Phenotypic Screening in C. elegans as a Tool for the Discovery of New Geroprotective Drugs


Abstract
1. Introduction
2. Target-Based Versus Phenotypic Screening
2.1. The Pros and Cons of Target-Based Versus Phenotypic Screening
2.2. Phenotypic Screening for Geroprotective Compounds
2.3. Target-Based Screens for Geroprotective Drugs
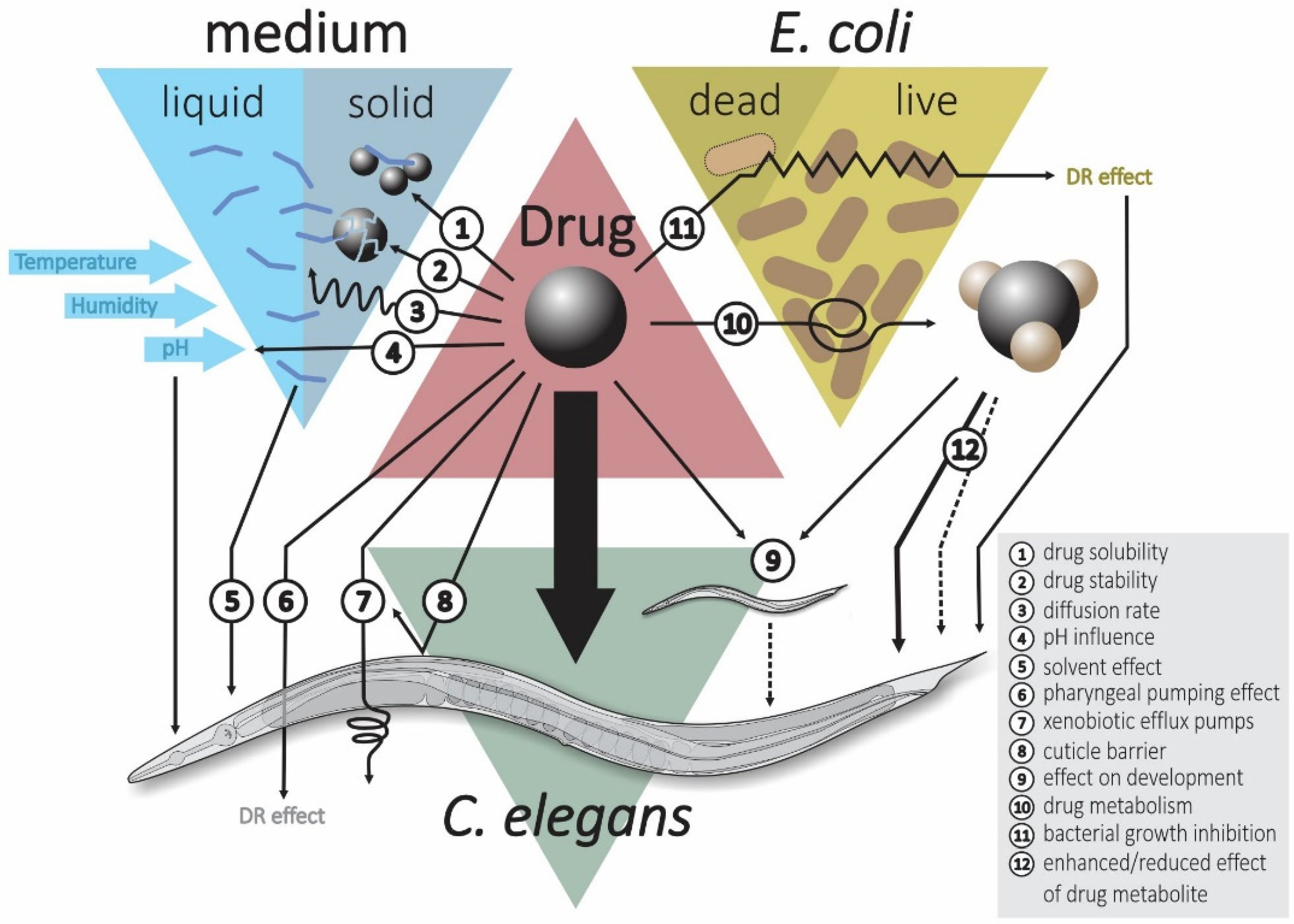
3. Important Considerations for Geroprotective Drug Screening in C. elegans
3.1. Pros and Cons of Drug Screening in C. elegans
3.2. Genetic Background of C. elegans
3.3. Effect of Bacteria on Administered Drugs
3.4. Drug Administration
3.5. Drug Stability
3.6. Age of First Drug Exposure
3.7. Drug Uptake
3.8. Food Intake
3.9. Abiotic Factors
3.10. Reproducibility and Plate-to-Plate Variability
4. Limitations of Manual Lifespan Assays
4.1. Agar Plate-Based Lifespan Assays
4.2. Liquid Culture-Based Lifespan Assays
4.3. Methods to Increase Throughput of Lifespan Assays
4.4. However, Manual Assays also Have Some Strengths
5. Automated Phenotyping and Lifespan Devices
5.1. Microfluidics-Based Platforms
5.2. Solid Medium-Based Platforms
5.3. Stress-Based Platforms
6. Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frieden, T.R. The future of public health. N. Engl. J. Med. 2015, 373, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Omran, A. La transición epidemiológica: Una teoría de la epidemiología del cambio poblacional. Milbank Mem. Fund Q. 1971, 49, 509–538. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, E.M. Lifespan and healthspan: Past, present, and promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef]
- Olshansky, S.; Carnes, B.; Désesquelles, A. Prospects for human longevity. Science 2001, 291, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef]
- Oeppen, J.; Vaupel, J. Broken limits to life expectancy. Science 2002, 296, 1029–1031. [Google Scholar] [CrossRef]
- Federal Interagency Forum on Ageing-Related Statistics. Older Americans 2016. Key Indicators of Well-Being; Federal Interagency Forum on Ageing-Related Statistics: Washington, DC, USA, 2016.
