Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
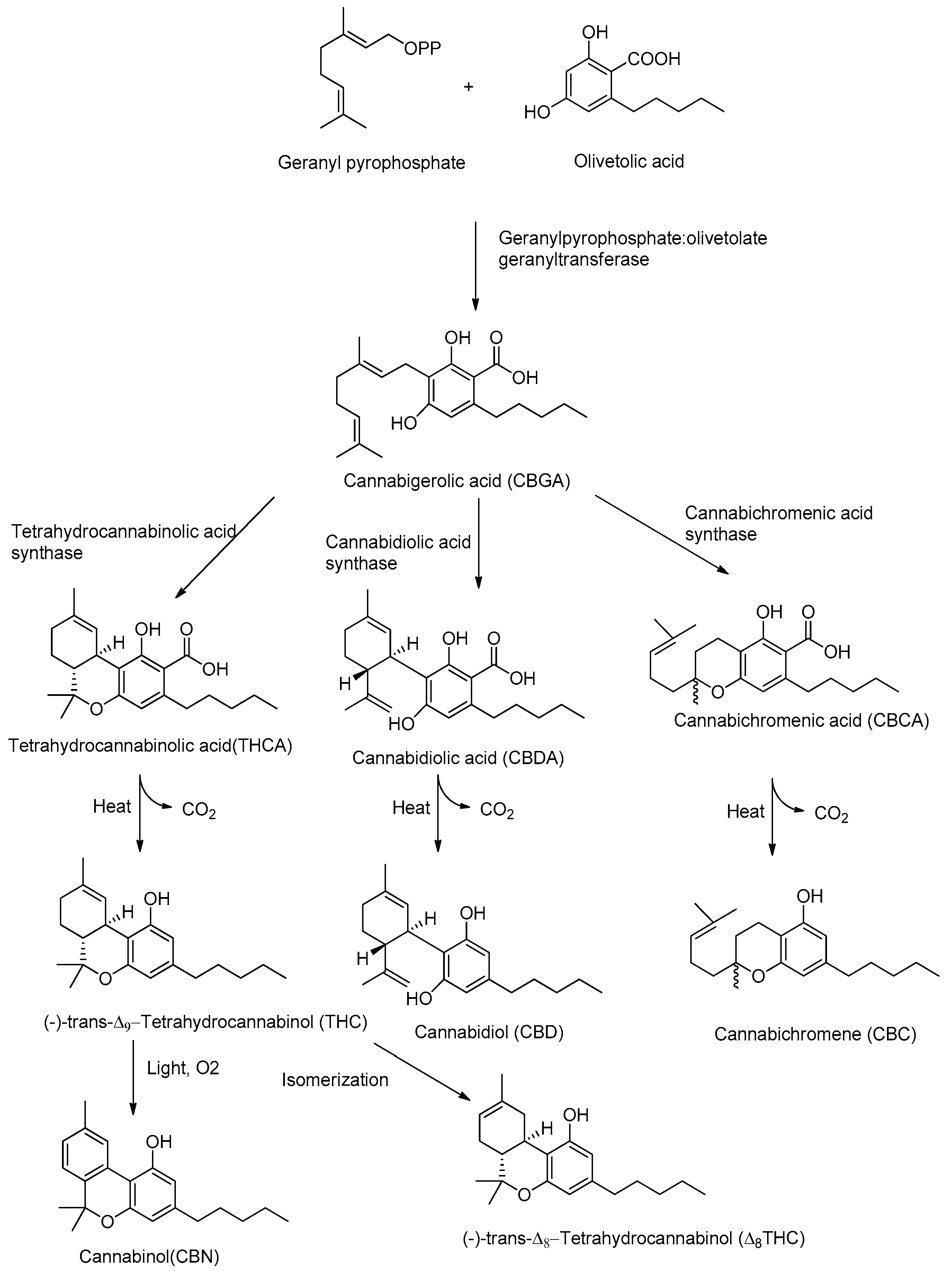
2. Cannabis sativa L. and Its Major Derivatives
3. A Brief Summary of the Endocannabinoid System (ECS)
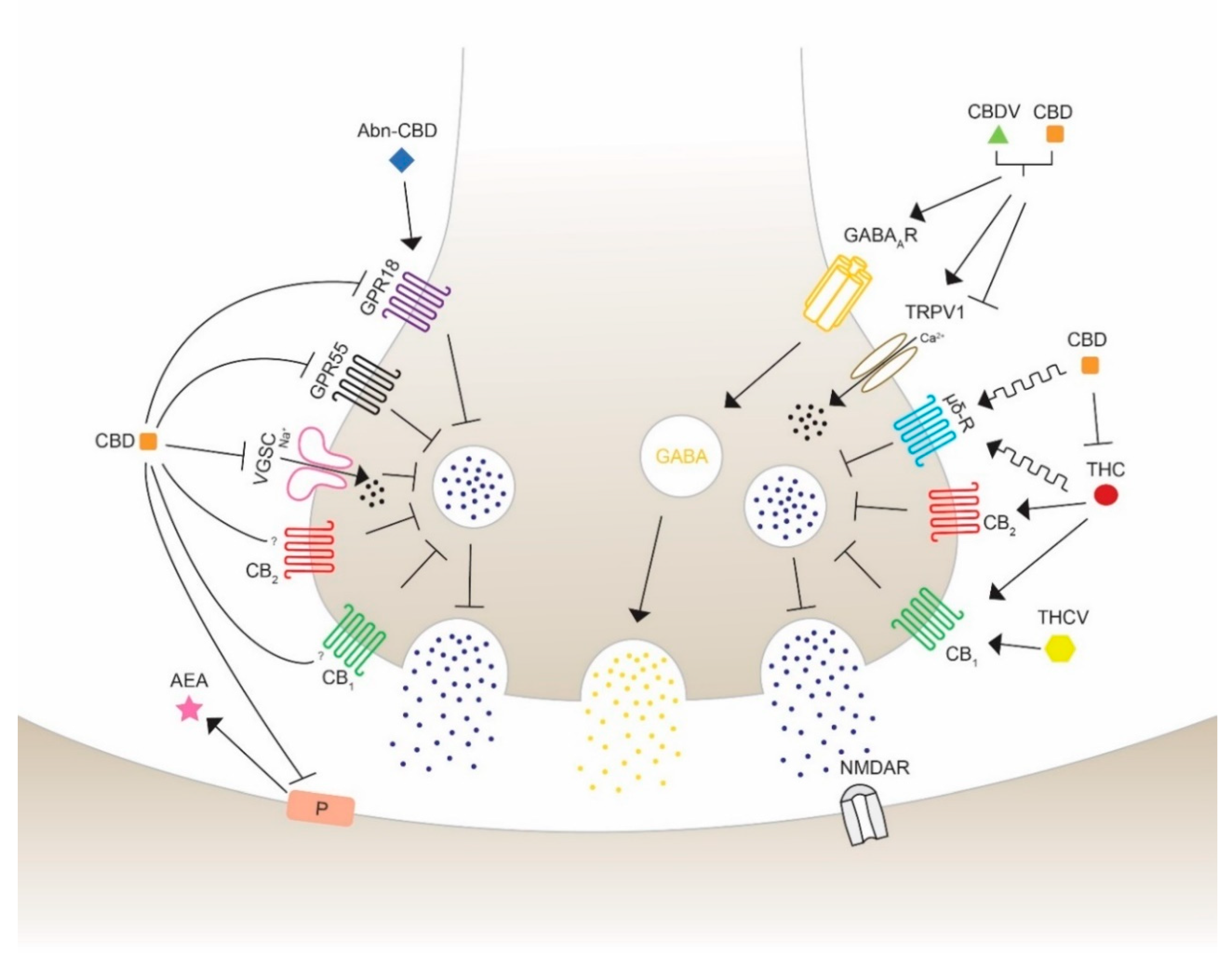
4. Anticonvulsant Effects of Phytocannabinoids on Diverse Targets
4.1. G Protein-Coupled Receptors
4.1.1. Cannabinoid Receptors CB1 & CB2
4.1.2. G Protein-Coupled Receptor 55 (GPR55)
4.1.3. G Protein-Coupled Receptor 18 (GPR18)
4.1.4. Opioid Receptor µ and δ
4.2. Transient Receptor Potential Vanilloid
4.3. GABAA Receptors
4.4. Voltage-Gated Sodium Channel (VGSC)
5. Isolated Phytocannabinoid versus Cannabis Extract: the “Entourage” Effect
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization (WHO). The World Health Report 2004: Changing History; Annex Table 3; World Health Organization: Geneva, Switzerland, 2004; Available online: http://www.who.int/whr/annex/topic/en/annex_3_en.pdf (accessed on 30 March 2020).
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy. Definitions proposed by the International League against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for classification of epilepsies and epileptic syndromes. Epilepsia 1981, 22, 489–501. [Google Scholar] [CrossRef]
- Shorvon, S.D.; Andermann, F.; Guerrini, R. The Causes of Epilepsy. Common and Uncommon Causes in Adults and Children, 1st ed.; Cambridge University Press: Cambridge, UK, 2011; ISBN 978-0521114479. [Google Scholar]
- Matsumoto, H.; Ajmonemarsan, C. Cellular mechanisms in experimental epileptic seizures. Science 1964, 144, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Walther, H.; Lambert, J.D.; Jones, R.S.; Heinemann, U.; Hamon, B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci. Lett. 1986, 69, 156–161. [Google Scholar] [CrossRef]
- George, A.L., Jr. Molecular basis of inherited epilepsy. Arch. Neurol. 2004, 61, 473–478. [Google Scholar] [CrossRef][Green Version]
- Wiechert, P.; Herbst, A. Provocation of cerebral seizures by derangement of the natural balance between glutamic acid and gamma-aminobutyric acid. J. Neurochem. 1966, 13, 59–64. [Google Scholar] [CrossRef]
- Croucher, M.J.; Collins, J.F.; Meldrum, B.S. Anticonvulsant action of excitatory amino acid antagonists. Science 1982, 216, 899–901. [Google Scholar] [CrossRef]
- Lothman, E.W.; Collins, R.C. Seizures and Epilepsy. In Neurobiology of Disease; Pearlman, A.L., Collins, R.C., Eds.; Oxford University Press: New York, NY, USA, 1990; pp. 276–298. [Google Scholar]
- Chisholm, D. Cost-effectiveness of first-line anti-epileptic drug treatments in the developing world: A population-level analysis. Epilepsia 2005, 46, 751–759. [Google Scholar] [CrossRef]
- Appleton, R.; Sweeney, A.; Choonara, I.; Robson, J.; Molyneux, E. Lorazepam versus diazepam in the acute treatment of epileptic seizures and status epilepticus. Dev. Med. Child Neurol. 1995, 37, 682–688. [Google Scholar] [CrossRef]
