Synthesis, Optimization, Antifungal Activity, Selectivity, and CYP51 Binding of New 2-Aryl-3-azolyl-1-indolyl-propan-2-ols

 , , ,
, , ,  , , , , , , , ,
, , , , , , , ,  and
and 
Abstract
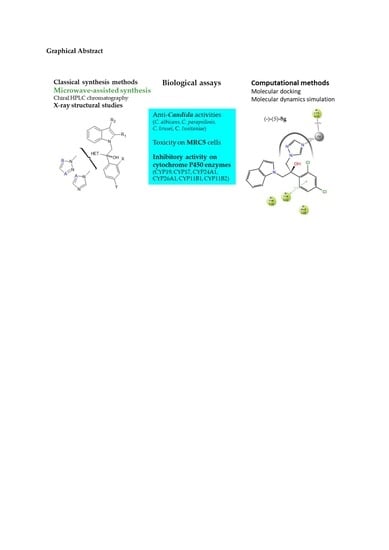
1. Introduction
2. Results and Discussion
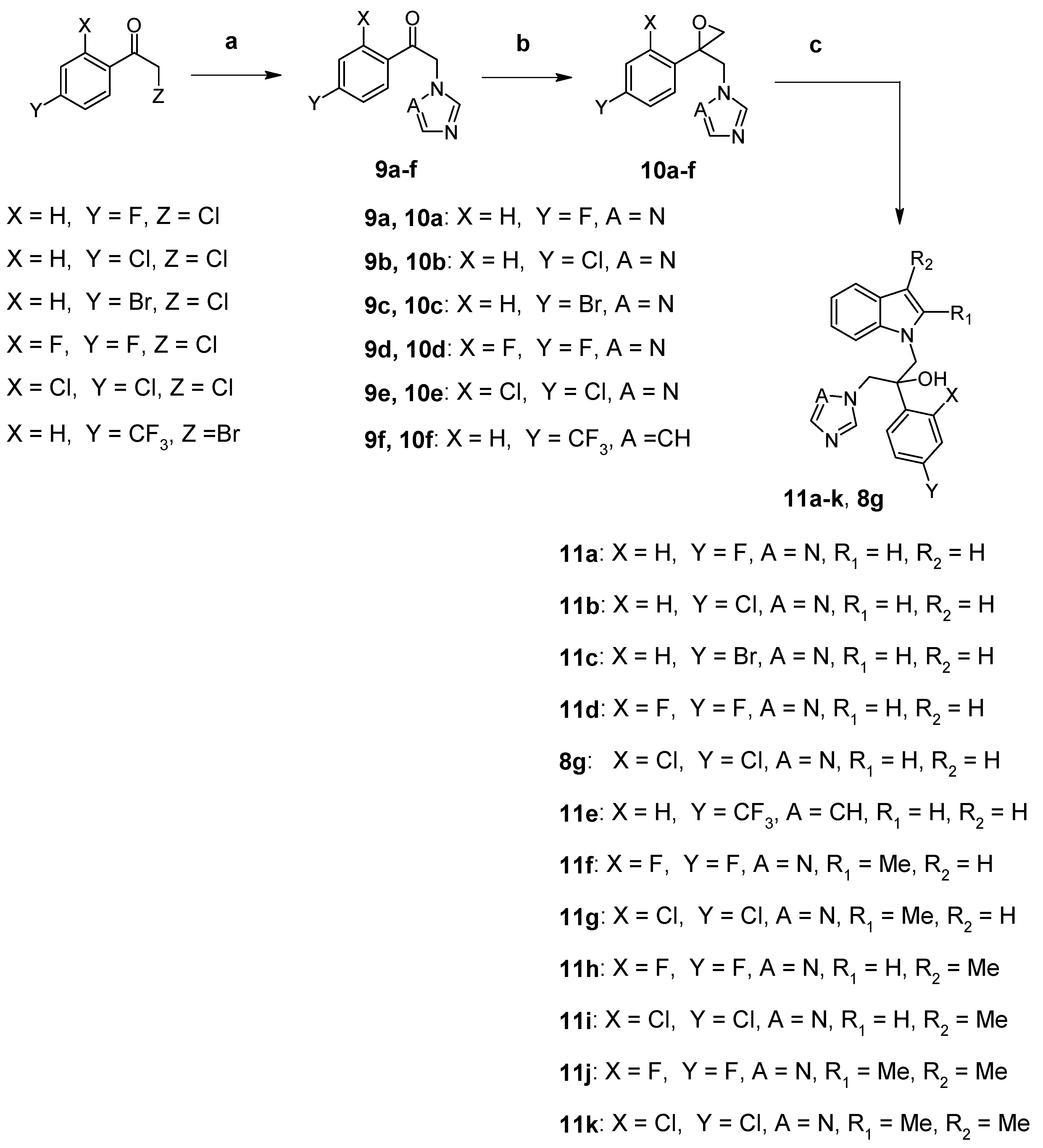
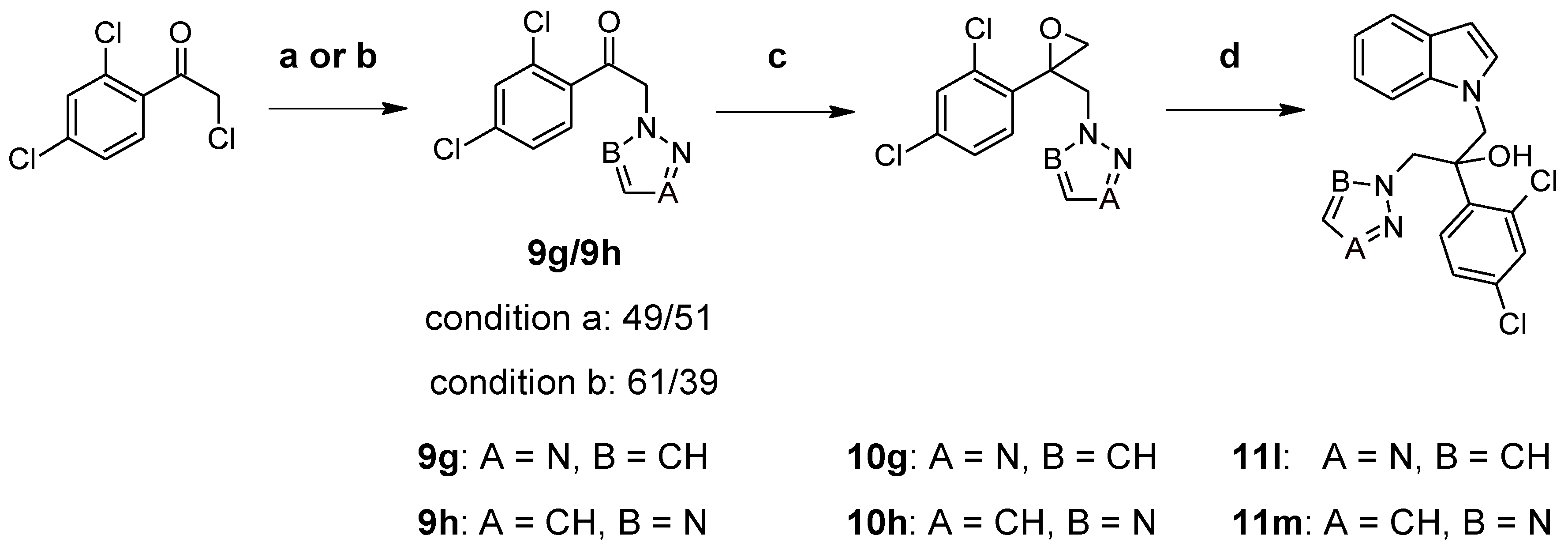
2.1. Chemistry
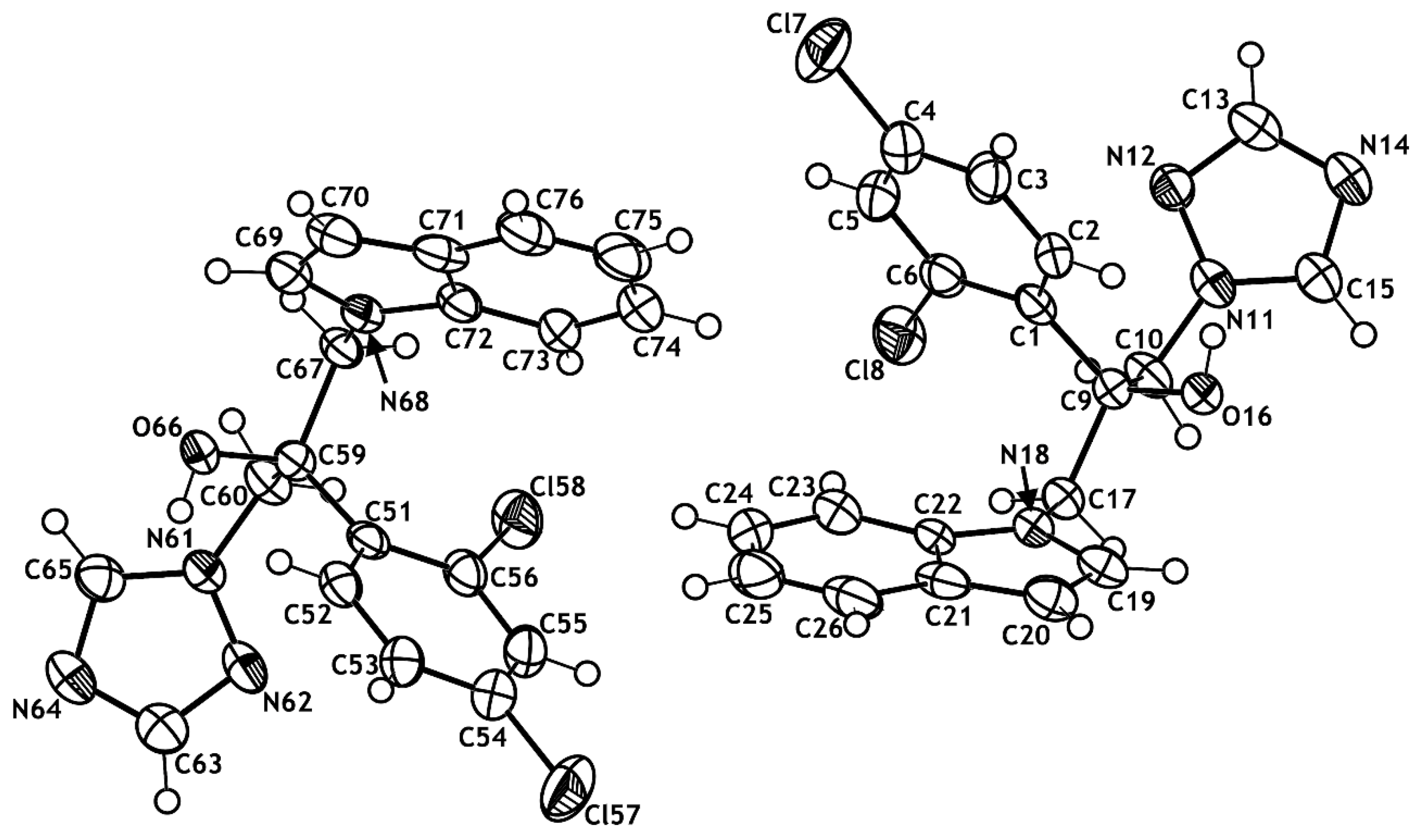
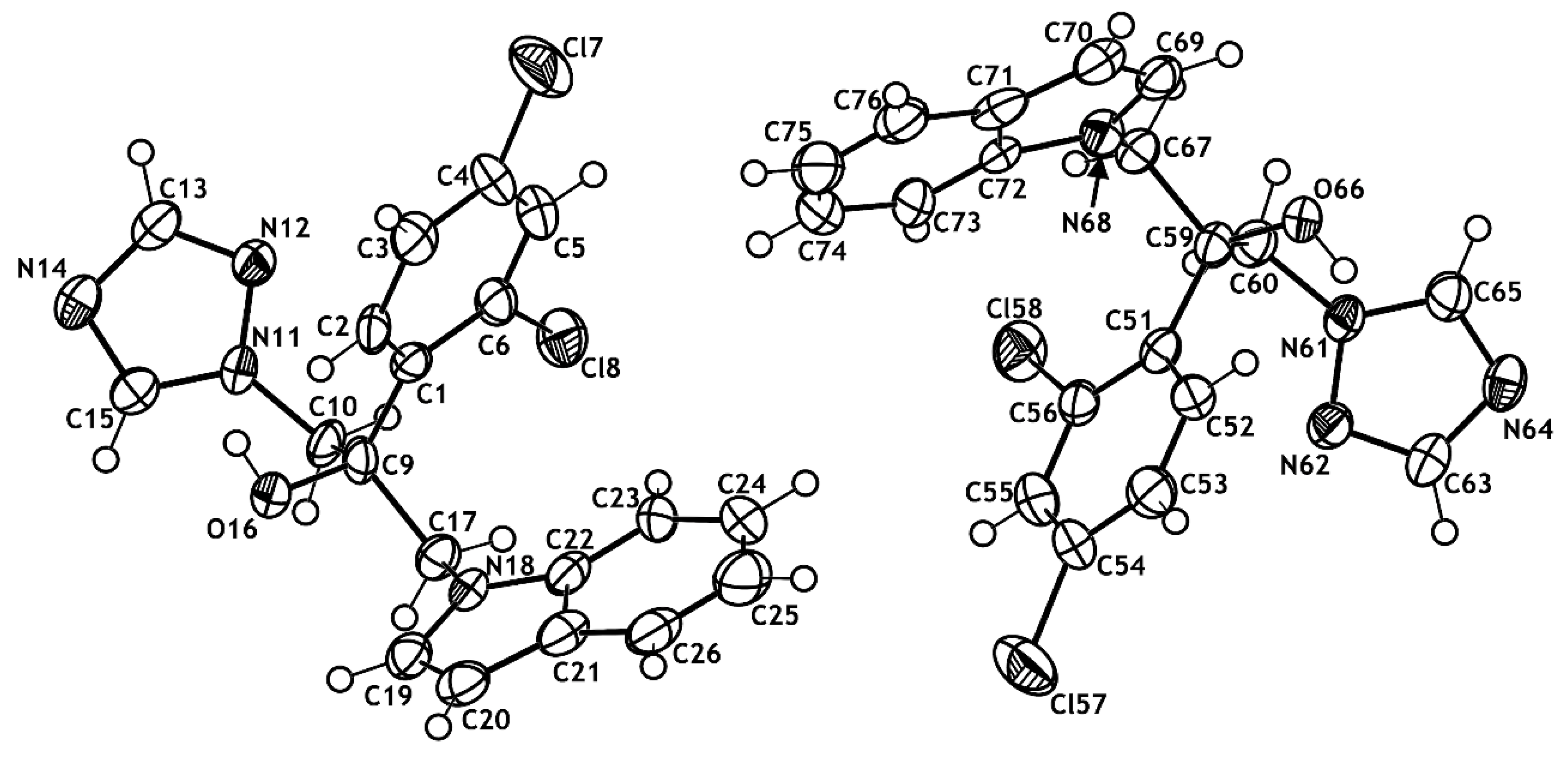
2.2. X-ray Structural Studies
2.3. Biological Results
2.3.1. In Vitro Anti-Candida Activity
2.3.2. Toxicity of Selected Compounds on Human Fetal Lung Fibroblast (MRC-5) Cells
2.3.3. Inhibitory Activity on Cytochrome P450 Enzymes
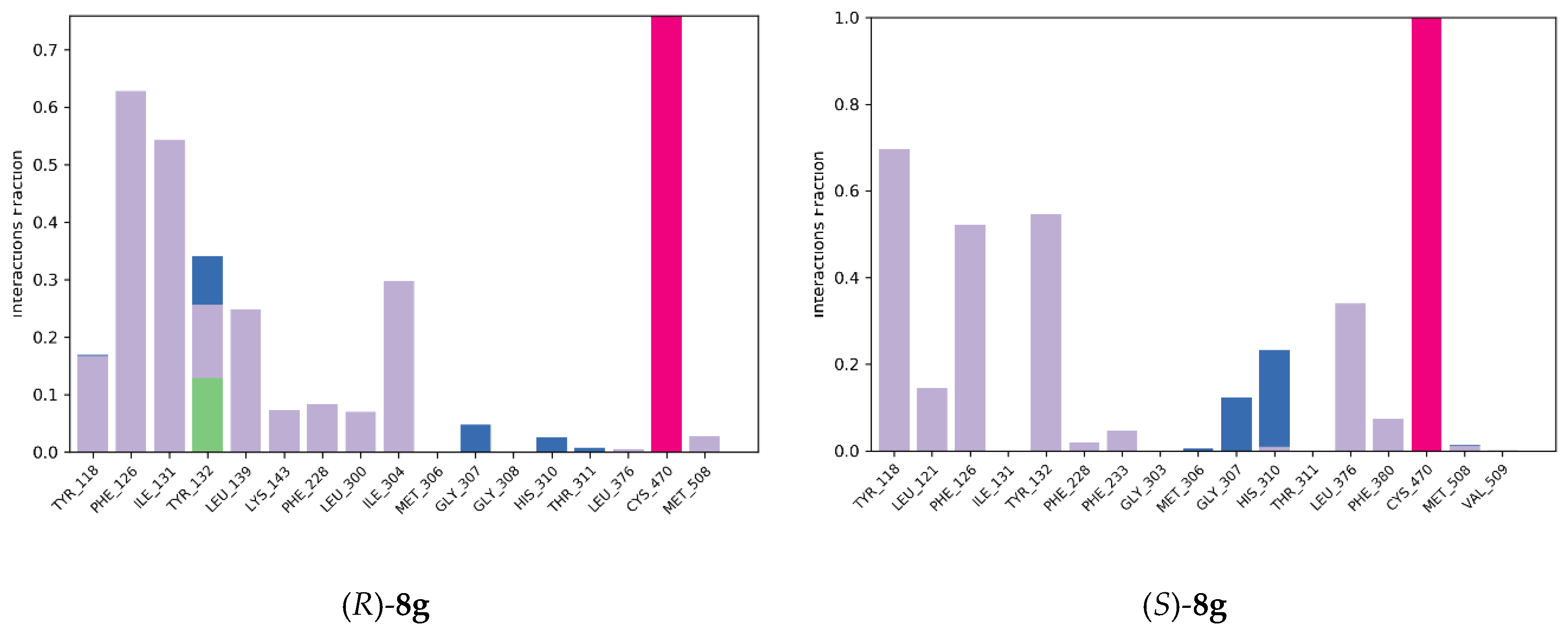
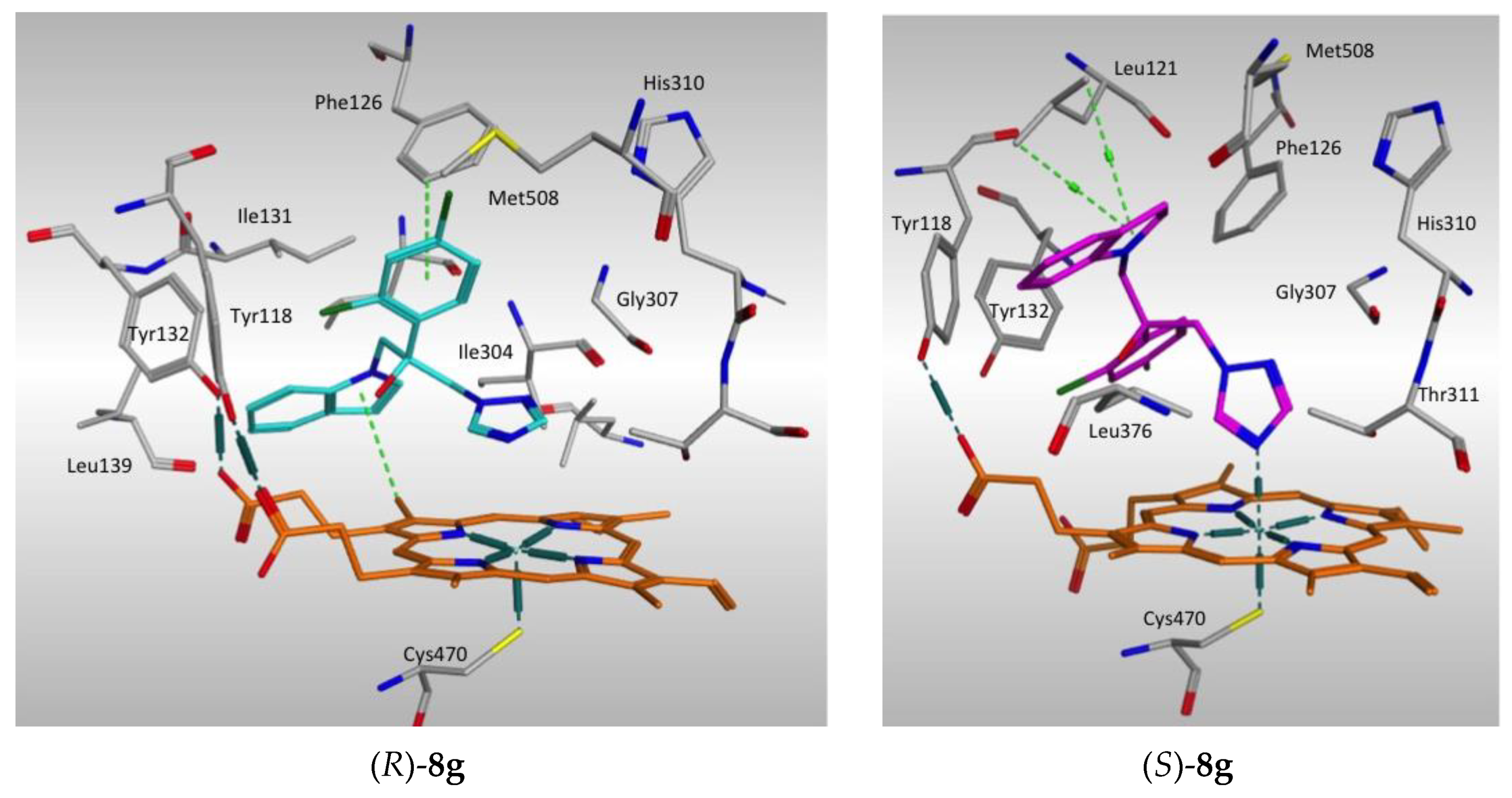
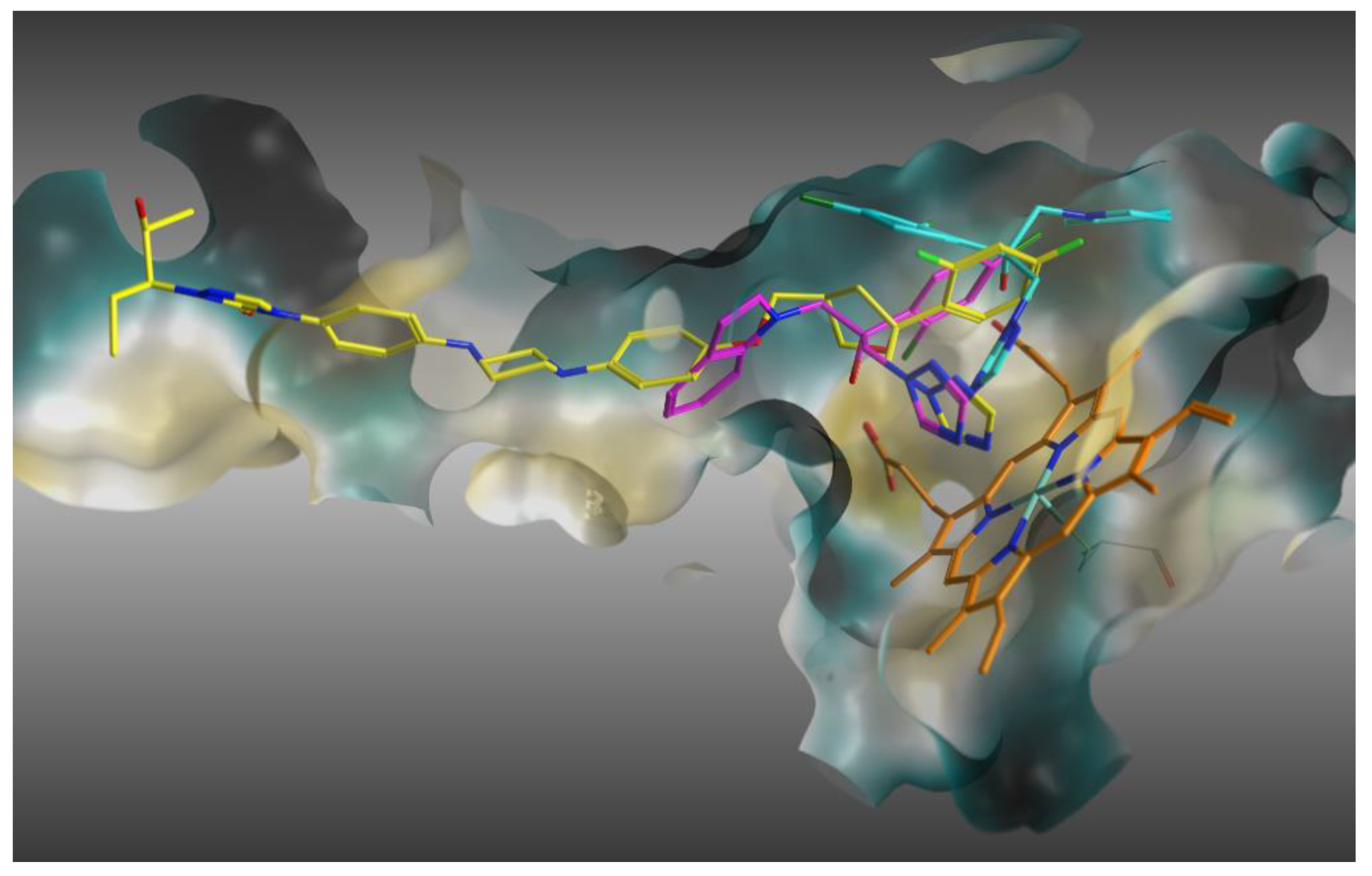
2.4. Molecular Modeling Studies
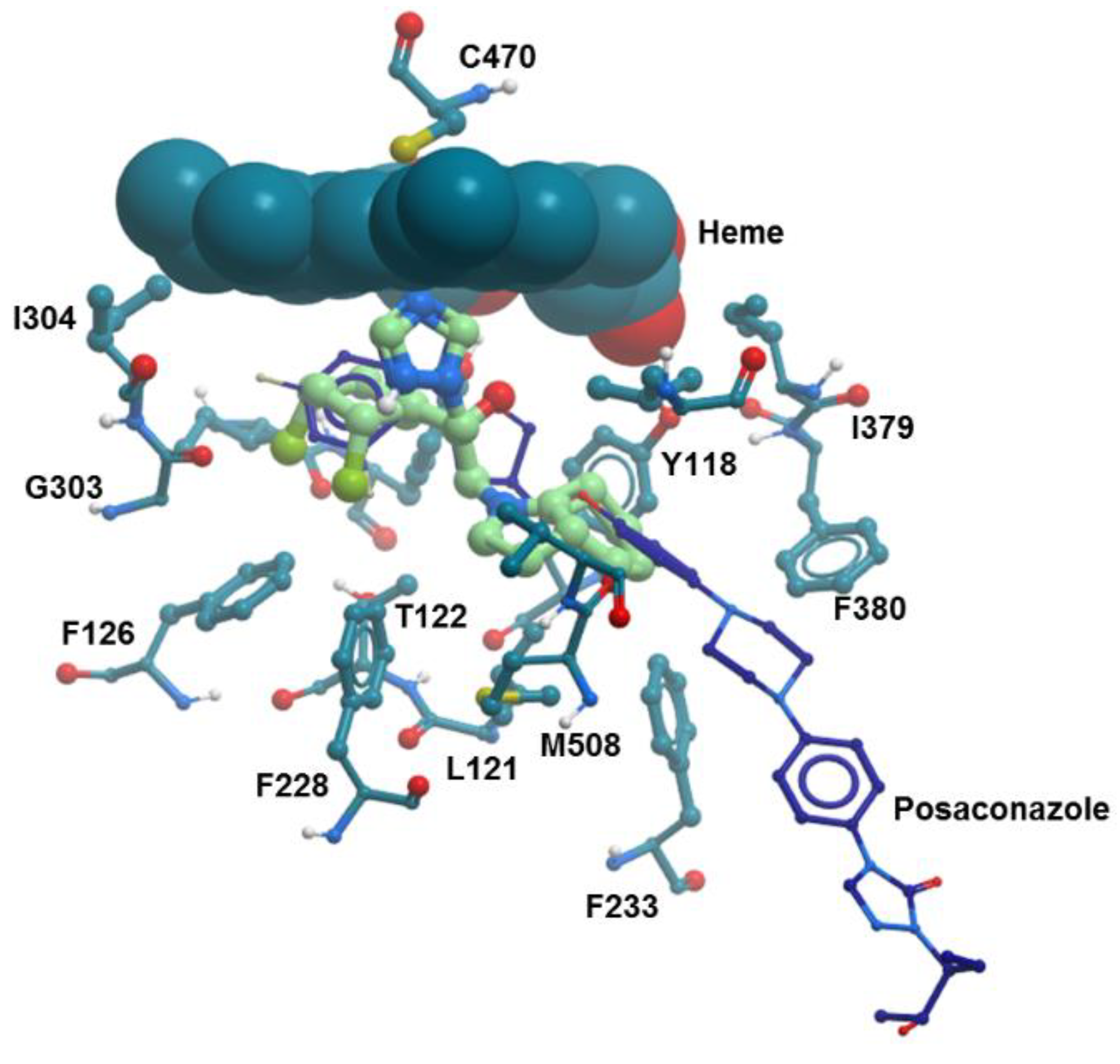
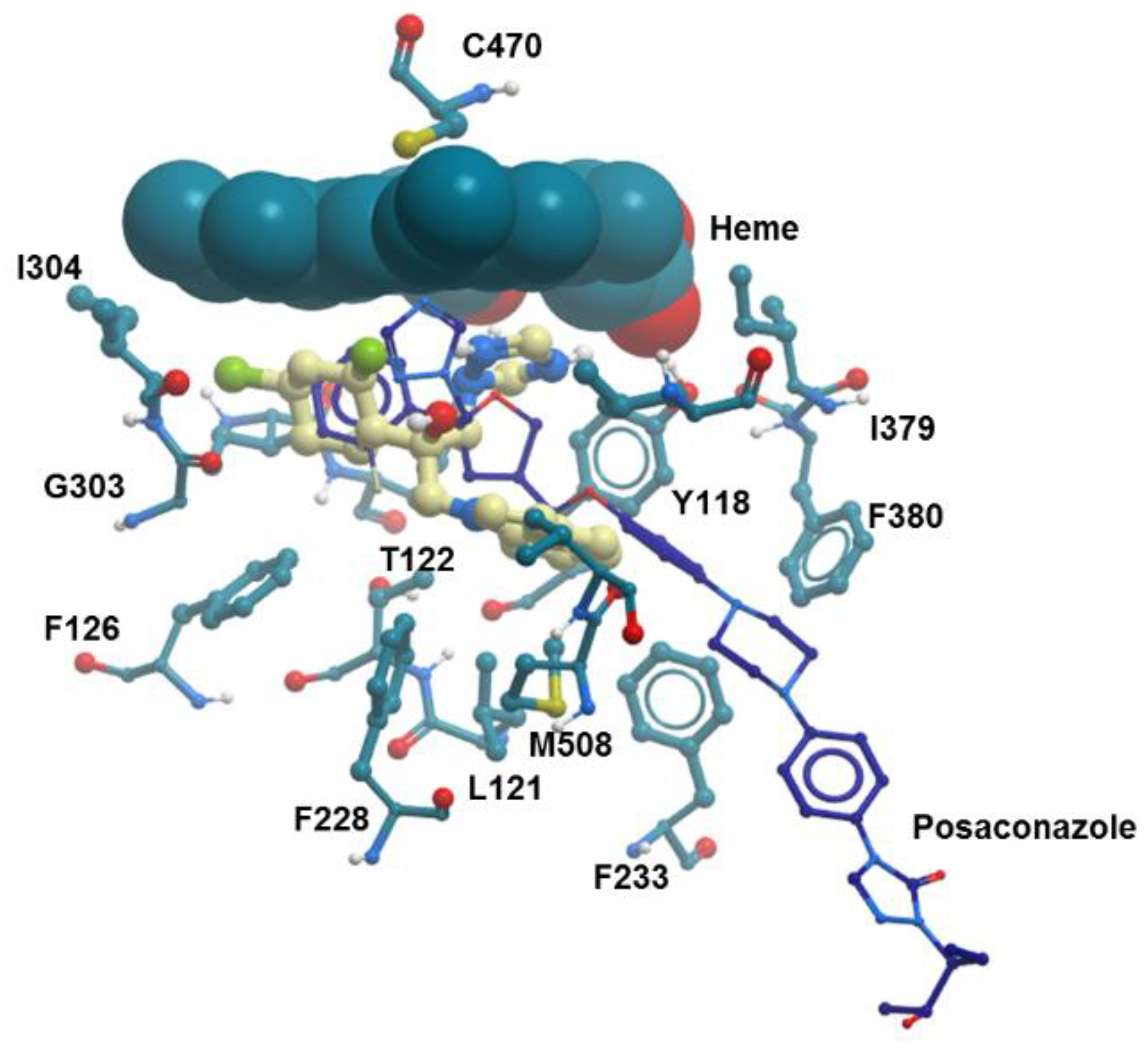
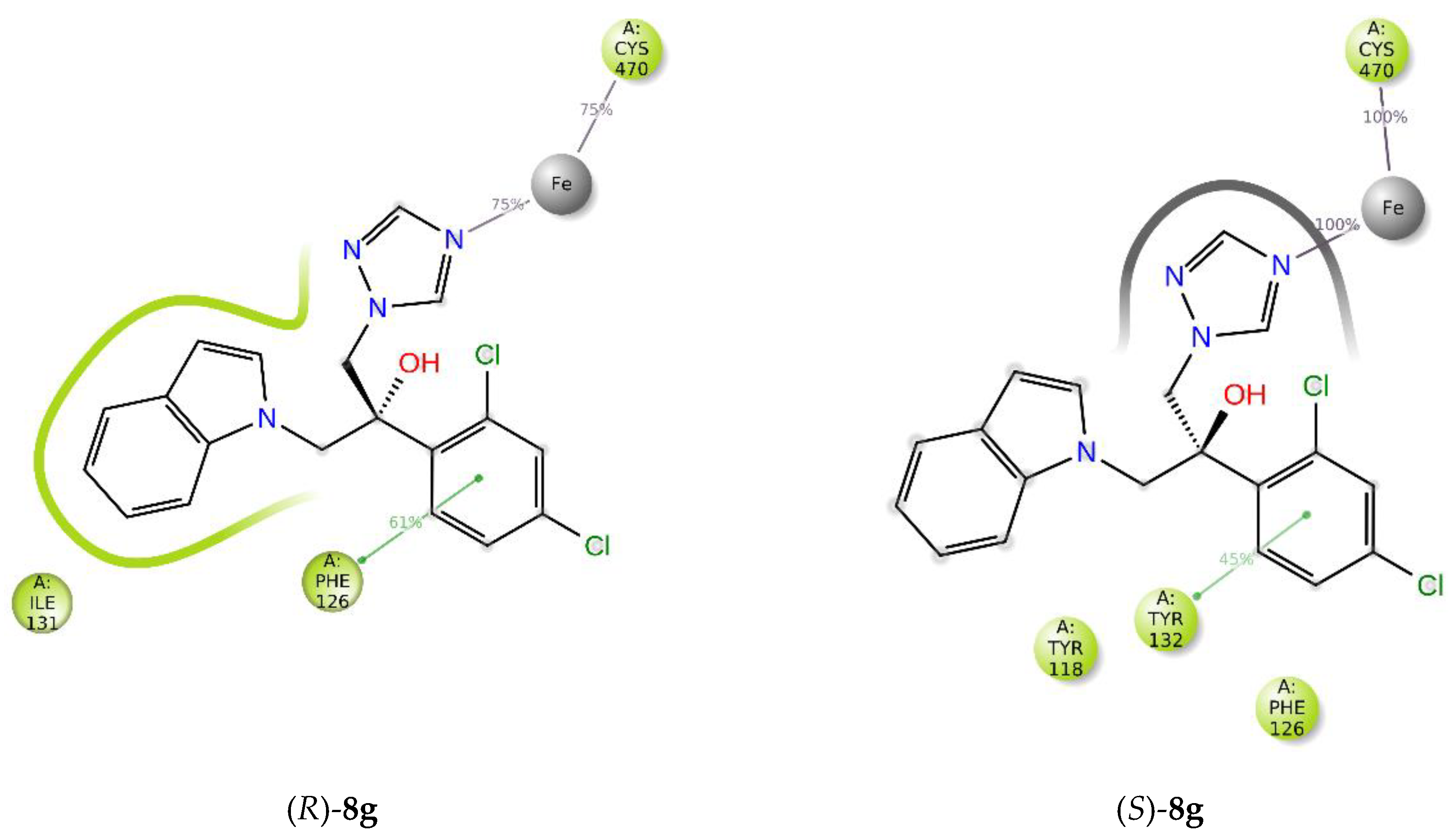
2.4.1. Molecular Docking
2.4.2. Molecular Dynamics Simulation
3. Materials and Methods
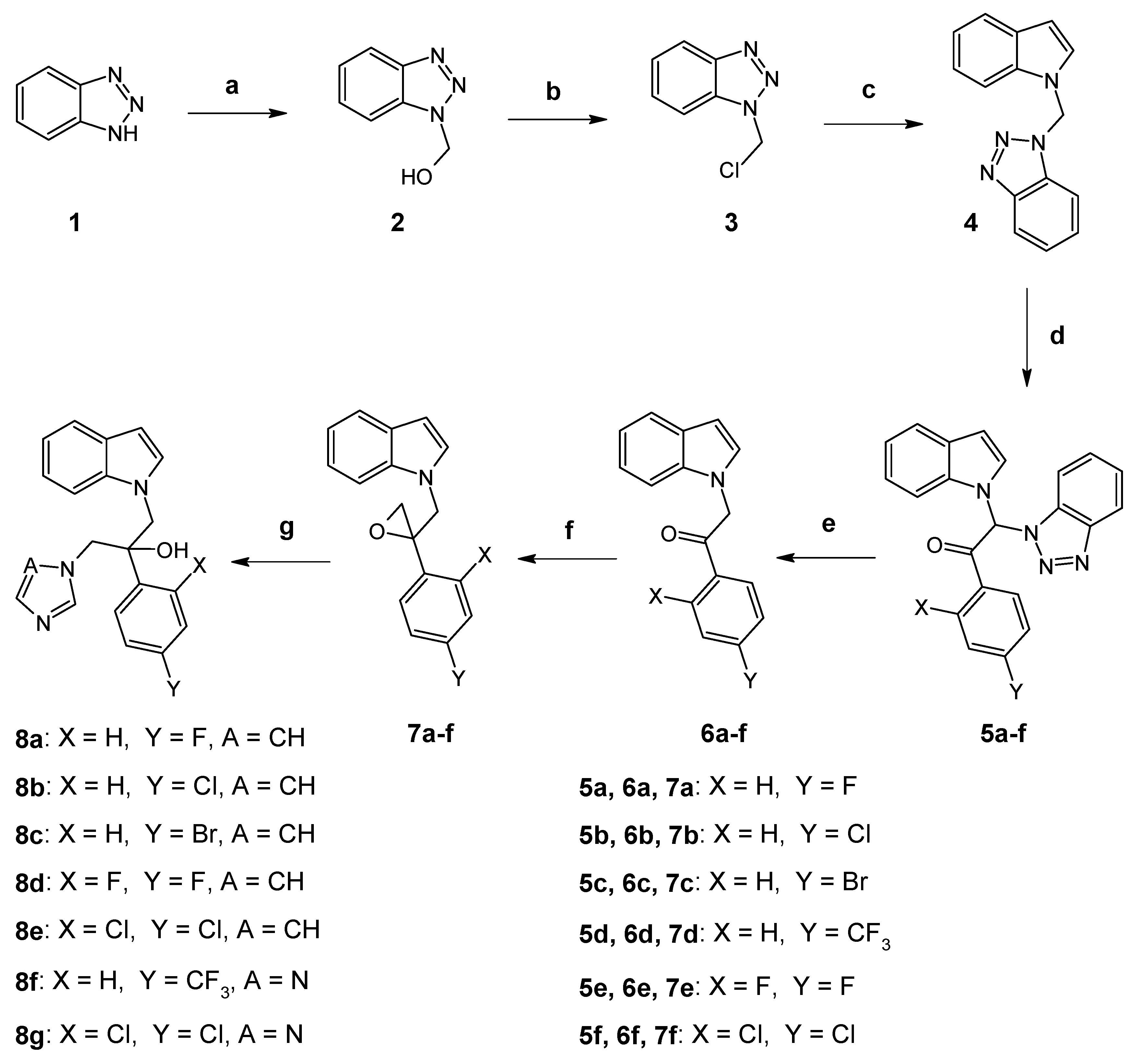
3.1. Chemistry
3.2. Synthesis of Compounds 8a–g (Route 1)
3.2.1. Procedure for the Synthesis of 1-Hydroxymethyl-1H-benzotriazole (2)
3.2.2. Procedure for the Synthesis of 1-Chloromethyl-1H-benzotriazole (3)
3.2.3. Procedure for the Synthesis of 1-(1H-Benzotriazol-1-yl-methyl)-1H-indole (4)
3.2.4. General Procedure for the Synthesis of Benzotriazole Derivatives 5a–f
3.2.5. General Procedure for the Synthesis of N-Substituted-1H-indoles 6a–f
3.2.6. General Procedure for the Synthesis of Oxiranes 7a–f
3.2.7. General Procedure for the Synthesis of Imidazole Derivatives 8a–e
3.2.8. General Procedure for the Synthesis of Triazole Derivatives 8f and 8g
3.3. Synthesis of Compounds 8g and 11a–k (Route 2)
3.3.1. Synthesis of 2-(1H-Imidazol-1-yl)-1-(4-trifluoromethylphenyl)ethanone (9f)
3.3.2. Synthesis of 3-(1H-imidazol-1-yl)-2-(4-trifluoromethylphenyl)-1,2-epoxypropane (10f)
3.3.3. General Procedure for the N-Alkylation of Indole Derivatives 8g and 11a–k
3.4. Synthesis of Compounds 11l and 11m (Route 2)
3.4.1. Procedure for the N-1 and N-2-alkylation of 1H-1,2,3-Triazole
3.4.2. General Procedure for the Synthesis of Oxiranes 10g and 10h
3.4.3. General Procedure for the Sysnthesis of Triazole Derivatives 11l and 11m
3.5. Chiral HPLC Chromatography
3.6. X-ray Crystallography Studies
3.7. Biological Assays
3.7.1. Anti-Candida In Vitro Assay
3.7.2. MRC5 Toxicity Assay
3.7.3. In Vitro Inhibition of Aromatase
3.7.4. In Vitro Inhibition of 17α-Hydroxylase/17,20-Lyase
3.7.5. MCF-7 Cell Culture and In Vitro Assay for Inhibition of Metabolism of All-trans Retinoic Acid (ATRA)
3.7.6. In Vitro Inhibition of CYP11B1 and CYP11B2
3.8. Computational Methods
3.8.1. Molecular Docking
