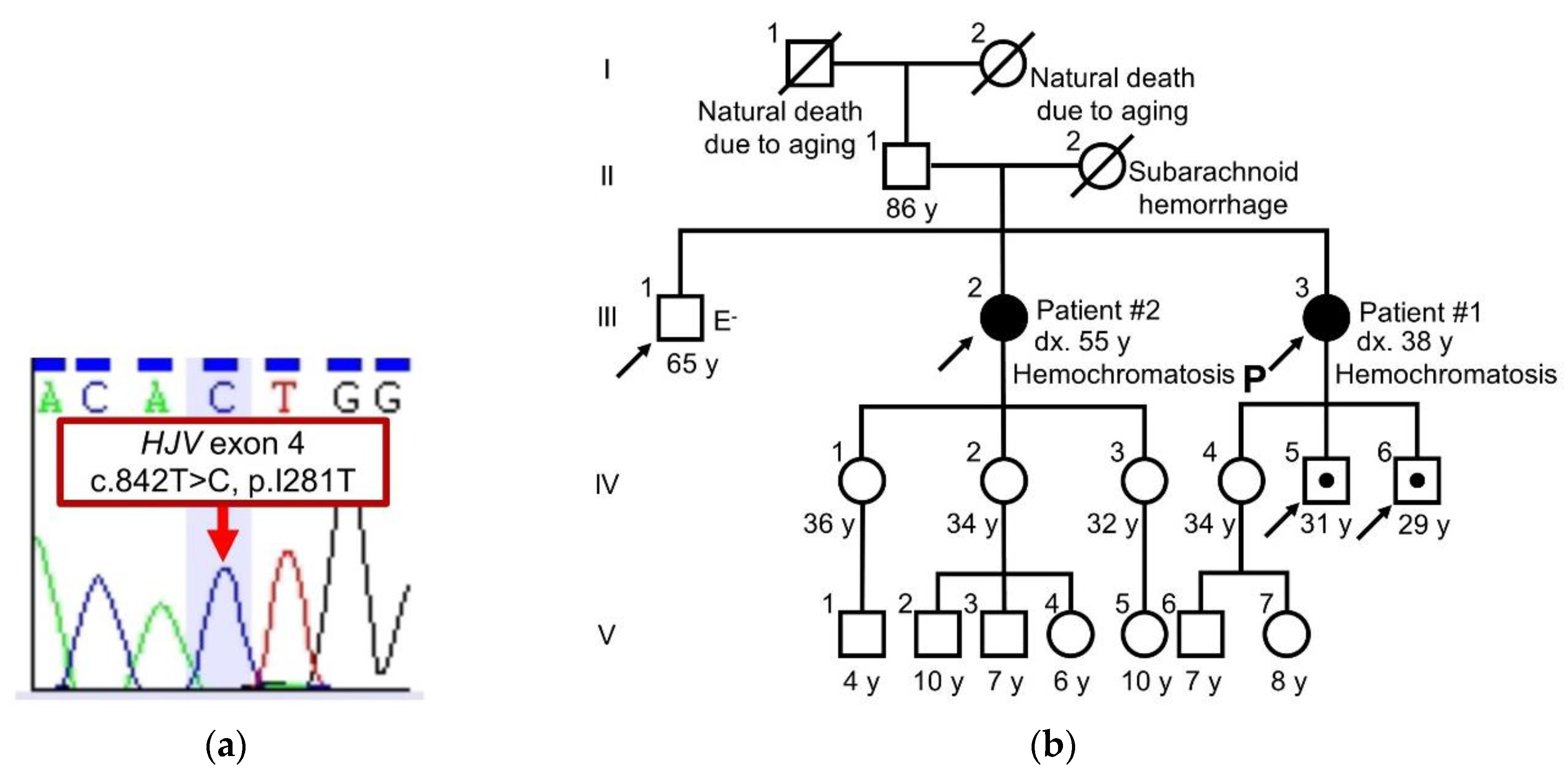
2.1. Case Report
2.2. Sequence Analysis of HJV and Family Tree
2.3. All Reported Cases of Juvenile Hemochromatosis
2.4. Characteristics of Juvenile Hemochromatosis with Homozygous Identical Mutations in HJV
2.5. Limitations
2.6. Strengths of the Present Study
3. Materials and Methods
3.1. Genetic Analysis for Hemochromatosis
3.2. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| JH | juvenile hemochromatosis |
| HJV | hemojuvelin |
| NMR | nuclear magnetic resonance; |
| Ref | reference |
| Dx | diagnosis |
| TSAT | transferrin saturation |
| F | female |
| M | male |
| + | present |
| - | absent |
| NA | not available |
| BDL | below detection limit |
| P | proband |
| 5’UTR | 5’prime untranslated region |
References
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loreal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Pantopoulos, K. Hepcidin Therapeutics. Pharmaceuticals (Basel) 2018, 11, 127. [Google Scholar] [CrossRef]
- Porto, G.; Cruz, E.; Teles, M.J.; de Sousa, M. HFE Related Hemochromatosis: Uncovering the Inextricable Link between Iron Homeostasis and the Immunological System. Pharmaceuticals (Basel) 2019, 12, 122. [Google Scholar] [CrossRef]
- Loreal, O.; Cavey, T.; Robin, F.; Kenawi, M.; Guggenbuhl, P.; Brissot, P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals (Basel) 2018, 11, 131. [Google Scholar] [CrossRef]
- Nowak, A.; Giger, R.S.; Krayenbuehl, P.A. Higher age at diagnosis of hemochromatosis is the strongest predictor of the occurrence of hepatocellular carcinoma in the Swiss hemochromatosis cohort: A prospective longitudinal observational study. Medicine (Baltimore) 2018, 97, e12886. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Biswas, K.; Rottman, J.; Allen, J.R.; Long, J.; Miranda, L.P.; Winters, A.; Arvedson, T.L. Identification of Antibody and Small Molecule Antagonists of Ferroportin-Hepcidin Interaction. Front. Pharmacol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Schmidt, P.J.; Meynard, D.; Garuti, C.; Montosi, G.; Chen, S.; Vukicevic, S.; Pietrangelo, A.; Lin, H.Y.; Babitt, J.L. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010, 139, 1721–1729. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.; Franchini, P.L.; Dube, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 2004, 36, 77–82. [Google Scholar] [CrossRef]
- Huang, F.W.; Rubio-Aliaga, I.; Kushner, J.P.; Andrews, N.C.; Fleming, M.D. Identification of a novel mutation (C321X) in HJV. Blood 2004, 104, 2176–2177. [Google Scholar] [CrossRef]