- Kirkland, J.L.; Stout, M.B.; Sierra, F. Resilience in Aging Mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 1407–1414. [Google Scholar] [CrossRef]
- Seals, D.R.; Justice, J.N.; Larocca, T.J. Physiological geroscience: Targeting function to increase healthspan and achieve optimal longevity. J. Physiol. 2016, 594, 2001–2024. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5. [Google Scholar] [CrossRef]
- Kaeberlein, M. Longevity and aging. F1000Prime Rep. 2013, 5, 5. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Pera, A.; Campos, C.; López, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Skirbekk, V.F.; Tyrovolas, S.; Kassebaum, N.J.; Dieleman, J.L. Measuring population ageing: An analysis of the Global Burden of Disease Study 2017. Lancet Public Health 2019, 4, e159–e167. [Google Scholar] [CrossRef]
- Kirkland, J.L. Translating the science of aging into therapeutic interventions. Cold Spring Harb. Perspect. Med. 2016, 6, a025908. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R.; Walford, R. The Retardation of Aging and Disease by Dietary Restriction; Thomas: Springfield, IL, USA, 1988. [Google Scholar]
- Spindler, S.R. Caloric restriction: From soup to nuts. Ageing Res. Rev. 2010, 9, 324–353. [Google Scholar] [CrossRef]
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span-from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Diaz-Ruiz, A.; Hauser, D.; Martinez-Romero, J.; Ferrucci, L.; Bernier, M.; de Cabo, R. The road ahead for health and lifespan interventions. Ageing Res. Rev. 2020, 59, 101037. [Google Scholar] [CrossRef]
- Partridge, L.; Fuentealba, M.; Kennedy, B.K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef]
- Singh, P.P.; Demmitt, B.A.; Nath, R.D.; Brunet, A. The Genetics of Aging: A Vertebrate Perspective. Cell 2019, 177, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Wang, A.M.; Bennett, C.F.; Kaeberlein, M. Biochemical genetic pathways that modulate aging in multiple species. Cold Spring Harb. Perspect. Med. 2015, 5, a025114. [Google Scholar] [CrossRef] [PubMed]
- Van Heemst, D. Insulin, IGF-1 and longevity. Aging Dis. 2010, 1, 147–157. [Google Scholar]
- Austad, S.N. The Geroscience Hypothesis: Is It Possible to Change the Rate of Aging. In Advances in Geroscience; Sierra, F., Kohanski, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–36. ISBN 978-3-319-23246-1. [Google Scholar]
- Sierra, F.; Kohanski, R. Geroscience and the trans-NIH Geroscience Interest Group, GSIG. GeroScience 2017, 39, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.W.; Polotsky, A.J. Can we live longer by eating less? A review of caloric restriction and longevity. Maturitas 2012, 71, 315–319. [Google Scholar] [CrossRef]
- Flanagan, E.; Most, J.; Mey, J.; Redman, L. Calorie Restriction and Aging in Humans. Annu. Rev. Nutr. 2020, 40. [Google Scholar] [CrossRef]
- Vaiserman, A.; De Falco, E.; Koliada, A.; Maslova, O.; Balistreri, C.R. Anti-ageing gene therapy: Not so far away? Ageing Res. Rev. 2019, 56, 100977. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359. [Google Scholar] [CrossRef]
- Bulterijs, S. Metformin as a geroprotector. Rejuvenation Res. 2011, 14. [Google Scholar] [CrossRef]
- Moskalev, A. Is anti-ageing drug discovery becoming a reality? Expert. Opin. Drug Discov. 2020, 15, 135–138. [Google Scholar] [CrossRef]
- Vaiserman, A.; Lushchak, O. Implementation of longevity-promoting supplements and medications in public health practice: Achievements, challenges and future perspectives. J. Transl. Med. 2017, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Piskovatska, V.; Strilbytska, O.; Koliada, A.; Vaiserman, A.; Lushchak, O. Health Benefits of Anti-aging Drugs. In Biochemistry and Cell Biology of Ageing: Part II Clinical Science; Harris, J., Korolchuk, V., Eds.; Springer Nature: Singapore, 2019; pp. 339–392. ISBN 978-981-13-3680-5. [Google Scholar]
- De Grey, A.D.N.J. TAME: A Genuinely Good Use of 75 Million Dollars. Rejuvenation Res. 2019, 22, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Al-Ali, H. The evolution of drug discovery: From phenotypes to targets, and back. Medchemcomm 2016, 7, 788–798. [Google Scholar] [CrossRef]
- Bosch, F.; Rosich, L. The contributions of paul ehrlich to pharmacology: A tribute on the occasion of the centenary of his nobel prize. Pharmacology 2008, 82, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Riethmiller, S. From atoxyl to salvarsan: Searching for the magic bullet. Chemotherapy 2005, 51, 234–242. [Google Scholar] [CrossRef]
- Sepkowitz, K. One Hundred Years of Salvarsan. N. Engl. J. Med. 2011, 365, 291–293. [Google Scholar] [CrossRef]
- Croston, G.E. The utility of target-based discovery. Expert Opin. Drug Discov. 2017, 12, 427–429. [Google Scholar] [CrossRef]
- Black, J. Drugs from Emasculated Hormones: The Principle of Syntopic Antagonism. Biosci Rep 2004, 24, 302–322. [Google Scholar] [CrossRef]
- Black, J. A Life in New Drug Research. Br. J. Pharmacol. 2010, 160, S15–S25. [Google Scholar] [CrossRef]
- Casy, A.; Parfitt, R. Opioid Analgesics: Chemistry and Receptors; Springer: New York, NY, USA, 1986; ISBN 978-1-4899-0585-7. [Google Scholar]
- Eggert, U.S. The why and how of phenotypic small-molecule screens. Nat. Chem. Biol. 2013, 9, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- Hertzberg, R.P.; Pope, A.J. High-throughput screening: New technology for the 21st century. Curr. Opin. Chem. Biol. 2000, 4, 445–451. [Google Scholar] [CrossRef]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int. J. Mol. Sci. 2012, 13, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A.; Williams, J.A. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712–713. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Loria, P.; Pregel, M.; Stanton, R.; Kitching, L.; Nocka, K.; Doyonnas, R.; Steppan, C.; Gilbert, A.; Schroeter, T.; et al. Developing predictive assays: The phenotypic screening “rule of 3”. Sci. Transl. Med. 2015, 7, 293ps15. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.; Ellis, L. Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Mencher, S.K.; Wang, L.G. Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol. 2005, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Sheffer, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef]