- Rogawski, M.A. Therapeutic potential of excitatory amino acid antagonists: Channel blockers and 2,3-benzodiazepines. Trends Pharm. Sci. 1993, 14, 325–331. [Google Scholar] [CrossRef]
- Gilad, R.; Izkovitz, N.; Dabby, R.; Rapoport, A.; Sadeh, M.; Weller, B.; Lampl, Y. Treatment of status epilepticus and acute repetitive seizures with i.v. valproic acid vs. phenytoin. Acta Neurol. Scand. 2008, 118, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Mattson, R.H.; Cramer, J.A.; Collins, J.F.; Smith, D.B.; Delgado-Escueta, A.V.; Browne, T.R.; Williamson, P.D.; Treiman, P.D.; McNamara, J.O.; McCutchen, C.B.; et al. Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondarily generalized tonic-clonic seizures. N. Engl. J. Med. 1985, 313, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V. On the Technique of Operations on the Central Nervous System. In Proceedings of the Seventy-Fourth Annual Meeting of the British Medical Association, Toronto, ON, Canada, 21–25 August 1906. [Google Scholar]
- Li, H.L. An archaeological and historical account of cannabis in China. Econ. Bot. 1973, 28, 437–448. [Google Scholar] [CrossRef]
- Mechoulam, R. The pharmacohistory of Cannabis sativa. In Cannabis as Therapeutic Agent; Mechoulam, R., Ed.; CRC Press: Boca Raton, FL, USA, 1986; pp. 1–19. [Google Scholar]
- Small, E.; Cronquist, A. A practical and natural taxonomy of Cannabis. Taxon 1976, 25, 405–435. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol. III. Reduction and cleavage. J. Am. Chem. Soc. 1940, 62, 735–737. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish-I: The structure of cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Jones, P.G.; Falvello, L.R.; Kennard, O.; Sheldrick, G.M.; Mechoulam, R. Cannabidiol. Acta Crystallogr. B 1977, 33, 3211–3214. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, structure and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Pacifico, D.; Miselli, F.; Carboni, A.; Moschella, A.; Mandolino, G. Time course of cannabinoid accumulation and chemotype development during the growth of Cannabis sativa L. Euphytica 2008, 160, 231–240. [Google Scholar] [CrossRef]
- Sirikantaramas, S.; Taura, F.; Tanaka, Y.; Ishikawa, Y.; Morimoto, S.; Shoyama, Y. Tetrahydrocannabinolic Acid Synthase, the Enzyme Controlling Marijuana Psychoactivity, is Secreted into the Storage Cavity of the Glandular Trichomes. Plant Cell Physiol. 2005, 46, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 1–36. [Google Scholar]