- Lv, T.; Zhang, W.; Xu, A.; Li, Y.; Zhou, D.; Zhang, B.; Li, X.; Zhao, X.; Wang, Y.; Wang, X.; et al. Non-HFE mutations in haemochromatosis in China: Combination of heterozygous mutations involving HJV signal peptide variants. J. Med. Genet. 2018, 55, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Yang, Q.; Jian, J.; Katz, S.; Abramson, S.B.; Huang, X. 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 2012, 153, 3170–3178. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakakis, C.; Bondy, C. Androgens and the breast. Breast Cancer Res 2009, 11, 212. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Kang, Y.; Li, C.X.; Zhang, Q. A rare case of juvenile hemochromatosis with mixed causes of hyperferritinemia. Kaohsiung J. Med. Sci. 2020, 36, 289–290. [Google Scholar] [CrossRef]
- Hamdi-Roze, H.; Ben Ali, Z.; Ropert, M.; Detivaud, L.; Aggoune, S.; Simon, D.; Pelletier, G.; Deugnier, Y.; David, V.; Bardou-Jacquet, E. Variable expressivity of HJV related hemochromatosis: "Juvenile" hemochromatosis? Blood Cells Mol. Dis. 2019, 74, 30–33. [Google Scholar] [CrossRef]
- Murugan, R.C.; Lee, P.L.; Kalavar, M.R.; Barton, J.C. Early age-of-onset iron overload and homozygosity for the novel hemojuvelin mutation HJV R54X (exon 3; c.160A-->T) in an African American male of West Indies descent. Clin. Genet. 2008, 74, 88–92. [Google Scholar] [CrossRef]
- Janosi, A.; Andrikovics, H.; Vas, K.; Bors, A.; Hubay, M.; Sapi, Z.; Tordai, A. Homozygosity for a novel nonsense mutation (G66X) of the HJV gene causes severe juvenile hemochromatosis with fatal cardiomyopathy. Blood 2005, 105, 432. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Ikuta, K.; Tatsumi, Y.; Toki, Y.; Hayashi, H.; Tonan, T.; Ohtake, T.; Hoshino, S.; Naito, M.; Kato, K.; et al. Identification of heterozygous p.Y150C and p.V274M mutations in the HJV gene in a Japanese patient with a mild phenotype of juvenile hemochromatosis: A case report. Hepatol. Res. 2020, 50, 144–150. [Google Scholar] [CrossRef]
- Lee, P.L.; Beutler, E.; Rao, S.V.; Barton, J.C. Genetic abnormalities and juvenile hemochromatosis: Mutations of the HJV gene encoding hemojuvelin. Blood 2004, 103, 4669–4671. [Google Scholar] [CrossRef]
- Lok, C.Y.; Merryweather-Clarke, A.T.; Viprakasit, V.; Chinthammitr, Y.; Srichairatanakool, S.; Limwongse, C.; Oleesky, D.; Robins, A.J.; Hudson, J.; Wai, P.; et al. Iron overload in the Asian community. Blood 2009, 114, 20–25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wallace, D.F.; Dixon, J.L.; Ramm, G.A.; Anderson, G.J.; Powell, L.W.; Subramaniam, N. Hemojuvelin (HJV)-associated hemochromatosis: Analysis of HJV and HFE mutations and iron overload in three families. Haematologica 2005, 90, 254–255. [Google Scholar] [PubMed]
- Lanzara, C.; Roetto, A.; Daraio, F.; Rivard, S.; Ficarella, R.; Simard, H.; Cox, T.M.; Cazzola, M.; Piperno, A.; Gimenez-Roqueplo, A.P.; et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood 2004, 103, 4317–4321. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, M.M.; Radmard, A.R.; Nouroozi, A.; Akbari, M.R.; Amini, M.; Navabakhsh, B.; Caleffi, A.; Pietrangelo, A.; Malekzadeh, R. Juvenile Hemochromatosis, Genetic Study and Long-term Follow up after Therapy. Middle East J. Dig. Dis. 2014, 6, 87–92. [Google Scholar]