- Childers, W.E.; Elokely, K.M.; Abou-Gharbia, M. The Resurrection of Phenotypic Drug Discovery. ACS Med. Chem. Lett. 2020, 3–11. [Google Scholar] [CrossRef]
- Giacomotto, J.; Ségalat, L. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 2010, 160, 204–216. [Google Scholar] [CrossRef]
- Szabo, M.; Akusjärvi, S.S.; Saxena, A.; Liu, J.; Chandrasekar, G.; Kitambi, S.S. Cell and small animal models for phenotypic drug discovery. Drug Des. Dev. Ther. 2017, 11, 1957–1967. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Khoshmanesh, K.; Akagi, J.; Williams, D.E.; Cooper, J.M. Wormometry-on-a-chip: Innovative technologies for in situ analysis of small multicellular organisms. Cytom. Part A 2011, 79A, 799–813. [Google Scholar] [CrossRef]
- Kapetanovic, I. Overview of Current Drug Discovery and Development with an Eye Towards the Future. In Drug Discovery and Development: Present and Future; IM, K., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol.-Cell Physiol. 2010, 298, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, A.; Bambade, T.; Au, C.; Tullet, J.M.A.; Monkhouse, J.; Dang, H.; Cetnar, K.; Chan, B.; Cabreiro, F.; Gems, D. New label-free automated survival assays reveal unexpected stress resistance patterns during C. elegans aging. Aging Cell 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Miller, E.A.; Morimoto, R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 2009, 106, 14914–14919. [Google Scholar] [CrossRef]
- Manière, X.; Krisko, A.; Pellay, F.X.; Di Meglio, J.M.; Hersen, P.; Matic, I. High transcript levels of heat-shock genes are associated with shorter lifespan of Caenorhabditis elegans. Exp. Gerontol. 2014, 60, 12–17. [Google Scholar] [CrossRef]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior proteome stability in the longest lived animal. Age 2014, 36, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Doonan, R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef]
- Guerville, F.; De Souto Barreto, P.; Ader, I.; Andrieu, S.; Casteilla, L.; Dray, C.; Fazilleau, N.; Guyonnet, S.; Langin, D.; Liblau, R.; et al. Revisiting the Hallmarks of Aging to Identify Markers of Biological Age. J. Prev. Alzheimers Dis. 2020, 7, 56–64. [Google Scholar] [CrossRef]
- Maglioni, S.; Arsalan, N.; Franchi, L.; Hurd, A.; Opipari, A.W.; Glick, G.D.; Ventura, N. An automated phenotype-based microscopy screen to identify pro-longevity interventions acting through mitochondria in C. elegans. Biochim. Biophys. Acta-Bioenerg. 2015, 1847, 1469–1478. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Recalde, U.; Lorenzo-Gómez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Domínguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 2019, 45, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhai, Y.; Cregg, J.; Ang, K.K.H.; Arkin, M.; Kenyon, C. Stress resistance screen in a human primary cell line identifies small molecules that affect aging pathways and extend Caenorhabditis elegans’ lifespan. G3 Genes Genomes Genet. 2020, 10, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Lujan, C.; Tyler, E.J.; Ecker, S.; Webster, A.P.; Stead, E.R.; Victoria, E.; Miguel, M.; Milligan, D.; Garbe, J.C.; Stampfer, M.R.; et al. A CellAgeClock for expedited discovery of anti-ageing compounds. bioRxiv 2020. [Google Scholar] [CrossRef]
- Myers, A.; Lithgow, G.J. Drugs that target ageing: How do we discover them? Expert Opin. Drug Discov. 2019, 14, 541–548. [Google Scholar] [CrossRef]
- Bansal, A.; Zhu, L.J.; Yen, K.; Tissenbaum, H.A. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc. Natl. Acad. Sci. USA 2015, 112, E277–E286. [Google Scholar] [CrossRef]
- Fischer, K.E.; Hoffman, J.M.; Sloane, L.B.; Gelfond, J.A.L.; Soto, V.Y.; Richardson, A.G.; Austad, S.N. A cross-sectional study of male and female C57BL/6Nia mice suggests lifespan and healthspan are not necessarily correlated. Aging 2016, 8, 2370–2391. [Google Scholar] [CrossRef]
- Rollins, J.A.; Howard, A.C.; Dobbins, S.K.; Washburn, E.H.; Rogers, A.N. Assessing health Span in Caenorhabditis elegans: Lessons from short-lived mutants. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 473–480. [Google Scholar] [CrossRef]
- Zhang, W.B.; Sinha, D.B.; Pittman, W.E.; Hvatum, E.; Stroustrup, N.; Pincus, Z. Extended Twilight among Isogenic C. elegans Causes a Disproportionate Scaling between Lifespan and Health. Cell Syst. 2016, 3, 333–345.e4. [Google Scholar] [CrossRef]
- Mattiazzi Usaj, M.; Styles, E.B.; Verster, A.J.; Friesen, H.; Boone, C.; Andrews, B.J. High-Content Screening for Quantitative Cell Biology. Trends Cell Biol. 2016, 26, 598–611. [Google Scholar] [CrossRef]
- Dorval, T.; Chanrion, B.; Cattin, M.E.; Stephan, J.P. Filling the drug discovery gap: Is high-content screening the missing link? Curr. Opin. Pharmacol. 2018, 42, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Partridge, L. Genetics of Longevity in Model Organisms: Debates and Paradigm Shifts. Annu. Rev. Physiol. 2013, 75, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Pan, H.; Finkel, T. Key proteins and pathways that regulate lifespan. J. Biol. Chem. 2017, 292, 6452–6460. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; Lamming, D.W.; Pentelute, B.L.; et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiang, Y. mTOR Inhibitors at a Glance. Mol. Cell. Pharmacol. 2015, 7, 15–20. [Google Scholar]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD + Metabolome Is Dysregulated in “normal” Aging. Rejuvenation Res. 2019, 22, 121–130. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, M.R.; Chellappa, K.; Baur, J.A. Age-related NAD+ decline. Exp. Gerontol. 2020, 134, 110888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; Amico, D.D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. Supplementary Materials for enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Young, E.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-enamorado, I.; Di Francesco, A.; et al. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 2018, 27, 667–676. [Google Scholar] [CrossRef]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Becherer, J.D.; Boros, E.E.; Carpenter, T.Y.; Cowan, D.J.; Deaton, D.N.; Haffner, C.D.; Jeune, M.R.; Kaldor, I.W.; Poole, J.C.; Preugschat, F.; et al. Discovery of 4-Amino-8-quinoline Carboxamides as Novel, Submicromolar Inhibitors of NAD-Hydrolyzing Enzyme CD38. J. Med. Chem. 2015, 58, 7021–7056. [Google Scholar] [CrossRef]