- Taura, F.; Sirikantaramas, S.; Shoyama, Y.; Shoyama, Y.; Morimoto, S. Phytocannabinoids in Cannabis sativa: Recent Studies on Biosynthetic Enzymes. Chem. Biodivers. 2007, 4, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Okamoto, K. Distribution of tetrahydrocannabinolic acid in fresh wild cannabis. Experientia 1970, 26, 819–820. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P. Effects of nabilone, a synthetic cannabinoid, on postoperative pain. Can. J. Anaesth. 2006, 53, 769–775. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- Brents, L.K.; Reichard, E.E.; Zimmermann, S.M.; Moran, J.H.; Fantegrossi, W.E.; Prather, P.L. Phase I hydroxylated metabolites of the K2 synthetic cannabinoid JWH-018 retain in vitro and in vivo cannabinoid 1 receptor affinity and activity. PLoS ONE 2011, 6, e21917. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, M.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Johnson, J.R.; Burnell-Nugent, M.; Lossignol, D.; Ganae-Motan, E.D.; Potts, R.; Fallon, M.T. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J. Pain Symptom. Manag. 2010, 39, 167–179. [Google Scholar] [CrossRef]
- Puffenbarger, R.A.; Boothe, A.C.; Cabral, G.A. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia 2000, 29, 58–69. [Google Scholar] [CrossRef]
- Panlilio, L.V.; Goldberg, S.R.; Justinova, Z. Cannabinoid abuse and addiction: Clinical and preclinical findings. Clin. Pharmacol. Ther. 2015, 6, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Jacobus, J.; Tapert, S.F. Effects of Cannabis on the Adolescent Brain. Curr. Pharm. Des. 2014, 13, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, S.; Engström, A.; Allebeck, P.; Rydberg, U. Cannabis and Schizophrenia A Longitudinal Study of Swedish Conscripts. Lancet 1987, 330, 1483–1486. [Google Scholar] [CrossRef]
- Izquierdo, I.; Orsingher, O.A.; Berardi, A.C. Effect of cannabidiol and of other Cannabis sativa compounds on hippocampal seizure discharges. Psychopharmacologia 1973, 28, 95–102. [Google Scholar] [CrossRef]
- Khan, A.A.; Shekh-Ahmad, T.; Khalil, A.; Walker, M.C.; Ali, A.B. Cannabidiol Exerts Antiepileptic Effects by Restoring Hippocampal Interneuron Functions in a Temporal Lobe Epilepsy Model. Br. J. Pharmacol. 2018, 175, 2097–2115. [Google Scholar] [CrossRef]
- Do Val-da Silva, A.R.; Peixoto-Santos, J.E.; Kandratavicius, L.; De Ross, J.B.; Esteves, I.; De Martinis, B.S.; Alves, M.N.R.; Scandiuzzi, R.C.; Hallak, J.E.C.; Zuardi, A.W.; et al. Protective Effects of Cannabidiol Against Seizures and Neuronal Death in a Rat Model of Mesial Temporal Lobe Epilepsy. Front. Pharmacol. 2017, 8, 131. [Google Scholar] [CrossRef]
- Nichols, J.M.; Kaplan, B.L.F. Immune responses regulated by cannabidiol. Cannabis Cannabinoid Res. 2019, 5, 12–31. [Google Scholar] [CrossRef]
- Alexander, S.P.H. Therapeutic potential of cannabis-related drugs. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 157–166. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves First Drug Comprised of an Active Ingredient from Marijuana to Treat Rare, Severe Forms of Epilepsy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-comprised-active-ingredient-derived-marijuana-treat-rare-severe-forms (accessed on 25 June 2018).