3.8.2. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Pianalto, K.M.; Alspaugh, J.A. New horizons in antifungal therapy. J. Fungi 2016, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L. Risk and prevention of tuberculosis and other serious opportunistic infections associated with the inhibition of tumor necrosis factor. Nat. Clin. Pract. Rheumatol. 2006, 2, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef]
- De Oliveira Santos, G.C.; Vasconcelos, C.C.; Lopes, A.J.O.; De Sousa Cartágenes, M.D.S.; Filho, A.K.D.B.; Do Nascimento, F.R.F.; Ramos, R.M.; Pires, E.R.R.B.; De Andrade, M.S.; Rocha, F.M.G.; et al. Candida infections and therapeutic strategies: Mechanisms of action for traditional and alternative agents. Front. Microbiol. 2018, 9, 1351. [Google Scholar] [CrossRef]
- Autmizguine, J.; Smith, P.B.; Prather, K.; Bendel, C.; Natarajan, G.; Bidegain, M.; Kaufman, D.A.; Burchfield, D.J.; Ross, A.S.; Pandit, P.; et al. Effect of fluconazole prophylaxis on Candida fluconazole susceptibility in premature infants. J. Antimicrob. Chemother. 2018, 73, 3482–3487. [Google Scholar] [CrossRef]
- Kaguelidou, F.; Pandolfini, C.; Manzoni, P.; Choonara, I.; Bonati, M.; Jacqz-Aigrain, E. European survey on the use of prophylactic fluconazole in neonatal intensive care units. Eur. J. Pediatr. 2012, 171, 439–445. [Google Scholar] [CrossRef]
- Mishra, M.; Agrawal, S.; Raut, S.; Kurhade, A.M.; Powar, R.M. Profile of yeasts isolated from urinary tracts of catheterized patients. J. Clin. Diagn. Res. 2014, 8, 44–46. [Google Scholar] [CrossRef]
- Chen, T.C.; Chen, Y.H.; Chen, Y.C.; Lu, P.L. Fluconazole exposure rather than clonal spreading is correlated with the emergence of Candida glabrata with cross-resistance to triazole antifungal agents. Kaohsiung J. Med. Sci. 2012, 28, 306–315. [Google Scholar] [CrossRef]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef]
- Zambrano-Huerta, A.; Cifuentes-Castañeda, D.D.; Bautista-Renedo, J.; Mendieta-Zerón, H.; Melgar-Fernández, R.C.; Pavón-Romero, S.; Morales-Rodríguez, M.; Frontana-Uribe, B.A.; González-Rivas, N.; Cuevas-Yañez, E. Synthesis and in vitro biological evaluation of 1,3-bis-(1,2,3-triazol-1-yl)-propan-2-ol derivatives as antifungal compounds fluconazole analogues. Med. Chem. Res. 2019, 28, 571–579. [Google Scholar] [CrossRef]
- Pongas, G.N.; Lewis, R.E.; Samonis, G.; Kontoyiannis, D.P. Voriconazole-associated Zygomycosis: A significant consequence of evolving antifungal prophylaxis and immunosuppression practices? Clin. Microbiol. Infect. 2009, 15, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U. Pharmacokinetics/pharmacodynamics of Echinocandins. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, V.W.; Santos, T.F.; Terra, L.; Santana, M.V.; Castro, H.C.; Rodrigues, C.R.; Abreu, P.A. Targeting CYP51 for drug design by the contributions of molecular modeling. Fundam. Clin. Pharmacol. 2017, 31, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 2007, 1770, 467–477. [Google Scholar] [CrossRef]