- Haffner, C.D.; Becherer, J.D.; Boros, E.E.; Cadilla, R.; Carpenter, T.; Cowan, D.; Deaton, D.N.; Guo, Y.; Harrington, W.; Henke, B.R.; et al. Discovery, synthesis, and biological evaluation of thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J. Med. Chem. 2015, 58, 3548–3571. [Google Scholar] [CrossRef] [PubMed]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef] [PubMed]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Academic Press: London, UK, 2017. [Google Scholar]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar]
- Boulin, T.; Hobert, O. From genes to function: The C. elegans genetic toolbox. WIREs Dev. Biol. 2012, 1, 114–137. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; de Jong, L.; Tavernarakis, N. Caenorhabditis elegans: A versatile platform for drug discovery. Biotechnol. J. 2006, 1, 1405–1418. [Google Scholar] [CrossRef]
- Hekimi, S. Crossroads of aging in the nematode Caenorhabditis elegans. In The Molecular Genetics of Aging; Hekimi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 81–112. [Google Scholar]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. Ortholist 2: A new comparative genomic analysis of human and Caenorhabditis elegans genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef]
- Silverman, G.A.; Luke, C.J.; Bhatia, S.R.; Long, O.S.; Vetica, A.C.; Perlmutter, D.H.; Pak, S.C. Modeling molecular and cellular aspects of human disease using the nematode Caenorhabditis elegans. Pediatr. Res. 2009, 65, 10–18. [Google Scholar] [CrossRef]
- Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.J.V. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Youngman, M.J.; Rogers, Z.N.; Kim, D.H. A decline in p38 MAPK signaling underlies immunosenescence in Caenorhabditis elegans. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human β-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Kawai, K.; Katsuno, M.; Huang, Z.; Jiang, Y.M.; Iguchi, Y.; Kobayashi, K.; Kimata, T.; Waza, M.; Tanaka, F.; et al. Dnc-1/dynactin 1 Knockdown Disrupts Transport of Autophagosomes and Induces Motor Neuron Degeneration. PLoS ONE 2013, 8, e54511. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.S.; Benian, G.M. Muscular dystrophy: The worm turns to genetic disease. Curr. Biol. 2000, 10, 795–797. [Google Scholar] [CrossRef]
- Gieseler, K.; Grisoni, K.; Ségalat, L. Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr. Biol. 2000, 10, 1092–1097. [Google Scholar] [CrossRef]
- Tsuda, Y.; Yamanaka, K.; Toyoshima, R.; Ueda, M.; Masuda, T.; Misumi, Y.; Ogura, T.; Ando, Y. Development of transgenic Caenorhabditis elegans expressing human transthyretin as a model for drug screening. Sci. Rep. 2018, 8, 17884. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent window into biology: A primer on Caenorhabditis elegans. WormBook 2015, 1–31. [Google Scholar] [CrossRef]
- Hunt, P.R. The C. elegans model in toxicity testing. J. Appl. Toxicol. 2017, 37, 50–59. [Google Scholar] [CrossRef]
- Burns, A.R.; Wallace, I.M.; Wildenhain, J.; Tyers, M.; Giaever, G.; Bader, G.D.; Nislow, C.; Cutler, S.R.; Roy, P.J. A predictive model for drug bioaccumulation and bioactivity in Caenorhabditis elegans. Nat. Chem. Biol. 2010, 6, 549–557. [Google Scholar] [CrossRef] [PubMed]
- McColl, G.; Killilea, D.W.; Hubbard, A.E.; Vantipalli, M.C.; Melov, S.; Lithgow, G.J. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 2008, 283, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Tam, Z.Y.; Gruber, J.; Ng, L.F.; Halliwell, B.; Gunawan, R. Effects of lithium on age-related decline in mitochondrial turnover and function in Caenorhabditis elegans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef]
- Anisimov, V.N. Effect of Buformin and Diphenylhydantoin on the Life Span, Estrous Function and Spontaneous Tumor Incidence in Rats. Vopr. Onkol. 1980, 26, 42–48. [Google Scholar]
- Anisimov, V.N. Effect of phenformin on life span, estrus function and spontaneous tumor incidence in rats. Farm. Toksikol 1982, 45, 127. [Google Scholar]
- Dilman, V.; Anisimov, V.N. Effect of Treatment With Phenformin, Diphenylhydantoin or L-dopa on Life Span and Tumour Incidence in C3H/Sn Mice. Gerontology 1980, 26, 241–246. [Google Scholar] [CrossRef]
- Bakaev, V. Effect of 1-butylbiguanide hydrochloride on the longevity in the nematoda Caenorhabditis elegans. Biogerontology 2002, 3, 23–24. [Google Scholar]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.E.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, 2501–2509. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Min, K.J. Drosophila melanogaster as a model system in the study of pharmacological interventions in aging. Transl. Med. Aging 2019, 3, 98–103. [Google Scholar] [CrossRef]
- Terzibasi, E.; Valenzano, D.R.; Cellerino, A. The short-lived fish Nothobranchius furzeri as a new model system for aging studies. Exp. Gerontol. 2007, 42, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Mack, H.I.D.; Heimbucher, T.; Murphy, C.T. The nematode Caenorhabditis elegans as a model for aging research. Drug Discov. Today Dis. Model. 2018, 27, 3–13. [Google Scholar] [CrossRef]