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Press, C.A.; Knupp, K.G.; Chapman, K.E. Parental reporting of response to oral cannabis extracts for treatment for refractory epilepsy. Epilepsy Behav. 2015, 45, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Citti, C.; Luongo, L.; Belardo, C.; Maione, S.; Vandelli, M.A.; Forni, F.; Gigli, G.; Laganà, A.; Montone, C.M.; et al. Isolation of a High-Affinity Cannabinoid for the Human CB1 Receptor from a Medicinal Cannabis sativa Variety: Δ9-Tetrahydrocannabutol, the Butyl Homologue of Δ9-Tetrahydrocannabinol. J. Nat. Prod. 2020, 83, 88–98. [Google Scholar] [CrossRef]
- Leighty, E.G.; Fentiman, A.F., Jr.; Foltz, R.L. Long-retained metabolites of Δ9- and Δ8-tetrahydrocannabinols identified as novel fatty acid conjugates. Res. Commun. Chem. Pathol. Pharmacol. 1976, 14, 13–28. [Google Scholar] [PubMed]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic Activity of Cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef] [PubMed]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.A.; Pertwee, R.G.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef]
- Vigli, D.; Cosentino, L.; Raggi, C.; Laviola, G.; Woolley-Roberts, M.; De Filippis, B. Chronic Treatment With the Phytocannabinoid Cannabidivarin (CBDV) Rescues Behavioural Alterations and Brain Atrophy in a Mouse Model of Rett Syndrome. Neuropharmacology 2018, 140, 121–129. [Google Scholar] [CrossRef]
- Cabral, G.A.; Raborn, E.S.; Ferreira, G.A. Phytocannabinoids and the immune system. In Handbook of Cannabis; Pertwee, R., Ed.; Oxford University Press: New York, NY, USA, 2014; pp. 261–279. [Google Scholar]
- Gugliandolo, A.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. In Vitro Model of Neuroinflammation: Efficacy of Cannabigerol, a Non-Psychoactive Cannabinoid. Int. J. Mol. Sci. 2018, 19, 1992. [Google Scholar] [CrossRef]
- Formukong, E.A.; Evans, A.T.; Evans, F.J. Analgesic and anti-inflammatory activity of the constituents of Cannabis sativa. Inflammation 1988, 12, 361–371. [Google Scholar] [CrossRef]
- Wirth, P.W.; Watson, E.S.; ElSohly, M.; Turner, C.E.; Murphy, J.C. Anti-inflammatory properties of cannabichromene. Life Sci. 1980, 26, 1991–1995. [Google Scholar] [CrossRef]
- Maione, S.; Piscitelli, F.; Gatta, L.; Vita, D.; De Petrocellis, L.; Palazzo, E.; de Novellis, V.; Di Marzo, V. Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br. J. Pharmacol. 2011, 162, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Singh, S.; Niyogi, R.G.; Lamont, G.J.; Wang, H.; Lamont, R.J.; Scott, D.A. Marijuana-Derived Cannabinoids Trigger a CB2/PI3K Axis of Suppression of the Innate Response to Oral Pathogens. Front. Immunol. 2019, 10, 2288. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E.; ElSohly, M.A.; Boeren, E.G. Constituents of Cannabis sativa L. XVII. A review of the natural constituents. J. Nat. Prod. 1980, 43, 169–234. [Google Scholar] [CrossRef]
- Wong, H.; Cairns, B.E. Cannabidiol, Cannabinol and Their Combinations Act as Peripheral Analgesics in a Rat Model of Myofascial Pain. Arch. Oral. Biol. 2019, 104, 33–39. [Google Scholar] [CrossRef]
- Monory, K.; Massa, F.; Egertiva, M.; Eder, M.; Blaudzun, H.; Westenbroek, R.; Kelsch, W.; Jacob, W.; Marsch, R.; Ekker, M.; et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 2006, 51, 455–466. [Google Scholar] [CrossRef]
- Mechoulam, R.; Spatz, M.; Shohami, E. Endocannabinoids and neuroprotection. Science Signal. 2002, 2002, re5. [Google Scholar] [CrossRef]
- Moldrich, G.; Wenger, T. Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides 2000, 21, 1735–1742. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commum. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid signaling in the brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.I.; Kunos, G.; Nicoll, R.A. Presynaptic Specificity of Endocannabinoid Signaling in the Hippocampus. Neuron 2001, 31, 453–462. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signaling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of Prostaglandin E2 Ethanolamide from Anandamide by Cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar] [CrossRef]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, R.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed]
- Ludányi, A.; Erőss, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 Cannabinoid Receptor and Related Molecular Elements of the Endocannabinoid System in Epileptic Human Hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef]