- Keniya, M.V.; Sabherwal, M.; Wilson, R.K.; Woods, M.A.; Sagatova, A.A.; Tyndall, J.D.A.; Monk, B.C. Crystal structures of full-length lanosterol 14α-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provide tools for antifungal discovery. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Onyewu, C.; Blankenship, J.R.; Del Poeta, M.; Heitman, J. Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 2003, 47, 956–964. [Google Scholar] [CrossRef]
- Lézé, M.-P.; Le Borgne, M.; Pinson, P.; Palusczak, A.; Duflos, M.; Le Baut, G.; Hartmann, R.W. Synthesis and biological evaluation of 5-[(aryl)(1H-imidazol-1-yl)methyl]-1H-indoles: Potent and selective aromatase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1134–1137. [Google Scholar] [CrossRef]
- Le Borgne, M.; Marchand, P.; Delevoye-Seiller, B.; Robert, J.M.; Le Baut, G.; Hartmann, R.W.; Palzer, M. New selective nonsteroidal aromatase inhibitors: Synthesis and inhibitory activity of 2, 3 or 5-(alpha-azolylbenzyl)-1H-indoles. Bioorg. Med. Chem. Lett. 1999, 9, 333–336. [Google Scholar] [CrossRef]
- Martínez-Matías, N.; Rodríguez-Medina, J.R. Fundamental concepts of azole compounds and triazole antifungals: A beginner’s review. P. R. Health Sci. J. 2018, 37, 135–142. [Google Scholar]
- Binjubair, F.A.; Parker, J.E.; Warrilow, A.G.; Puri, K.; Braidley, P.J.; Tatar, E.; Kelly, S.L.; Kelly, D.E.; Simons, C. Small molecule inhibitors targeting sterol 14α-demethylase (CYP51): Synthesis, molecular modelling and evaluation against Candida albicans. ChemMedChem 2020, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.M.; Le Borgne, M.; Pagniez, F.; Le Baut, G.; Le Pape, P. Synthesis and antifungal activity of new 1-halogenobenzyl-3-imidazolylmethylindole derivatives. Eur. J. Med. Chem. 2003, 38, 75–87. [Google Scholar] [CrossRef]
- Pagniez, F.; Le Borgne, M.; Marchand, P.; Na, Y.M.; Le Baut, G.; Robert-Piessard, S.; Le Pape, P. In vitro activity of a new antifungal azolyl-substituted indole against Aspergillus fumigatus. J. Enzyme Inhib. Med. Chem. 2002, 17, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Guillon, R.; Giraud, F.; Logé, C.; Le Borgne, M.; Picot, C.; Pagniez, P.; Le Pape, P. Design of new antifungal agents: Synthesis and evaluation of 1-[(1H-indol-5-ylmethyl)amino]-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols. Bioorg. Med. Chem. Lett. 2009, 19, 5833–5836. [Google Scholar] [CrossRef] [PubMed]
- Lebouvier, N.; Pagniez, F.; Duflos, M.; Le Pape, P.; Na, Y.M.; Le Baut, G.; Le Borgne, M. Synthesis and antifungal activities of new fluconazole analogues with azaheterocycle moiety. Bioorg. Med. Chem. Lett. 2007, 17, 3686–3689. [Google Scholar] [CrossRef] [PubMed]
- Pagniez, F.; Lebouvier, N.; Na, Y.M.; Le Borgne, M.; Pfalzer, M.; Hartmann, R.W.; Le Pape, P. Broad spectrum anti-Candida activities of a new indolyl-triazole, NL114. In Proceedings of the 17th European Congress of Clinical Microbiology and Infectious Diseases and 25th International Congress of Chemotherapy, Munich, Germany, 31 March–3 April 2007; Volume 29, p. 474. [Google Scholar]