- McGhee, J.D. The C. elegans intestine. WormBook 2007, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Ausubel, F.M. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 2012, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Raices, M.; Maruyama, H.; Dillin, A.; Kariseder, J. Uncoupling of longevity and telomere length in C. elegans. PLoS Genet. 2005, 1, e30. [Google Scholar] [CrossRef]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef]
- Dmitrieva, N.I.; Burg, M.B. High NaCl promotes cellular senescence. Cell Cycle 2007, 6, 3108–3113. [Google Scholar] [CrossRef]
- Takauji, Y.; Wada, T.; Takeda, A.; Kudo, I.; Miki, K.; Fujii, M.; Ayusawa, D. Restriction of protein synthesis abolishes senescence features at cellular and organismal levels. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.J.; Grainger, D.W. Senescence and quiescence induced compromised function in cultured macrophages. Biomaterials 2012, 33, 7497–7507. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Avelar, R.A.; Ortega, J.G.; Tacutu, R.; Tyler, E.J.; Bennett, D.; Binetti, P.; Budovsky, A.; Chatsirisupachai, K.; Johnson, E.; Murray, A.; et al. A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol. 2020, 21, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.V.; Mani, K.; Fay, D.S. Cancer models in Caenorhabditis elegans. Dev. Dyn. 2010, 239, 1413–1448. [Google Scholar] [CrossRef]
- Johnson, T.E. Advantages and disadvantages of Caenorhabditis elegans for aging research. Exp. Gerontol. 2003, 38, 1329–1332. [Google Scholar] [CrossRef]
- Christensen, M.; Estevez, A.; Yin, X.; Fox, R.; Morrison, R.; Mcdonnell, M.; Gleason, C.; Iii, D.M.M.; Strange, K. Neurotechnique for Functional Analysis of C. elegans Neurons and Muscle Cells. Neuron 2002, 33, 503–514. [Google Scholar] [CrossRef]
- Sangaletti, R.; Bianchi, L. A method for culturing embryonic C. elegans cells. J. Vis. Exp. 2013, e50649. [Google Scholar] [CrossRef]
- Zhang, S.; Banerjee, D.; Kuhn, J.R. Isolation and culture of larval cells from C. elegans. PLoS ONE 2011, 6, e19505. [Google Scholar] [CrossRef]
- Kroetz, M.B.; Zarkower, D. Cell-specific mRNA profiling of the Caenorhabditis elegans somatic gonadal precursor cells identifies suites of sex-biased and gonad-enriched transcripts. G3 Genes Genomes Genet. 2015, 5, 2831–2841. [Google Scholar] [CrossRef]
- Taylor, S.; Santpere, G.; Reilly, M.; Glenwinkel, L.; Poff, A.; McWhirter, R.; Xu, C.; Weinreb, A.; Basavaraju, M.; Cook, S.; et al. Expression profiling of the mature C. elegans nervous system by single-cell RNA-Sequencing. bioRxiv 2019, 737577. [Google Scholar] [CrossRef]
- Hadwiger, G.; Dour, S.; Arur, S.; Fox, P.; Nonet, M.L. A monoclonal antibody Toolkit for C. elegans. PLoS ONE 2010, 5, e10161. [Google Scholar] [CrossRef] [PubMed]
- Frézal, L.; Félix, M.A.C. C. elegans outside the Petri dish. Elife 2015, 4, 1–14. [Google Scholar] [CrossRef]
- Schulenburg, H.; Félix, M.A. The natural biotic environment of Caenorhabditis elegans. Genetics 2017, 206, 55–86. [Google Scholar] [CrossRef] [PubMed]
- Galimov, E.R.; Gems, D. Shorter life and reduced fecundity can increase colony fitness in virtual Caenorhabditis elegans. Aging Cell 2020, 19, 1–15. [Google Scholar] [CrossRef]
- Galimov, E.R.; Lohr, J.N.; Gems, D. When and How Can Death Be an Adaptation? Biochemistry (Moscow) 2019, 84, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.N.; Galimov, E.R.; Gems, D. Does senescence promote fitness in Caenorhabditis elegans by causing death? Ageing Res. Rev. 2019, 50, 58–71. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Poole, R.J.; Gems, D. A fln-2 mutation affects lethal pathology and lifespan in C. elegans. Nat. Commun. 2019, 10, 5087. [Google Scholar] [CrossRef]
- Lucanic, M.; Plummer, W.T.; Harke, J.; Lucanic, M.; Chen, E.; Bhaumik, D.; Harinath, G.; Coleman-Hulbert, A.; Dumas, K.; Onken, B.; et al. Standardized Protocols from the Caenorhabditis Intervention Testing Program 2013-2016: Conditions and Assays used for Quantifying the Development, Fertility and Lifespan of Hermaphroditic Caenorhabditis Strains. Protoc. Exch. 2017, 1–30. [Google Scholar] [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. WormBook 2006, 1–11. [Google Scholar] [CrossRef]
- Zečić, A.; Dhondt, I.; Braeckman, B.P. The nutritional requirements of Caenorhabditis elegans. Genes Nutr. 2019, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Diot, C.; Garcia-Gonzalez, A.P.; Walhout, A.J.M. C. elegans and its bacterial diet: An interspecies model to explore the effects of microbiota on drug response. Drug Discov. Today Dis. Model. 2018, 28, 21–26. [Google Scholar] [CrossRef]
- Gruber, J.; Ng, L.F.; Poovathingal, S.K.; Halliwell, B. Deceptively simple but simply deceptive-Caenorhabditis elegans lifespan studies: Considerations for aging and antioxidant effects. FEBS Lett. 2009, 583, 3377–3387. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Ding, A.J.; Li, G.P.; Wu, G.S.; Luo, H.R. Drug Absorption Efficiency in Caenorhbditis elegans Delivered by Different Methods. PLoS ONE 2013, 8, e56877. [Google Scholar] [CrossRef]
- Gruber, J.; Soon, Y.T.; Halliwell, B. Evidence for a trade-off between survival and fitness caused by resveratrol treatment of Caenorhabditis elegans. Ann. N. Y. Acad. Sci. 2007, 1100, 530–542. [Google Scholar] [CrossRef]
- Schaffer, S.; Gruber, J.; Ng, L.F.; Fong, S.; Wong, Y.T.; Tang, S.Y.; Halliwell, B. The effect of dichloroacetate on health- and lifespan in C. elegans. Biogerontology 2011, 12, 195–209. [Google Scholar] [CrossRef]
- Huang, X.B.; Mu, X.H.; Wan, Q.L.; He, X.M.; Wu, G.S.; Luo, H.R. Aspirin increases metabolism through germline signalling to extend the lifespan of Caenorhabditis elegans. PLoS ONE 2017, 12, e0184027. [Google Scholar] [CrossRef]
- Qi, B.; Kniazeva, M.; Han, M. A vitamin-B2-sensing mechanism that regulates gut protease activity to impact animal’s food behavior and growth. Elife 2017, 6, e26243. [Google Scholar] [CrossRef]
- Gems, D.; Riddle, D.L. Genetic, behavioral and environmental determinants of male longevity in Caenorhabditis elegans. Genetics 2000, 154, 1597–1610. [Google Scholar]