- Daraio, F.; Ryan, E.; Gleeson, F.; Roetto, A.; Crowe, J.; Camaschella, C. Juvenile hemochromatosis due to G320V/Q116X compound heterozygosity of hemojuvelin in an Irish patient. Blood Cells Mol. Dis. 2005, 35, 174–176. [Google Scholar] [CrossRef]
- Gehrke, S.G.; Pietrangelo, A.; Kascak, M.; Braner, A.; Eisold, M.; Kulaksiz, H.; Herrmann, T.; Hebling, U.; Bents, K.; Gugler, R.; et al. HJV gene mutations in European patients with juvenile hemochromatosis. Clin. Genet. 2005, 67, 425–428. [Google Scholar] [CrossRef]
- Van Dijk, B.A.; Kemna, E.H.; Tjalsma, H.; Klaver, S.M.; Wiegerinck, E.T.; Goossens, J.P.; Slee, P.H.; Breuning, M.H.; Swinkels, D.W. Effect of the new HJV-L165X mutation on penetrance of HFE. Blood 2007, 109, 5525–5526. [Google Scholar] [CrossRef]
- Ka, C.; Le Gac, G.; Letocart, E.; Gourlaouen, I.; Martin, B.; Ferec, C. Phenotypic and functional data confirm causality of the recently identified hemojuvelin p.r176c missense mutation. Haematologica 2007, 92, 1262–1263. [Google Scholar] [CrossRef]
- Aguilar-Martinez, P.; Lok, C.Y.; Cunat, S.; Cadet, E.; Robson, K.; Rochette, J. Juvenile hemochromatosis caused by a novel combination of hemojuvelin G320V/R176C mutations in a 5-year old girl. Haematologica 2007, 92, 421–422. [Google Scholar] [CrossRef]
- Koyama, C.; Hayashi, H.; Wakusawa, S.; Ueno, T.; Yano, M.; Katano, Y.; Goto, H.; Kidokoro, R. Three patients with middle-age-onset hemochromatosis caused by novel mutations in the hemojuvelin gene. J. Hepatol. 2005, 43, 740–742. [Google Scholar] [CrossRef]
- Lee, P.L.; Barton, J.C.; Brandhagen, D.; Beutler, E. Hemojuvelin (HJV) mutations in persons of European, African-American and Asian ancestry with adult onset haemochromatosis. Br. J. Haematol. 2004, 127, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Koshy, A.; Mukkada, R.J.; Chettupuzha, A.P.; Francis, J.V.; Kandathil, J.C.; Mahadevan, P. Hemochromatosis in India: First Report of Whole Exome Sequencing With Review of the Literature. J. Clin. Exp. Hepatol. 2020, 10, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi, J.; Lueff, S.; Szucs, N.; Pozsonyi, Z.; Toth, A.; Karadi, I.; Pietrangelo, A. Hemochromatosis and hemojuvelin G320V homozygosity in a Hungarian woman. Acta. Haematol. 2010, 123, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Cherfane, C.; Lee, P.; Guerin, L.; Brown, K. A late presentation of a fatal disease: Juvenile hemochromatosis. Case Rep. Med. 2013, 2013, 875093. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, G.; Pelucchi, S.; Mariani, R.; Silvestri, L.; Camaschella, C.; Piperno, A. A severe hemojuvelin mutation leading to late onset of HFE2-hemochromatosis. Dig. Liver. Dis. 2018, 50, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, B.K.; Chopra, G.; Jamwal, M.; Chandak, G.R.; Duseja, A.; Malhotra, P.; Chawla, Y.K.; Garewal, G.; Das, R. Adult onset hereditary hemochromatosis is associated with a novel recurrent Hemojuvelin (HJV) gene mutation in north Indians. Blood Cells Mol. Dis. 2018, 73, 14–21. [Google Scholar] [CrossRef]