- Lebouvier, N.; Giraud, F.; Corbin, T.; Na, Y.M.; Le Baut, G.; Marchand, P.; Le Borgne, M. Efficient microwave-assisted synthesis of 1-(1H-indol-1-yl)-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols as antifungal agents. Tetrahedron Lett. 2006, 47, 6479–6483. [Google Scholar] [CrossRef]
- Pagniez, F.; Lebouvier, N.; Na, Y.M.; Ourliac-Garnier, I.; Picot, C.; Le Borgne, M.; Le Pape, P. Biological exploration of a novel 1,2,4-triazole-indole hybrid molecule as antifungal agent. J. Enzyme Inhib. Med. Chem. 2020, 35, 398–403. [Google Scholar] [CrossRef]
- Hutschenreuter, T.U.; Ehmer, P.B.; Hartmann, R.W. Synthesis of hydroxy derivatives of highly potent non-steroidal CYP 17 inhibitors as potential metabolites and evaluation of their activity by a non cellular assay using recombinant human enzyme. J. Enzyme Inhib. Med. Chem. 2004, 19, 17–32. [Google Scholar] [CrossRef]
- Kragie, L.; Turner, S.D.; Patten, C.J.; Crespi, C.L.; Stresser, D.M. Assessing pregnancy risks of azole antifungals using a high throughput aromatase inhibition assay. Endocr. Res. 2002, 28, 129–140. [Google Scholar] [CrossRef]
- Trösken, E.R.; Fischer, K.; Völkel, W.; Lutz, W.K. Inhibition of human CYP19 by azoles used as antifungal agents and aromatase inhibitors, using a new LC-MS/MS method for the analysis of estradiol product formation. Toxicology 2006, 219, 33–40. [Google Scholar] [CrossRef]
- Lézé, M.P.; Palusczak, A.; Hartmann, R.W.; Le Borgne, M. Synthesis of 6-or 4-functionalized indoles via a reductive cyclization approach and evaluation as aromatase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4713–4715. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, M.; Marchand, P.; Nourrisson, M.R.; Loquet, D.; Palzer, M.; Le Baut, G.; Hartmann, R.W. Synthesis and biological evaluation of 3-(azolylmethyl)-1 H-indoles and 3-(α-azolylbenzyl)-1H-indoles as selective aromatase inhibitors. J. Enzyme Inhib. Med. Chem. 2007, 22, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Marchand, P.; Le Borgne, M.; Palzer, M.; Le Baut, G.; Hartmann, R.W. Preparation and pharmacological profile of 7-(α-azolylbenzyl)-1H-indoles and indolines as new aromatase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1553–1555. [Google Scholar] [CrossRef]
- Le Borgne, M.; Marchand, P.; Duflos, M.; Delevoye-Seiller, B.; Piessard-Robert, S.; Le Baut, G.; Hartmann, R.W.; Palzer, M. Synthesis and in vitro evaluation of 3-(1-azolylmethyl)-1H-indoles and 3-(1-azolyl-1-phenylmethyl)-1H-indoles as inhibitors of P450 arom. Arch. Pharm. 1997, 330, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, M.; Marchand, P.; Duflos, M.; Robert-Piessard, S.; Le Baut, G.; Ahmadi, M.; Hartmann, R.W.; Palzer, M. Comparison of the in-vitro aromatase inhibitory activity of 3-(azolylmethyl)-1H-indoles. Pharm. Pharmacol. Commun. 1997, 3, 279–281. [Google Scholar]