- Saul, N.; Pietsch, K.; Menzel, R.; Stürzenbaum, S.R.; Steinberg, C.E.W. Catechin induced longevity in C. elegans: From key regulator genes to disposable soma. Mech. Ageing Dev. 2009, 130, 477–486. [Google Scholar] [CrossRef]
- Win, M.T.T.; Yamamoto, Y.; Munesue, S.; Han, D.; Harada, S.I.; Yamamoto, H. Validated liquid culture monitoring system for lifespan extension of Caenorhabditis elegans through genetic and dietary manipulations. Aging Dis. 2013, 4, 178–185. [Google Scholar] [PubMed]
- Wilson, I.; Nicholson, J. The role of gut microbiota in drug response. Curr. Pharm. Des. 2009, 15, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Yehye, W.A.; Abd Rahman, N.; Tan, M.W.; Nathan, S. Discovery of potential anti-infectives against Staphylococcus aureus using a Caenorhabditis elegans infection model. BMC Complement. Altern. Med. 2014, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.K.; Leroi, A.M.; Bundy, J.G. Fluorodeoxyuridine affects the identification of metabolic responses to daf-2 status in Caenorhabditis elegans. Mech. Ageing Dev. 2012, 133, 46–49. [Google Scholar] [CrossRef]
- Mitchell, D.; Stiles, J.; Santelli, J.; Sanadi, D. Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. J. Gerontol. 1979, 34, 28–36. [Google Scholar] [CrossRef]
- Bharill, P.; Ayyadevara, S.; Alla, R.; Shmookler Reis, R.J. Extreme depletion of PIP3 accompanies the increased life span and stress tolerance of PI3K-null C. elegans mutants. Front. Genet. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Lucanic, M.; Plummer, W.T.; Chen, E.; Harke, J.; Foulger, A.C.; Onken, B.; Coleman-Hulbert, A.L.; Dumas, K.J.; Guo, S.; Johnson, E.; et al. Impact of genetic background and experimental reproducibility on identifying chemical compounds with robust longevity effects. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Clay, K.; Petrascheck, M. Design and analysis of pharmacological studies in aging. In Aging: Methods and Protocols; Springer: New York, NY, USA, 2020; pp. 77–89. ISBN 978-1-0716-0592-9. [Google Scholar]
- Wang, X.; Wang, X.; Li, L.; Wang, D. Lifespan extension in Caenorhabditis elegans by DMSO is dependent on sir-2.1 and daf-16. Biochem. Biophys. Res. Commun. 2010, 400, 613–618. [Google Scholar] [CrossRef]
- Frankowski, H.; Alavez, S.; Spilman, P.; Mark, K.A.; Nelson, J.D.; Mollahan, P.; Rao, R.V.; Chen, S.F.; Lithgow, G.J.; Ellerby, H.M. Dimethyl sulfoxide and dimethyl formamide increase lifespan of C. elegans in liquid. Mech. Ageing Dev. 2013, 134, 69–78. [Google Scholar] [CrossRef]
- Calvert, S.; Tacutu, R.; Sharifi, S.; Teixeira, R.; Ghosh, P.; de Magalhães, J.P. A network pharmacology approach reveals new candidate caloric restriction mimetics in C. elegans. Aging Cell 2016, 15, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Hart, A. Behavior of the C. elegans. WormBook 2006, 1–67. [Google Scholar] [CrossRef]
- Viswanathan, M.; Kim, S.K.; Berdichevsky, A.; Guarente, L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell 2005, 9, 605–615. [Google Scholar] [CrossRef]
- Kozikowski, B.A.; Burt, T.M.; Tirey, D.A.; Williams, L.E.; Kuzmak, B.R.; Stanton, D.T.; Morand, K.L.; Nelson, S.L. The effect of freeze/thaw cycles on the stability of compounds in DMSO. J. Biomol. Screen. 2003, 8, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Assay Development: Fundamentals and Practices; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ilouga, P.E.; Winkler, D.; Kirchhoff, C.; Schierholz, B.; Wölcke, J. Investigation of 3 industry-wide applied storage conditions for compound libraries. J. Biomol. Screen. 2007, 12, 21–32. [Google Scholar] [CrossRef]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.S.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–230. [Google Scholar] [CrossRef]
- Lewis, L.K.; Robson, M.H.; Vecherkina, Y.; Ji, C.; Beall, G.W. Interference with spectrophotometric analysis of nucleic acids and proteins by leaching of chemicals from plastic tubes. Biotechniques 2010, 48, 297–302. [Google Scholar] [CrossRef]
- McDonald, G.R.; Hudson, A.L.; Dunn, S.M.J.; You, H.; Baker, G.B.; Whittal, R.M.; Martin, J.W.; Jha, A.; Edmondson, D.E.; Holt, A. Bioactive contaminants leach from disposable laboratory plasticware. Science 2008, 322, 917. [Google Scholar] [CrossRef]
- Olivieri, A.; Degenhardt, O.S.; Mcdonald, G.R.; Narang, D.; Paulsen, I.M.; Kozuska, J.L.; Holt, A. On the disruption of biochemical and biological assays by chemicals leaching from disposable laboratory plasticware. Can. J. Physiol. Pharmacol. 2012, 90, 697–703. [Google Scholar] [CrossRef]
- Evason, K.; Collins, J.J.; Huang, C.; Hughes, S.; Kornfeld, K. Valproic acid extends Caenorhabditis elegans lifespan. Aging Cell 2008, 7, 305–317. [Google Scholar] [CrossRef]
- Solis, G.M.; Kardakaris, R.; Valentine, E.R.; Bar-Peled, L.; Chen, A.L.; Blewett, M.M.; McCormick, M.A.; Williamson, J.R.; Kennedy, B.; Cravatt, B.F.; et al. Translation attenuation by minocycline enhances longevity and proteostasis in old post-stress-responsive organisms. Elife 2018, 7, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Espada, L.; Dakhovnik, A.; Chaudhari, P.; Martirosyan, A.; Miek, L.; Poliezhaieva, T.; Schaub, Y.; Nair, A.; Döring, N.; Rahnis, N.; et al. Late life metformin treatment limits cell survival and shortens lifespan by triggering an aging-associated failure of energy metabolism. bioRxiv 2019, 863357. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Berstein, L.M.; Popovich, I.G.; Zabezhinski, M.A.; Egormin, P.A.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Kovalenko, I.G.; et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging 2011, 3, 148–157. [Google Scholar] [CrossRef]