- Zareie, P.; Sadegh, M.; Palizvan, M.R.; Moradi-Chameh, H. Anticonvulsive effects of endocannabinoids; an investigation to determine the role of regulatory components of endocannabinoid metabolism in the Pentylenetetrazol induced tonic-clonic seizures. Metab. Brain Dis. 2018, 33, 939–948. [Google Scholar] [CrossRef]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, F.; Zannino, S.; Corte, F.; et al. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef]
- Mikheeva, I.B.; Shubina, L.; Matveeva, N.; Pavlik, L.L.; Kitchigina, V.F. Fatty acid amide hydrolase inhibitor URB597 may protect against kainic acid-induced damage to hippocampal neurons: Dependence on the degree of injury. Epilepsy Res. 2017, 137, 84–94. [Google Scholar] [CrossRef]
- Vilela, L.R.; Gomides, F.L.; David, B.A.; Antunes, M.M.; Diniz, A.B.; de Araújo Moreira, F.; Menezes, G.B. Cannabidiol rescues acute hepatic toxicity and seizure induced by cocaine. Mediat. Inflamm. 2015, 2015, 523418. [Google Scholar] [CrossRef] [PubMed]
- Crippa, J.A.; Hallak, J.E.C.; Abílio, V.C.; Tavares de Lacerda, A.L.; Zuardi, A.W. Cannabidiol and Sodium Nitroprusside: Two Novel Neuromodulatory Pharmacological Interventions to Treat and Prevent Psychosis. CNS Neurol. Disord. Drug Targets 2015, 8, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Katona, I.; Sperlágh, B.; Maglóczky, Z.; Sántha, E.; Köfalvi, A.; Czirják, S.; Mackie, K.; Vizi, E.S.; Freund, T.F. GABAeric interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience 2000, 100, 797–804. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef]
- Katona, I.; Urbán, G.R.; Wallace, M.; Ledent, C.; Jung, K.-M.; Piomelli, D.; Mackie, K.; Freund, T.F. Molecular composition of the endocannabinoid system at glutamatergic synapses. J. Neurosci. 2006, 26, 5628–5637. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- McPartland, J.M.; Glass, M. Functional mapping of cannabinoid receptor homologs in mammals, other vertebrates, and invertebrates. Gene 2003, 312, 297–303. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Atwood, B.K.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Ray, A.; Dittel, B.N. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J. Immunol. 2011, 187, 5720–5732. [Google Scholar] [CrossRef] [PubMed]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Hill, T.D.; Cascio, M.-G.; Romano, B.; Duncam, M.; Pertwee, R.G.; Williams, C.M.; Whalley, B.J.; Hill, A.J. Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br. J. Pharmacol. 2013, 170, 679–692. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Pertwee, R.G. The ring test: A quantitative method for assessing the “cataleptic” effect of cannabis in mice. Br. J. Pharmacol. 1972, 46, 753. [Google Scholar] [CrossRef]
- Hill, A.J.; Weston, S.E.; Jones, N.A.; Smith, I.; Bevan, S.A.; Williamson, E.M.; Stephens, G.J.; Williams, C.M.; Whalley, B.J. Δ⁹-Tetrahydrocannabivarin Suppresses in Vitro Epileptiform and in Vivo Seizure Activity in Adult Rats. Epilepsia 2010, 51, 1522–1532. [Google Scholar] [CrossRef]
- Huffman, J.W.; Liddle, J.; Yu, S.; Aung, M.M.; Abood, M.E.; Wiley, J.L.; Martin, B.R. 3-(1′,1’-Dimethylbutyl)-1-deoxy-Δ8-THC and related compounds: Synthesis of selective ligands for the CB2 receptor. Bioorganic Med. Chem. 1999, 7, 2905–2914. [Google Scholar] [CrossRef]
- Izquierdo, I.; Tannhauser, M. Letter: The effect of cannabidiol on maximal electroshock seizures in rats. J. Pharm. Pharmacol. 1973, 25, 916–917. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Sills, R.C.; Braun, A.G.; Haseman, J.K.; Bucher, J.R. Toxicity and carcinogenicity of Δ9-tetrahydrocannabinol in Fischer rats and B6C3F1 mice. Fundam. Appl. Toxicol. 1996, 30, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, B.K.; Lindamood, C., III; Craig, C.R. Effects of marihuana cannabinoids on seizure activity in cobalt-epileptic rats. Pharmacol. Biochem. Behav. 1982, 16, 573–578. [Google Scholar] [CrossRef]
- Karler, R.; Cely, W.; Turkanis, S.A. The anticonvulsant activity of cannabidiol and cannabinol. Life Sci. 1973, 13, 1527–1531. [Google Scholar] [CrossRef]
- MacLennan, S.J.; Reynen, P.H.; Kwan, J.; Bonhaus, D.W. Evidence for inverse agonism of SR141716A at human recombinant cannabinoid CB1 and CB2 receptors. Br. J. Pharmacol. 1998, 124, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Jones, N.A.; Smith, I.; Hill, C.L.; Williams, C.M.; Stephens, G.J.; Whalley, B.J. Voltage-gated Sodium (NaV) Channel Blockade by Plant Cannabinoids Does Not Confer Anticonvulsant Effects Per Se. Neurosci. Lett. 2014, 566, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.M.; Hatoum, N.S. Neurobehavioral Actions of Cannabichromene and Interactions With Delta 9-tetrahydrocannabinol. Gen. Pharmacol. 1983, 14, 247–252. [Google Scholar] [CrossRef]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Méndez-Díaz, M.; Prospéro-García, O. Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. 2018, 78, 41–50. [Google Scholar] [CrossRef]