- Li, C.X.; Zhang, L.; Wang, P.; Sun, L. Clinicopathological diagnosis and treatment of juvenile hemochromatosis. Chin. Med. J. (Engl.) 2019, 132, 3018–3020. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Promrat, K.; Mallette, C.; Flynn, M.; Beutler, E. A juvenile hemochromatosis patient homozygous for a novel deletion of cDNA nucleotide 81 of hemojuvelin. Acta. Haematol. 2006, 115, 123–127. [Google Scholar] [CrossRef]
- Santiago de Sousa Azulay, R.; Magalhaes, M.; Tavares, M.D.G.; Dualibe, R.; Barbosa, L.; Sa Gaspar, S.; Faria, A.M.; Nascimento, G.C.; Damianse, S.; Rocha, V.C.C.; et al. Novel Mutation in the Hemojuvelin Gene (HJV) in a Patient with Juvenile Hemochromatosis Presenting with Insulin-dependent Diabetes Mellitus, Secondary Hypothyroidism and Hypogonadism. Am. J. Case Rep. 2020, 21, e923108. [Google Scholar] [CrossRef]
- Huang, F.W.; Pinkus, J.L.; Pinkus, G.S.; Fleming, M.D.; Andrews, N.C. A mouse model of juvenile hemochromatosis. J. clin. investig. 2005, 115, 2187–2191. [Google Scholar] [CrossRef]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Investig. 2005, 115, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Fazi, C.; Gerardi, G.; Levi, S.; Arosio, P.; Camaschella, C. Defective targeting of hemojuvelin to plasma membrane is a common pathogenetic mechanism in juvenile hemochromatosis. Blood 2007, 109, 4503–4510. [Google Scholar] [CrossRef]
- Pagani, A.; Silvestri, L.; Nai, A.; Camaschella, C. Hemojuvelin N-terminal mutants reach the plasma membrane but do not activate the hepcidin response. Haematologica 2008, 93, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Brune, M.; Rossander, L.; Hallberg, L. Iron absorption and phenolic compounds: Importance of different phenolic structures. Eur. J. Clin. Nutr. 1989, 43, 547–557. [Google Scholar]
- Ikuta, K.; Hatayama, M.; Addo, L.; Toki, Y.; Sasaki, K.; Tatsumi, Y.; Hattori, A.; Kato, A.; Kato, K.; Hayashi, H.; et al. Iron overload patients with unknown etiology from national survey in Japan. Int. J. Hematol. 2017, 105, 353–360. [Google Scholar] [CrossRef]
- Tomosugi, N.; Kawabata, H.; Wakatabe, R.; Higuchi, M.; Yamaya, H.; Umehara, H.; Ishikawa, I. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood 2006, 108, 1381–1387. [Google Scholar] [CrossRef]


{kind=link}
| No. | 1 | 2 | 3 |
|---|---|---|---|
| Family no. | 1 | 2 | 2 |
| Ref | [9] | Current case | Current case |
| Origin | Greece | Japan | Japan |
| Allele | I281T/I281T | I281T/I281T | I281T/I281T |
| Age at onset | 39 | 37 | 52 |