- Denner, K.; Vogel, R.; Schmalix, W.; Doehmer, J.; Bernhardt, R. Cloning and stable expression of the human mitochondrial cytochrome P45011B1cDNA V79 Chinese harmster cells and their application for testing of potential inhibitors. Pharmacogenetics 1995, 5, 89–96. [Google Scholar] [CrossRef]
- Hakki, T.; Hübel, K.; Waldmann, H.; Bernhardt, R. The development of a whole-cell based medium throughput screening system for the discovery of human aldosterone synthase (CYP11B2) inhibitors: Old drugs disclose new applications for the therapy of congestive heart failure, myocardial fibrosis and hypertension. J. Steroid Biochem. Mol. Biol. 2011, 125, 120–128. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14 alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); 2015.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Schrödinger Release 2020-1; Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2020. Available online: https://www.schrodinger.com/Desmond/ (accessed on 11 June 2020).
- Burckhalter, J.H.; Stephens, V.C.; Hall, L.A.R. Proof of structures derived from the hydroxy- and amino-methylation of benzotriazole. J. Am. Chem. Soc. 1952, 74, 3868–3870. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Drewniak-Deyrup, M.; Lan, X.; Brunner, F. Preparation, lithiation and transformation of N-(benzotriazol-1-ylmethyl) heterocycles. J. Heterocycl. Chem. 1989, 26, 829–836. [Google Scholar] [CrossRef]
- Crystallographic data (excluding structure factors) for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC–237961 ((R)-8g) and no. CCDC–237960 ((S)-8g). Copies of the Data can be Obtained Free of Charge on Application to CCDC; University Chemical Lab.: 12 Union Road, Cambridge, UK, 2004. [Google Scholar]
- North, A.C.T.; Phillips, D.C.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Crystallogr. 1968, A24, 351–359. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS86. In Crystallographic Computing 3: Data Collection, Structure Determination, Proteins and Databases, 1st ed.; Sheldrick, G.M., Kröger, C., Goddard, R., Eds.; Oxford University Press: New-York, NY, USA, 1985; pp. 175–189. [Google Scholar]
- Sheldrick, G.M. SHELXL-93. In A Program for the Refinement of the Crystal Structures from Diffraction Data; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Pagniez, F.; Le Pape, P. New fluorometric screening test for possible antifungal drugs. J. Mycol. Med. 2001, 11, 73–78. [Google Scholar]
- Thompson, E.A., Jr.; Siiteri, P.K. Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J. Biol. Chem. 1974, 249, 5364–5372. [Google Scholar] [PubMed]
- Graves, P.E.; Salhanick, H.A. Stereoselective inhibition of aromatase by enantiomers of aminoglutethimide. Endocrinology 1979, 105, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.B.; Jarman, M.; Leung, C.S.; Rowlands, M.G.; Taylor, G.N. Analogues of aminoglutethimide: Selective inhibition of cholesterol side-chain cleavage. J. Med. Chem. 1983, 26, 50–54. [Google Scholar] [CrossRef]