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; van der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflamm. 2018, 15, 322. [Google Scholar] [CrossRef] [PubMed]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 Signaling Promotes Proliferation of Pancreatic Cancer Cells and Tumour Growth in Mice, and Its Inhibition Increases Effects of Gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Anavi-Goffer, S.; Baillie, G.; Irving, A.J.; Gertsch, J.; Greig, I.R.; Pertwee, R.G.; Ross, R.A. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J. Biol. Chem. 2012, 287, 91–104. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Gantz, I.; Muraoka, A.; Yang, Y.K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef]
- Wang, X.; Sumida, H.; Cyster, J.G. GPR18 is required for a normal CD8αα intestinal intraepithelial lymphocyte compartment. J. Exp. Med. 2014, 211, 2351–2359. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S17–S129. [Google Scholar] [CrossRef]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef]
- Rajaraman, G.; Simcocks, A.; Hryciw, D.H.; Hutchinson, D.S.; McAinch, A.J. G protein coupled receptor 18: A potential role for endocannabinoid signaling in metabolic dysfunction. Mol. Nutr. Food Res. 2015, 60, 92–102. [Google Scholar] [CrossRef]
- Kohno, M.; Hasegawa, H.; Inoue, A.; Muraoka, M.; Miyazaki, T.; Oka, K.; Yasukawa, M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Verdegaal, E.M.E.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.B.; Joseph, W.R.; Grimsey, N.L.; Glass, M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-Arachidonoyl Glycine. PeerJ 2016, 4, e1835. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, R.; Inoue, K.; Kambe, Y.; Miyata, A. N-arachidonoyl glycine induces macrophage apoptosis via GPR18. Biochem. Biophys. Res. Commun. 2012, 418, 366–371. [Google Scholar] [CrossRef]
- Miller, S.; Leishman, E.; Oehler, O.; Daily, L.; Murataeva, N.; Wager-Miller, J.; Bradshaw, H.; Straiker, A. Evidence for a GPR18 role in diurnal Regulation of intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6419–6426. [Google Scholar] [CrossRef]
- McHugh, D.; Roskowski, D.; Xie, S.; Bradshaw, H.B. Δ9-THC and N-arachidonoyl glycine regulate BV-2 microglial morphology and cytokine release plasticity: Implications for signaling at GPR18. Front. Pharmacol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Neal, C.R., Jr.; Mansour, A.; Reinscheid, R.; Nothacker, H.P.; Civelli, O.; Akil, H.; Watson, S.J., Jr. Opioid Receptor-Like (ORL1) Receptor Distribution in the Rat Central Nervous System: Comparison of ORL1 Receptor mRNA Expression With (125)I-[(14)Tyr]-orphanin FQ Binding. J. Comp. Neurol. 1999, 412, 563–605. [Google Scholar] [CrossRef]
- Lutz, P.E.; Kieffer, B.L. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci. 2013, 36, 195–206. [Google Scholar] [CrossRef]
- Pasternak, G.W. Opiate pharmacology and relief of pain. J. Clin. Oncol. 2014, 32, 1655–1661. [Google Scholar] [CrossRef]
- Kreek, M.J.; Levran, O.; Reed, B.; Schlussman, S.D.; Zhou, Y.; Butelman, E.R. Opiate addiction and cocaine addiction: Underlying molecular neurobiology and genetics. J. Clin. Investig. 2012, 122, 3387–3393. [Google Scholar] [CrossRef] [PubMed]
- Kathmann, M.; Flau, K.; Redmer, A.; Tränkle, C.; Schlicker, E. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Garzón, J. Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 2018, 9, 23373–23389. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Muñoz, M.; Onetti, Y.; Cortés-Montero, E.; Garzón, J.; Sánchez-Blázquez, P. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol. Brain. 2018, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef]
- Sun, F.J.; Guo, W.; Zheng, D.H. Increased expression of TRPV1 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J. Mol. Neurosci. 2013, 49, 182–193. [Google Scholar] [CrossRef]
- Nichol, K.; Stott, C.; Jones, N.; Bazelot, M.; Whalley, B.J. The proposed multimodal mechanism of action of cannabidiol in epilepsy: Modulation of intracellular calcium and adenosine-mediated signaling. In Proceedings of the American Epilepsy Society Annual Meeting, New Orleans, LA, USA, 30 November–4 December 2018; Available online: https://www.aesnet.org/meetings_events/annual_meeting_abstracts/view/554059 (accessed on 9 April 2020).