| Age at Dx | 49 | 38 | 55 |
| Sex | NA | F | F |
| Years post-Dx | NA | >19 | >6 |
| Serum ferritin (ng/mL) | 4127 | 2274 | 4340 |
| TSAT | 90% | 93% | 92% |
| Hepcidin (ng/mL) | NA | BDL | BDL |
| Hypogonadism | + | + | + |
| Arthropathy | + | + | - |
| Skin pigmentation | + | + | + |
| Glucose intolerance | - | + | + |
| Heart disease | - | - | + |
| Hepatic fibrosis | + | + | + |
| No. | Family No. | Ref | Origin | Allele | Age at Onset | Age at Dx | Sex |
|---|---|---|---|---|---|---|---|
| 1 | 1 | [9] | Greece | I281T/I281T | 39 | 49 | NA |
| 2 | 2 | Current case | Japan | I281T/I281T | 37 | 38 | F |
| 3 | 2 | Current case | Japan | I281T/I281T | 52 | 55 | F |
| 4 | 3 | [10] | China | I281T/C321X | 14 | 19 | F |
| 5 | 4 | [11] | China | I281T/C321X | 26 | 26 | M |
| 6 | 4 | [11] | China | I281T/C321X | 27 | 27 | M |
| 7 | 5 | [15] | China | I281T/C208X/R6S | 35 | 36 | F |
| 8 | 6 | [9] | Canada | I222N/G320V | 7 | 7 | NA |
| 9 | 7 | [9] | Greece | G320V/G320V | 21 | 25 | NA |
| 10 | 8 | [9] | Greece | G320V/G320V | 32 | 39 | NA |
| 11 | 9 | [9] | Greece | G320V/G320V | 25 | 32 | NA |
| 12 | 10 | [9] | Greece | G320V/G320V | 20 | 21 | NA |
| 13 | 11 | [9] | Greece | C361fsX366/C361fsX366 | 26 | 33 | NA |
| 14 | 12 | [9] | Greece | G99V/G99V | 28 | 33 | NA |
| 15 | 13 | [9] | Greece | G320V/G320V | 21 | 25 | NA |
| 16 | 14 | [9] | Greece | G320V/R326X | 33 | 37 | NA |
| 17 | 15 | [9] | Greece | G320V/G320V | 29 | 31 | NA |
| 18 | 16 | [9] | France | G320V/G320V | 16 | 23 | NA |
| 19 | 17 | [11] | China | C321X/H104R | 18 | NA | M |
| 20 | 18 | [11] | China | C321X/V274M | 57 | NA | M |
| 21 | 19 | [11] | China | Q312X/Q312X | 22 | NA | F |
| 22 | 20 | [11] | China | F103L/F103L | 36 | NA | F |
| 23 | 21 | [16] | France (Caucasian) | G320V/G320V | 28 | NA | M |
| 24 | 22 | [16] | France (Caucasian) | G320V/G320V | 31 | NA | M |
| 25 | 23 | [16] | France (North African) | R385X/R385X | 8 | NA | M |
| 26 | 24 | [16] | France (Caucasian) | H180R/L101P | 60 | NA | F |
| 27 | 25 | [16] | France (Caucasian) | A384V/R288W | 32 | NA | F |
| 28 | 26 | [16] | France (Caucasian) | G320V/G320V | 16 | NA | F |
| 29 | 27 | [17] | African American | R54X/R54X | 4 | 23 | M |
| 30 | 28 | [18] | Romania | G66X/G66X | 25 | 25 | M |
| 31 | 29 | [19] | Japan | Y150C/V274M | 25 | 39 | M |
| 32 | 30 | [20] | United States | C80R/L101P | 18 | 23 | F |
| 33 | 31 | [20] | United States | C80R/L101P | 17 | 21 | F |
| 34 | 31 | [20] | United States | L101P/L101P | 13 | 23 | F |
| 35 | 31 | [20] | United States | L101P/L101P | 15 | 21 | M |
| 36 | 31 | [20] | United States | L101P/L101P | 12 | 18 | F |
| 37 | 31 | [20] | United States | L101P/L101P | 8 | 8 | F |
| 38 | 32 | [20] | United States | I222N/G320V | 17 | 23 | F |
| 39 | 33 | [21] | Bangladesh | C80Y/G320V | NA | 19 | F |
| 40 | 34 | [21] | Pakistan | G99R/G99R | NA | 26 | M |