- Ehmer, P.B.; Jose, J.; Hartmann, R.W. Development of a simple and rapid assay for the evaluation of inhibitors of human 17α-hydroxylase-C(17,20)-lyase (P450c17) by coexpression of P450c17 with NADPH-cytochrome-P450-reductase in Escherichia coli. J. Steroid Biochem. Mol. Biol. 2000, 75, 57–63. [Google Scholar] [CrossRef]
- Gomaa, M.S.; Bridgens, C.E.; Illingworth, N.A.; Veal, G.J.; Redfern, C.P.F.; Brancale, A.; Armstrong, J.L.; Simons, C. Novel retinoic acid 4-hydroxylase (CYP26) inhibitors based on a 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-(4-(phenylamino)phenyl)propyl scaffold. Bioorg. Med. Chem. 2012, 20, 4201–4207. [Google Scholar] [CrossRef]
- Bureik, M.; Hübel, K.; Dragan, C.A.; Scher, J.; Becker, H.; Lenz, L.; Bernhardt, R. Development of test systems for the discovery of selective human aldosterone synthase (CYP11B2) and 11β-hydroxylase (CYP11B1) inhibitors. Discovery of a new lead compound for the therapy of congesitve heart failure, myocardial fibrosis and hypertension. Mol. Cell. Endocrinol. 2004, 217, 249–254. [Google Scholar] [CrossRef]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012, 40, D535–D540. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Cavling Arendrup, M.; Patterson, T.F. Multidrug-resistant Candida: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 2017, 216, S445–S451. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.J.; Denisov, I.G. Spectroscopic studies of the cytochrome P450 reaction mechanisms. Biochim. Biophys. Acta 2018, 1866, 178–204. [Google Scholar] [CrossRef] [PubMed]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand access channels in cytochrome P450 enzymes: A review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Madrona, Y.; Kandel, S.; Lampe, J.N.; Nishida, C.R.; Ortiz de Montellano, P.R. Analysis of cytochrome P450 CYP119 ligand-dependent conformational dynamics by two-dimensional NMR and x-ray crystallography. J. Biol. Chem. 2015, 290, 10000–10017. [Google Scholar] [CrossRef] [PubMed]
- Raingeval, C.; Cala, O.; Brion, B.; Le Borgne, M.; Hubbard, R.E.; Krimm, I. 1D NMR WaterLOGSY as an efficient method for fragment-based lead discovery. J. Enzyme Inhib. Med. Chem. 2019, 34, 1218–1225. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Rachakonda, G.; Blobaum, A.L.; Fisher, P.; Melo de Oliveira, G.; França da Silva, C.; De Nazaré, C.; Soeiro, M.; Nes, W.D.; et al. Sterol 14α-demethylase structure-based optimization of drug candidates for human infections with the protozoan Trypanosomatidae. J. Med. Chem. 2018, 61, 10910–10921. [Google Scholar] [CrossRef]