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef]
- Huizenga, M.H.; Sepulveda-Rodriguez, A.; Forcelli, P.A. Preclinical Safety and Efficacy of Cannabidivarin for Early Life Seizures. Neuropharmacology 2019, 148, 189–198. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allar, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol 2011, 163, 1479. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Orlando, P.; Schiano Moriello, A.; Aviello, G.; Stott, C.; Izzo, A.A.; Di Marzo, V. Cannabinoid actions at TRPV channels: Effects on TRPV3 TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol. 2011, 2, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Frankel, S. gamma-aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63. [Google Scholar] [PubMed]
- Hevers, W.; Lüddens, H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol. Neurobiol. 1998, 18, 35–86. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. GABAA receptor pharmacology. Pharmacol. Ther. 1996, 69, 173–198. [Google Scholar] [CrossRef]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M.; Stiegelbaum, S.A.; Hudspeth, A.J. Principles of Neural Science, 5th ed.; McGraw-Hill Education: New York, NY, USA, 2012; ISBN 9780071390118. [Google Scholar]
- Sieghart, W.; Fuchs, K.; Tretter, V.; Ebert, V.; Jechlinger, M.; Hoger, H.; Adamiker, D. Structure and subunit composition of GABAA receptors. Neurochem. Int. 1999, 34, 379–385. [Google Scholar] [CrossRef]
- Alam, S.; Laughton, D.L.; Walding, A.; Wolstenholme, A.J. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 2006, 43, 1432–1442. [Google Scholar] [CrossRef]
- Jones-Davis, D.M.; Macdonald, R.L. GABA(A) Receptor Function and Pharmacology in Epilepsy and Status Epilepticus. Curr. Opin. Pharmacol. 2003, 3, 12–18. [Google Scholar] [CrossRef]
- Wassef, A.; Baker, J.; Kochan, L.D. GABA and schizophrenia: A review of basic science and clinical studies. J. Chin. Psychopharmacol. 2003, 23, 601–640. [Google Scholar] [CrossRef]
- Sinkkonen, S.T.; Homanics, G.E.; Korpi, E.R. Mouse models of Angelman syndrome, a neurodevelopmental disorder, display different brain regional GABA(A) receptor alterations. Neurosci. Lett. 2003, 340, 205–208. [Google Scholar] [CrossRef]
- Raol, Y.H.; Lund, I.V.; Bandyopadhyay, S.; Zhang, G.; Roberts, D.S.; Wolfe, J.H.; Russek, S.J.; Brooks-Kayal, A.R. Enhancing GABA(A) Receptor Alpha 1 Subunit Levels in Hippocampal Dentate Gyrus Inhibits Epilepsy Development in an Animal Model of Temporal Lobe Epilepsy. J. Neurosci. 2006, 26, 11342–11346. [Google Scholar] [CrossRef]
- Loup, F.; Wieser, H.G.; Yonekawa, Y.; Aguzzi, A.; Fritschy, J.M. Selective Alterations in GABAA Receptor Subtypes in Human Temporal Lobe Epilepsy. J. Neurosci. 2000, 20, 5401–5419. [Google Scholar] [CrossRef] [PubMed]
- Bakas, T.; Van Nieuwenhuijzen, P.; Devenish, S.; McGregor, I.S.; Arnold, J.C.; Chebib, M. The direct actions of cannabidiol and 2-arachidonyl glycerol on GABA-A receptors. Pharmacol. Res. 2017, 119, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Absalom, N.L.; Abelev, S.V.; Low, I.K.; Doohan, P.T.; Martin, L.J.; Chebib, M.; McGregor, I.S.; Arnold, J.C. Coadministered cannabidiol and clobazam: Preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia 2019, 60, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, G.; Cifelli, P.; Roseti, C.; Thom, M.; van Vliet, E.A.; Limatola, C.; Aronica, E.; Palma, E. A novel GABAergic dysfunction in human Dravet syndrome. Epilepsia 2018, 59, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Morano, A.; Cifelli, P.; Nencini, P.; Antonilli, L.; Fattouch, J.; Ruffolo, G.; Roseti, C.; Aronica, E.; Limatola, C.; Di Bonaventura, C.; et al. Cannabis in epilepsy: From clinical practice to basic research focusing on the possible role of cannabidivarin. Epilepsia Open 2016, 1, 145–151. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef]
- Agnew, W.S.; Levinson, S.R.; Brabson, J.S.; Raftery, M.A. Purification of the tetrodotoxin-binding component associated with the voltage-sensitive sodium channel from Electrophorus electricus electroplax membranes. Proc. Natl. Acad. Sci. USA 1978, 75, 2606–2610. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Sun, H.; Liu, X.; Yang, X.; Xiong, H.; Jiang, Y.; Bao, X.; Wang, S.; Yang, Z.; et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. 2014, 36, 676–681. [Google Scholar] [CrossRef]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef]
- Ghovanloo, M.R.; Aimar, K.; Ghadiry-Tavi, R.; Yu, A.; Ruben, P.C. Physiology and pathophysiology of sodium channel inactivation. Curr. Top. Membr. 2015, 78, 479–509. [Google Scholar] [CrossRef]
- Veeramah, K.R.; O’Brien, J.E.; Meisler, M.H.; Cheng, X.; Dib-Hajj, S.D.; Waxman, S.G.; Talwar, D.; Girirajan, S.; Eichler, E.E.; Restifo, L.L.; et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 2012, 90, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; Gasser, A.; Dib-Hajj, S.D.; Waxman, S.G. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp. Neurol. 2010, 224, 362–368. [Google Scholar] [CrossRef]
- Ghovanloo, M.R.; Shuart, N.G.; Mezeyova, J.; Dean, R.A.; Ruben, P.C.; Goodchild, S.J. Inhibitory effects of cannabidiol on voltage-gated sodium currents. J. Biol. Chem. 2018, 293, 16546–16558. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Barbosa, C.; Brustovetsky, T.; Brustovetsky, N.; Cummins, T.R. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 2016, 139, 2164–2181. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Marinol (Dronabinol); U.S. Food and Drug Administration: New Hampshire, MD, USA, 2004.