| 41 | 35 | [21] | Pakistan | G99R/G99R | NA | 11 | F |
| 42 | 36 | [21] | Pakistan | P192L/P192L | NA | 23 | M |
| 43 | 37 | [21] | Pakistan | L194P/L194P | NA | 32 | M |
| 44 | 38 | [21] | Sri Lanka | A343PfsX23/A343PfsX23 | NA | 17 | M |
| 45 | 39 | [22] | Australia | G320V/G320V | NA | 12 | F |
| 46 | 40 | [22] | Australia | C80R/R326X | NA | 18 | F |
| 47 | 41 | [22] | Australia | G320V/G320V | NA | 32 | F |
| 48 | 42 | [23] | Italy | R385X/R385X | 15 | NA | F |
| 49 | 43 | [23] | Italy | F170S/F170S | 20 | NA | F |
| 50 | 44 | [23] | Italy | W191C/W191C | 21 | NA | F |
| 51 | 45 | [23] | Italy | R385X/R385X | 20 | NA | M |
| 52 | 46 | [23] | Italy | D149fsX245/D149fsX245 | 20 | NA | F |
| 53 | 47 | [23] | Italy | S205R/G250V | 21 | NA | F |
| 54 | 48 | [23] | Italy | F170S/F170S | 14 | NA | F |
| 55 | 49 | [23] | Italy | V74fsX113/N269fsX311 | 24 | NA | F |
| 56 | 49 | [23] | Italy | D149fsX245/D149fsX245 | 21 | NA | M |
| 57 | 50 | [23] | Italy | R131fsX245/R131fsX245 | 20 | NA | F |
| 58 | 51 | [23] | Canada/Italy | G320V/G320V | 29 | NA | M |
| 59 | 52 | [23] | Italy | S85P/S85P | 30 | NA | F |
| 60 | 53 | [23] | France | R288W/R288W | 26 | NA | F |
| 61 | 54 | [23] | Italy | D172E/G319fsX341 | 20 | NA | F |
| 62 | 55 | [23] | Australia/English | A168D/A168D | 28 | NA | M |
| 63 | 56 | [23] | Albania | L101P/G99R | 26 | NA | F |
| 64 | 57 | [23] | Italy | D149fsX245/D149fsX245 | 22 | NA | M |
| 65 | 58 | [23] | Canada/Italy | G320V/G320V | 27 | NA | F |
| 66 | 59 | [24] | Iran | C89R/C89R | 26 | 26 | M |
| 67 | 59 | [24] | Iran | C89R/C89R | 30 | 30 | F |
| 68 | 60 | [25] | English/Ireland | G320V/Q116X | 25 | NA | F |
| 69 | 61 | [26] | Croatia | G320V/G320V | NA | 24 | M |
| 70 | 62 | [26] | Germany | G320V/G320V | NA | 24 | M |
| 71 | 63 | [26] | Germany | G320V/G320V | NA | 24 | M |
| 72 | 64 | [26] | Slovakia | G320V/S328fsX337 | NA | 25 | M |
| 73 | 64 | [26] | Slovakia | G320V/S328fsX337 | NA | 16 | F |
| 74 | 65 | [26] | Germany | G320V/G320V | NA | 28 | F |
| 75 | 66 | [26] | Germany | C119F/C119F | NA | 25 | M |
| 76 | 67 | [27] | Netherland | L165X/L165X | NA | 16 | M |
| 77 | 68 | [28] | France (Caucasian) | R176C/R176C | NA | 17 | F |
| 78 | 69 | [29] | France | G320V/R176C | 5 | 5 | F |
| 79 | 70 | [30] | Japan | D249H/D249H | 48 | 48 | M |
| 80 | 71 | [30] | Japan | Q312X/Q312X | 51 | 51 | M |
| 81 | 72 | [30] | Japan | Q312X/Q312X | 51 | 51 | F |
| 82 | 73 | [31] | Caucasian | G320V/C321W | 23 | 30 | F |
| 83 | 74 | [32] | India | D355Y/D355Y | 35 | 42 | M |
| 84 | 74 | [32] | India | D355Y/D355Y | 28 | 32 | M |
| 85 | 75 | [33] | Romania | G320V/G320V | 27 | 31 | F |
| 86 | 76 | [34] | Caucasian | G320V/G320V | 20 | 39 | F |
| 87 | 77 | [35] | Italy | C317S/C317S | 35 | 68 | F |
| 88 | 78 | [36] | India | G336X/G336X | NA | 45 | M |
| 89 | 79 | [36] | India | G336X/G336X | NA | 49 | F |