- Debnath, A.; Calvet, C.M.; Jennings, G.; Zhou, W.; Aksenov, A.; Luth, M.R.; Abagyan, R.; Nes, W.D.; McKerrow, J.H.; Podust, L.M. CYP51 is an essential drug target for the treatment of primary amoebic meningoencephalitis (PAM). PLoS Negl. Trop. Dis. 2017, 11, e0006104. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | A | B | C | D | R1 | R2 | X | Y | MIC 1 (µg/mL) C. albicans CA98001 |
| 8a | CH | CH | N | CH | H | H | H | F | 0.03 |
| 8b | CH | CH | N | CH | H | H | H | Cl | 0.02 |
| 8c | CH | CH | N | CH | H | H | H | Br | 0.02 |
| 8d | CH | CH | N | CH | H | H | F | F | 0.00035 |
| 8e | CH | CH | N | CH | H | H | Cl | Cl | 0.062 |
| 11e | CH | CH | N | CH | H | H | H | CF3 | 0.23 |
| 11a | N | CH | N | CH | H | H | H | F | 0.21 |
| 11b | N | CH | N | CH | H | H | H | Cl | 0.024 |
| 11c | N | CH | N | CH | H | H | H | Br | 0.027 |
| 11d | N | CH | N | CH | H | H | F | F | 0.0198 |
| 8g | N | CH | N | CH | H | H | Cl | Cl | 0.000259 |
| (+)-(R)-8g | N | CH | N | CH | H | H | Cl | Cl | 0.023 |
| (−)-(S)-8g | N | CH | N | CH | H | H | Cl | Cl | 0.000256 |
| 8f | N | CH | N | CH | H | H | H | CF3 | 0.009 |
| 11f | N | CH | N | CH | CH3 | H | F | F | 0.022 |
| 11g | N | CH | N | CH | CH3 | H | Cl | Cl | 0.0058 |
| 11h | N | CH | N | CH | H | CH3 | F | Cl | 0.0011 |
| 11i | N | CH | N | CH | H | CH3 | Cl | Cl | 0.007 |
| 11j | N | CH | N | CH | CH3 | CH3 | F | F | 0.157 |
| 11k | N | CH | N | CH | CH3 | CH3 | Cl | Cl | 1.246 |
| 11l | N | N | CH | CH | H | H | Cl | Cl | >100 |
| 11m | N | CH | CH | N | H | H | Cl | Cl | >100 |
| KTC | 0.005 | ||||||||
| FLC | 0.02 | ||||||||
| Compound | ATCC2091 | CA98002 | CA98005 | ATCC6258 | CK98002 | CK98003 | CL98001 | ATCC90018 |
|---|---|---|---|---|---|---|---|---|
| 8a | 0.131 | 0.205 | 0.148 | >33.5 | 12.761 | 28.597 | 0.231 | 2.11 |
| 8b | 0.215 | 0.165 | 0.106 | >35.2 | 2.723 | >35.183 | 0.222 | 1.3 |
| 8c | 0.357 | 0.25 | 0.21 | >39.6 | 5.076 | 36.565 | 0.25 | 1.84 |
| 8d | 0.726 | 0.609 | 0.439 | 3.863 | 5.692 | 5.205 | 0.068 | 0.614 |
| 8e | 0.19 | 0.251 | 0.274 | 1.2 | 1.8 | 0.28 | 0.116 | 0.24 |
| 11e | 2.12 | 0.32 | 0.258 | 22.121 | 18.306 | 16.225 | 0.173 | 2.466 |
| 11a | 0.437 | 0.404 | 0.437 | 19.24 | 21.426 | 18.668 | 0.259 | 2.523 |
| 11b | 0.258 | 0.261 | 0.222 | 2.434 | 12.666 | 2.434 | 0.208 | 2.046 |
| 11c | 0.477 | 0.338 | 0.286 | 1.51 | 2.463 | 2.503 | 0.215 | 2.026 |
| 11d | 0.021 | 0.026 | 0.064 | 2.181 | 1.55 | 24.05 | 0.021 | 0.201 |
| 8g | <0.004 | <0.004 | <0.004 | 0.08 | 2.788 | 3.563 | <0.004 | 0.15 |
| (+)-(R)-8g | 1.007 | 2.75 | 2.707 | 24.398 | 22.074 | 19.363 | 9.682 | >38.726 |
| (−)-(S)-8g | <0.039 | <0.039 | 0.026 | 0.167 | 0.022 | 0.039 | 0.024 | 0.027 |
| 8f | 0.247 | 0.233 | 0.749 | 19.091 | 10.557 | 3.068 | 0.199 | 2 |
| 11f | 0.199 | 0.302 | 0.516 | 17.572 | 7.589 | 5.452 | 0.232 | 2.505 |
| 11g | 0.526 | 0.321 | 0.269 | >40 | >40 | >40 | 0.181 | >40 |
| 11h | 0.136 | 0.033 | 0.265 | 1.4 | 0.81 | 0.7 | 0.023 | 0.309 |
| 11i | 0.026 | 0.044 | 0.03 | 3.01 | 0.803 | 1.685 | 0.025 | 0.245 |
| 11j | 0.256 | 0.382 | 2.409 | 17.97 | 11.472 | 13.92 | 0.256 | 4.015 |