- Desjardins, D.; Cacho-Valadez, B.; Liu, J.L.; Wang, Y.; Yee, C.; Bernard, K.; Khaki, A.; Breton, L.; Hekimi, S. Antioxidants reveal an inverted U-shaped dose-response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans. Aging Cell 2017, 16, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Partridge, F.A.; Tearle, A.W.; Gravato-Nobre, M.J.; Schafer, W.R.; Hodgkin, J. The C. elegans glycosyltransferase BUS-8 has two distinct and essential roles in epidermal morphogenesis. Dev. Biol. 2008, 317, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Pears, C.; Woollard, A. An enhanced C. elegans based platform for toxicity assessment. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Lindblom, T.; Dodd, A. Xenobiotic detoxification in the nematode Caenorhabditis elegans. J. Exp. Zool. A Comp. Exp. Biol. 2006, 305, 720–730. [Google Scholar] [CrossRef]
- Shibamura, A.; Ikeda, T.; Nishikawa, Y. A method for oral administration of hydrophilic substances to Caenorhabditis elegans: Effects of oral supplementation with antioxidants on the nematode lifespan. Mech. Ageing Dev. 2009, 130, 652–655. [Google Scholar] [CrossRef]
- Spindler, S.R. Review of the literature and suggestions for the design of rodent survival studies for the identification of compounds that increase health and life span. Age 2012, 34, 111–120. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, C.; Lu, M.; Dong, Q.; Wang, Z.; Wang, Z.; Xiong, W.; Zhang, N.; Zhou, J.; Liu, Q.; et al. Calorie restriction is the most reasonable anti-ageing intervention: A meta-analysis of survival curves. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Mair, W.; Dillin, A. Aging and Survival: The Genetics of Life Span Extension by Dietary Restriction. Annu. Rev. Biochem. 2008, 77, 727–754. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Amaro, R.L.; Valentine, E.R.; Carretero, M.; Leboeuf, S.E.; Rangaraju, S.; Broaddus, C.D.; Solis, G.M.; Williamson, J.R.; Petrascheck, M. Measuring food intake and nutrient absorption in Caenorhabditis elegans. Genetics 2015, 200, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Klass, M.R. Aging in the nematode Caenorhabditis elegans: Major biological and environmental factors influencing life span. Mech. Ageing Dev. 1977, 6, 413–429. [Google Scholar] [CrossRef]
- Zhao, B.; Khare, P.; Feldman, L.; Dent, J.A. Reversal frequency in Caenorhabditis elegans represents an integrated response to the state of the animal and its environment. J. Neurosci. 2003, 23, 5319–5328. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes Filho, C.D.; Henriquez, B.; Seah, N.E.; Evans, R.M.; Lapierre, L.R.; Dillin, A. Visible light reduces C. elegans longevity. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Ludewig, A.H.; Gimond, C.; Judkins, J.C.; Thornton, S.; Pulido, D.C.; Micikas, R.J.; Dö Ring, F.; Antebi, A.; Braendle, C.; Schroeder, F.C. Larval crowding accelerates C. elegans development and reduces lifespan and Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases. PLoS Genet. 2017, 13, e1006717. [Google Scholar] [CrossRef]
- Shinn-Thomas, J.H.; Scanga, S.E.; Spica, P.S.; Nariya, H.K.; Klempic, E.; Brockett, M.R. Wrapping culture plates with Parafilm M® increases Caenorhabditis elegans growth. BMC Res. Notes 2019, 12, 818. [Google Scholar] [CrossRef]
- Miller, H.; Fletcher, M.; Primitivo, M.; Leonard, A.; Sutphin, G.L.; Rintala, N.; Kaeberlein, M.; Leiser, S.F. Genetic interaction with temperature is an important determinant of nematode longevity. Aging Cell 2017, 16, 1425–1429. [Google Scholar] [CrossRef]
- Park, H.E.H.; Jung, Y.; Lee, S.J.V. Survival assays using Caenorhabditis elegans. Mol. Cells 2017, 40, 90–99. [Google Scholar] [CrossRef]
- Banse, S.A.; Lucanic, M.; Sedore, C.A.; Coleman-Hulbert, A.L.; Plummer, W.T.; Chen, E.; Kish, J.L.; Hall, D.; Onken, B.; Presley, M.P.; et al. Automated lifespan determination across Caenorhabditis strains and species reveals assay-specific effects of chemical interventions. GeroScience 2019, 41, 945–960. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kümmel, A.; Gubler, H.; Gehin, P.; Beibel, M.; Gabriel, D.; Parker, C. Integration of Multiple Readouts Into the Z’ Factor for Assay Quality Assessment. J. Biomol. Screen. 2010, 15, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Burt, S.M.; Carter, T.J.N.; Kricka, L.J. Thermal characteristics of microtitre plates used in immunological assays. J. Immunol. Methods 1979, 31, 231–236. [Google Scholar] [CrossRef]
- Faessel, H.M.; Levasseur, L.M.; Slocum, H.K.; Greco, W.R. Parabolic Growth Patterns in 96-Well Plate Cell Growth Experiments. Vitr. Cell Dev. Biol. Anim. 1999, 35, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.G.; Sanders, A.H.; Douglas Hogg, R.; Woods Hellman, J. Thermal gradients in microtitration plates. Effects on enzyme-linked immunoassay. J. Immunol. Methods 1981, 42, 195–201. [Google Scholar] [CrossRef]
- Lundholt, B.K.; Scudder, K.M.; Pagliaro, L. A simple technique for reducing edge effect in cell-based assays. J. Biomol. Screen. 2003, 8, 566–570. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, Z.Y. Transgenerational effects of different sequential exposure to 2,2′,4,4′-tetra-brominated diphenyl ether (BDE47) and lead (Pb) on Caenorhabditis elegans. Environ. Sci. Eur. 2020, 32. [Google Scholar] [CrossRef]
- Caraus, I.; Alsuwailem, A.A.; Nadon, R.; Makarenkov, V. Detecting and overcoming systematic bias in highthroughput screening technologies: A comprehensive review of practical issues and methodological solutions. Brief. Bioinform. 2015, 16, 974–986. [Google Scholar] [CrossRef]
- Bray, M.; Carpenter, A. Imaging Platform, Broad Institute of MIT and Harvard. Advanced Assay Development Guidelines for Image-Based High Content Screening and Analysis. In Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2017; pp. 1–31. [Google Scholar]
- Lithgow, G.J.; Driscoll, M.; Phillips, P. A long journey to reproducible results. Nature 2017, 548, 387–388. [Google Scholar] [CrossRef]
- Pho, K.B.; MacNeil, L.T. Biology is the root of variability: Cautionary tales in Caenorhabditis elegans biology. Biochem. Soc. Trans. 2019, 47, 887–896. [Google Scholar] [CrossRef]