- Boggs, D.L.; Nguyen, J.D.; Morgenson, D.; Taffe, M.A.; Ranganathan, M. Clinical and Preclinical Evidence for Functional Interactions of Cannabidiol and Δ9-Tetrahydrocannabinol. Neuropsychopharmacology 2018, 43, 142–154. [Google Scholar] [CrossRef]
- Carlini, E.A.; Karniol, I.G.; Renault, P.F.; Schuster, C.R. Effects of marihuana in laboratory animals and man. Brit. J. Pharmacol. 1974, 50, 299–309. [Google Scholar] [CrossRef]
- Russo, E.B.; McPartland, J.M. Cannabis is more than simply Delta(9)-tetrahydrocannabinol. Psychopharmacology 2003, 165, 431–432. [Google Scholar] [CrossRef]
- Ilan, A.B.; Gevins, A.; Coleman, M.; ElSohly, M.A.; de Wit, H. Neurophysiological and subjective profile of marijuana with varying concentrations of cannabinoids. Behav. Pharmacol. 2005, 16, 487–496. [Google Scholar] [CrossRef]
- Morgan, C.J.; Schafer, G.; Freeman, T.P.; Curran, H.V. Impact of cannabidiol on the acute memory and psychotomimetic effects of smoked cannabis: Naturalistic study: Naturalistic study [corrected]. Br. J. Psychiatry. 2010, 4, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Mishima, K.; Hazekawa, M.; Sano, K.; Irie, K.; Orito, K.; Egawa, T.; Kitamura, Y.; Uchida, N.; Nishimura, R.; et al. Cannabidiol potentiates pharmacological effects of Δ9-tetrahydrocannabinol via CB1 receptor-dependent mechanism. Brain Res. 2008, 1188, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Osborne, A.L.; Solowij, N.; Weston-Green, K. A systematic review of the effect of cannabidiol on cognitive function: Relevance to schizophrenia. Neurosci. Biobehav. Rev. 2017, 72, 310–324. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.; Russo, E.B. Cannabis and Cannabis extracts: Greater than the sum of their parts? J. Cannabis. Ther. 2001, 3, 103–132. [Google Scholar] [CrossRef]
- King, K.M.; Myers, A.M.; Soroka-Monzo, A.J.; Tuma, R.F.; Tallarida, R.J.; Walker, E.A.; Ward, S.J. Single and combined effects of Δ9-tetrahydrocannabinol and cannabidiol in a mouse model of chemotherapy-induced neuropathic pain. Br. J. Pharmacol. 2017, 174, 2832–4281. [Google Scholar] [CrossRef] [PubMed]
- Karniol, I.G.; Carlini, E.A. Pharmacological interaction between cannabidiol and delta 9-tetrahydrocannabinol. Psychopharmacologia 1973, 33, 53–70. [Google Scholar] [CrossRef]
- Russo, E.B.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef]
- Englund, A.; Morrison, P.D.; Nottage, J.; Hague, D.; Kane, F.; Bonaccorso, S.; Stone, J.M.; Reichenberg, A.; Brennesien, R.; Holt, D.; et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J. Psychopharmacol. 2013, 27, 19–27. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2005, 172, 4790–4805. [Google Scholar] [CrossRef]
- DAC/NRF 2016/2 C-052. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf (accessed on 12 June 2020).
- Pamplona, F.A.; Rolim da Silva, L.; Coan, A.C. Potential Clinical Benefits of CBD-Rich Cannabis Extracts Over Purified CBD in Treatment-Resistant Epilepsy: Observational Data Meta-analysis. Front. Neurol. 2018, 9, 759. [Google Scholar] [CrossRef]
- Citti, C.; Battisti, U.M.; Braghiroli, D.; Ciccarella, G.; Schmid, M.; Vandelli, M.A.; Cannazza, G. A Metabolomic Approach Applied to a Liquid Chromatography Coupled to High-Resolution Tandem Mass Spectrometry Method (HPLC-ESI-HRMS/MS): Towards the Comprehensive Evaluation of the Chemical Composition of Cannabis Medicinal Extracts. Phytochem. Anal. 2018, 29, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Thiele, E.A.; Marsh, E.D.; French, J.A.; Mazurkiewicz-Beldzinska, M.; Benbadis, S.R.; Joshi, C.; Lyons, P.D.; Taylor, A.; Roberts, C.; Sommerville, K.; et al. Cannabidiol in patients with seizures associated with lennox-gastaut syndrome (GWPCARE4): A randomised double-blind, placebo-controlled Phase 3 trial. Lancet 2018, 391, 1085–1096. [Google Scholar] [CrossRef]
- Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, I.; Maione, S.; Laganà, A.; Capriotti, A.L.; Fiorni, F.; Vandelli, M.A.; et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphirol. Sci. Rep. 2019, 9, 20335. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senn, L.; Cannazza, G.; Biagini, G. Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy. Pharmaceuticals 2020, 13, 174. https://doi.org/10.3390/ph13080174
Senn L, Cannazza G, Biagini G. Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy. Pharmaceuticals. 2020; 13(8):174. https://doi.org/10.3390/ph13080174
Chicago/Turabian StyleSenn, Lara, Giuseppe Cannazza, and Giuseppe Biagini. 2020. "Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy" Pharmaceuticals 13, no. 8: 174. https://doi.org/10.3390/ph13080174
APA StyleSenn, L., Cannazza, G., & Biagini, G. (2020). Receptors and Channels Possibly Mediating the Effects of Phytocannabinoids on Seizures and Epilepsy. Pharmaceuticals, 13(8), 174. https://doi.org/10.3390/ph13080174