| 11k | 2.741 | 1.121 | 2.99 | 25.293 | 20.226 | >41.532 | 2.866 | 9.76 |
| KTC | 0.008 | 0.009 | 0.006 | 0.079 | 0.37 | 0.43 | 0.007 | 0.064 |
| FLC | 0.07 | 1 | 0.57 | 7.5 | 2 | 6.7 | 0.55 | 0.6 |
| Compound | IC50 1 (µM) |
|---|---|
| 8a | 50 |
| 8b | 37 |
| 8c | 31 |
| 8d | 34 |
| 8e | 31 |
| 11e | 36 |
| 11a | >100 |
| 11b | >100 |
| 11c | 38 |
| 11d | 197.2 |
| 8g | 35.0 2 |
| (+)-(R)-8g | 32 |
| (−)-(S)-8g | 30 |
| 8f | 76 |
| 11f | 190.7 |
| 11g | 157 |
| 11h | 105.4 |
| 11i | 19 |
| 11j | 25 |
| 11k | 97.3 |
| KTC | 69.1 |
| FLC | >100 |
| Compound | CYP19 1 IC50 (µM) (% Inhibition) | CYP17 2 IC50 (µM) (% Inhibition) | CYP26A1 3 IC50 (µM) | CYP11B1 4 IC50 (µM) (Inhibition Effect) | CYP11B2 4 IC50 (µM) (Inhibtion Effect) |
|---|---|---|---|---|---|
| 11d | 3.58 | - | - | - | - |
| (89) | (no inhibition) | ||||
| 8g | - | - | - | - | - |
| (27) | (<10%) | no inhibition | slight inhibition | ||
| (+)-(R)-8g | - | - | 34 | - | - |
| (51) | (<5% inhibition) | - | no inhibition | slight inhibition | |
| (−)-(S)-8g | - | - | 18 | - | - |
| (72) | (<5% inhibition) | - | no inhibition | no inhibition | |
| AG | 29.75 | - | - | - | - |
| fadrozole | 0.030 | not active | - | - | - |
| anastrozole | 0.163 | - | - | - | - |
| letrozole | 0.025 | - | - | - | - |
| liarozole | - | - | 7 | - | - |
| BW19 | - | 0.15 | - | - | - |
| KTC | - | 4.5 | 10 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebouvier, N.; Pagniez, F.; Na, Y.M.; Shi, D.; Pinson, P.; Marchivie, M.; Guillon, J.; Hakki, T.; Bernhardt, R.; Yee, S.W.; et al. Synthesis, Optimization, Antifungal Activity, Selectivity, and CYP51 Binding of New 2-Aryl-3-azolyl-1-indolyl-propan-2-ols. Pharmaceuticals 2020, 13, 186. https://doi.org/10.3390/ph13080186
Lebouvier N, Pagniez F, Na YM, Shi D, Pinson P, Marchivie M, Guillon J, Hakki T, Bernhardt R, Yee SW, et al. Synthesis, Optimization, Antifungal Activity, Selectivity, and CYP51 Binding of New 2-Aryl-3-azolyl-1-indolyl-propan-2-ols. Pharmaceuticals. 2020; 13(8):186. https://doi.org/10.3390/ph13080186
Chicago/Turabian StyleLebouvier, Nicolas, Fabrice Pagniez, Young Min Na, Da Shi, Patricia Pinson, Mathieu Marchivie, Jean Guillon, Tarek Hakki, Rita Bernhardt, Sook Wah Yee, and et al. 2020. "Synthesis, Optimization, Antifungal Activity, Selectivity, and CYP51 Binding of New 2-Aryl-3-azolyl-1-indolyl-propan-2-ols" Pharmaceuticals 13, no. 8: 186. https://doi.org/10.3390/ph13080186
APA StyleLebouvier, N., Pagniez, F., Na, Y. M., Shi, D., Pinson, P., Marchivie, M., Guillon, J., Hakki, T., Bernhardt, R., Yee, S. W., Simons, C., Lézé, M.-P., Hartmann, R. W., Mularoni, A., Le Baut, G., Krimm, I., Abagyan, R., Le Pape, P., & Le Borgne, M. (2020). Synthesis, Optimization, Antifungal Activity, Selectivity, and CYP51 Binding of New 2-Aryl-3-azolyl-1-indolyl-propan-2-ols. Pharmaceuticals, 13(8), 186. https://doi.org/10.3390/ph13080186