| 90 | 80 | [36] | India | G336X/G336X | NA | 38 | F |
| 91 | 81 | [36] | India | G336X/G336X | NA | 47 | M |
| 92 | 82 | [36] | India | 5’UTR -358 (G>A)/ 5’UTR -36 (G>A) | NA | 43 | M |
| 93 | 83 | [36] | India | 5’UTR -358 (G>A)/ 5’UTR -36 (G>A) | NA | 32 | F |
| 94 | 84 | [37] | China | C321X/Q6H | 23 | 31 | M |
| 95 | 85 | [38] | English/Ireland | L28SfsX24/ L28SfsX24 | 25 | 25 | F |
| 96 | 86 | [39] | Brazil | Q233fsX245/Q233fsX245 | 26 | 26 | F |
| Total, F/M | 49 (58%)/35 (42%) | ||||||
| Median age (total) | 25 | 26 | |||||
| Median age (F) | 23 | 26 | |||||
| Range (F) | 5–60 | 5–68 | |||||
| Median age (M) | 26 | 26 | |||||
| Range (M) | 4–57 | 16–51 | |||||
| P value at age (F vs. M) | 0.47 | 0.48 | |||||
| Standard deviation (total) | 11 | 12 | |||||
| Standard deviation (F) | 11 | 14 | |||||
| Standard deviation (M) | 12 | 10 | |||||
| Group Number | Allele | Origin (Number of Cases) | Number of Cases | Sex, F/M | Median Age, y | Minimum, y | Maximum, y | p Value at Age vs. G320V /G320V |
|---|---|---|---|---|---|---|---|---|
| 1 | G320V/G320V | Greece (6), France (4), Germany (3), Canada (2), Australia (2), Romania (1), Croatia (1), Caucasian (1) | 20 | 7/6 | 26 | 16 | 32 | Reference |
| 2 | G336X/G336X | India (4) | 4 | 2/2 | NA | NA | NA | NA |
| 3 | L101P/L101P | United states (4) | 4 | 3/1 | 13 | 8 | 15 | 0.004 |
| 4 | I281T/I281T | Japan (2), Greece (1) | 3 | 2/0 | 39 | 37 | 52 | 0.010 |
| 5 | Q312X/Q312X | Japan (2), China (1) | 3 | 2/1 | 51 | 22 | 51 | 0.105 |
| 6 | D149fsX245/D149fsX245 | Italy (3) | 3 | 1/2 | 21 | 20 | 22 | 0.498 |
| 7 | G99R/G99R | Pakistan (2), Greece (1) | 3 | 1/1 | 28 | 28 | 28 | 0.708 |
| 8 | R385X/R385X | Italy (2), France (1) | 3 | 1/2 | 15 | 8 | 20 | 0.031 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takami, A.; Tatsumi, Y.; Sakai, K.; Toki, Y.; Ikuta, K.; Oohigashi, Y.; Takagi, J.; Kato, K.; Takami, K. Juvenile Hemochromatosis: A Case Report and Review of the Literature. Pharmaceuticals 2020, 13, 195. https://doi.org/10.3390/ph13080195
Takami A, Tatsumi Y, Sakai K, Toki Y, Ikuta K, Oohigashi Y, Takagi J, Kato K, Takami K. Juvenile Hemochromatosis: A Case Report and Review of the Literature. Pharmaceuticals. 2020; 13(8):195. https://doi.org/10.3390/ph13080195
Chicago/Turabian StyleTakami, Akiyoshi, Yasuaki Tatsumi, Katsuhisa Sakai, Yasumichi Toki, Katsuya Ikuta, Yuka Oohigashi, Junko Takagi, Koichi Kato, and Kazuhisa Takami. 2020. "Juvenile Hemochromatosis: A Case Report and Review of the Literature" Pharmaceuticals 13, no. 8: 195. https://doi.org/10.3390/ph13080195
APA StyleTakami, A., Tatsumi, Y., Sakai, K., Toki, Y., Ikuta, K., Oohigashi, Y., Takagi, J., Kato, K., & Takami, K. (2020). Juvenile Hemochromatosis: A Case Report and Review of the Literature. Pharmaceuticals, 13(8), 195. https://doi.org/10.3390/ph13080195