- Gosai, S.J.; Kwak, J.H.; Luke, C.J.; Long, O.S.; King, D.E.; Kovatch, K.J.; Johnston, P.A.; Shun, T.Y.; Lazo, J.S.; Perlmutter, D.H.; et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS ONE 2010, 5, e15460. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.N.; Strait, N.L.; Vayndorf, E.M.; Blue, B.W.; Tran, C.H.; Davis, B.E.M.; Huang, K.; Johnson, B.J.; Lim, K.M.; Liu, S.; et al. WormBot, an open-source robotics platform for survival and behavior analysis in C. elegans. GeroScience 2019, 41, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Arantes-Oliveira, N.; Apfeld, J.; Dillin, A.; Kenyon, C. Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 2002, 295, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.R.; Kenyon, C. Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 2006, 124, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Golegaonkar, S.; Tabrez, S.S.; Pandit, A.; Sethurathinam, S.; Jagadeeshaprasad, M.G.; Bansode, S.; Sampathkumar, S.G.; Kulkarni, M.J.; Mukhopadhyay, A. Rifampicin reduces advanced glycation end products and activates DAF-16 to increase lifespan in Caenorhabditis elegans. Aging Cell 2015, 14, 463–473. [Google Scholar] [CrossRef]
- Wan, Q.L.; Zheng, S.Q.; Wu, G.S.; Luo, H.R. Aspirin extends the lifespan of Caenorhabditis elegans via AMPK and DAF-16/FOXO in dietary restriction pathway. Exp. Gerontol. 2013, 48, 499–506. [Google Scholar] [CrossRef]
- Wan, Q.L.; Fu, X.; Meng, X.; Luo, Z.; Dai, W.; Yang, J.; Wang, C.; Wang, H.; Zhou, Q. Hypotaurine promotes longevity and stress tolerance: Via the stress response factors DAF-16/FOXO and SKN-1/NRF2 in Caenorhabditis elegans. Food Funct. 2020, 11, 347–357. [Google Scholar] [CrossRef]
- Amrit, F.R.G.; Ratnappan, R.; Keith, S.A.; Ghazi, A. The C. elegans lifespan assay toolkit. Methods 2014, 68, 465–475. [Google Scholar] [CrossRef]
- Angeli, S.; Klang, I.; Sivapatham, R.; Mark, K.; Zucker, D.; Bhaumik, D.; Lithgow, G.J.; Andersen, J.K. A DNA synthesis inhibitor is protective against proteotoxic stressors via modulation of fertility pathways in Caenorhabditis elegans. Aging 2013, 5, 759–769. [Google Scholar] [CrossRef]
- Aitlhadj, L.; Stürzenbaum, S.R. The use of FUdR can cause prolonged longevity in mutant nematodes. Mech. Ageing Dev. 2010, 131, 364–365. [Google Scholar] [CrossRef]
- van Raamsdonk, J.M.; Hekimi, S. FUdR causes a twofold increase in the lifespan of the mitochondrial mutant gas-1. Mech. Ageing Dev. 2011, 132, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; Corkins, M.E.; Li, J.C.; Singh, K.; Parsons, S.; Tucey, T.M.; Sorkaç, A.; Huang, H.; Dimitriadi, M.; Sinclair, D.A.; et al. C. elegans lifespan extension by osmotic stress requires FUdR, base excision repair, FOXO, and sirtuins. Mech. Ageing Dev. 2016, 154, 30–42. [Google Scholar] [CrossRef]
- Petrascheck, M.; Ye, X.; Buck, L.B. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature 2007, 450, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Lev, I.; Bril, R.; Liu, Y.; Ceré, L.I.; Rechavi, O. Inter-generational consequences for growing Caenorhabditis elegans in liquid. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374. [Google Scholar] [CrossRef] [PubMed]
- Hulme, S.E.; Shevkoplyas, S.S.; McGuigan, A.P.; Apfeld, J.; Fontana, W.; Whitesides, G.M. Lifespan-on-a-chip: Microfluidic chambers for performing lifelong observation of C. elegans. Lab Chip 2010, 10, 589–597. [Google Scholar] [CrossRef]
- Çelen, I.; Doh, J.H.; Sabanayagam, C.R. Effects of liquid cultivation on gene expression and phenotype of C. elegans. BMC Genom. 2018, 19, 562. [Google Scholar] [CrossRef]
- Laranjeiro, R.; Harinath, G.; Burke, D.; Braeckman, B.P.; Driscoll, M. Single swim sessions in C. elegans induce key features of mammalian exercise. BMC Biol. 2017, 15, 30. [Google Scholar] [CrossRef]
- Chuang, H.S.; Kuo, W.J.; Lee, C.L.; Chu, I.H.; Chen, C.S. Exercise in an electrotactic flow chamber ameliorates age-related degeneration in Caenorhabditis elegans. Sci. Rep. 2016, 6, 28064. [Google Scholar] [CrossRef]
- Hartman, J.H.; Smith, L.L.; Gordon, K.L.; Laranjeiro, R.; Driscoll, M.; Sherwood, D.R.; Meyer, J.N. Swimming Exercise and Transient Food Deprivation in Caenorhabditis elegans Promote Mitochondrial Maintenance and Protect Against Chemical-Induced Mitotoxicity. Sci. Rep. 2018, 8, 8359. [Google Scholar] [CrossRef]
- Sun, T.; Wu, H.; Cong, M.; Zhan, J.; Li, F. Meta-analytic evidence for the anti-aging effect of hormesis on Caenorhabditis elegans. Aging 2020, 12, 2723–2746. [Google Scholar] [CrossRef]
- Houthoofd, K.; Braeckman, B.P.; Lenaerts, I.; Brys, K.; De Vreese, A.; Van Eygen, S.; Vanfleteren, J.R. No reduction of metabolic rate in food restricted Caenorhabditis elegans. Exp. Gerontol. 2002, 37, 1359–1369. [Google Scholar] [CrossRef]
- Zarse, K.; Ristow, M. Antidepressants of the serotonin-antagonist type increase body fat and decrease lifespan of adult Caenorhabditis elegans. PLoS ONE 2008, 3, e4062. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, A.; Samuelson, A. Analysis of Lifespan in C. elegans: Low- And High-Throughput Approaches. In Aging: Methods and Protocols; Curran, S., Ed.; Springer: New York, NY, USA, 2020; pp. 7–27. [Google Scholar]
- Lucanic, M.; Garrett, T.; Yu, I.; Calahorro, F.; Asadi Shahmirzadi, A.; Miller, A.; Gill, M.S.; Hughes, R.E.; Holden-Dye, L.; Lithgow, G.J. Chemical activation of a food deprivation signal extends lifespan. Aging Cell 2016, 15, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Lucanic, M.; Garrett, T.; Gill, M.S.; Lithgow, G.J. A simple method for high throughput chemical screening in Caenorhabditis elegans. J. Vis. Exp. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pieper, A.A.; Xie, S.; Capota, E.; Estill, S.J.; Zhong, J.; Long, J.M.; Becker, G.L.; Huntington, P.; Goldman, S.E.; Shen, C.H.; et al. Discovery of a Proneurogenic, Neuroprotective Chemical. Cell 2010, 142, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Chung, T. Screen compounds singly: Why muck it up? J. Biomol. Screen. 1998, 171–173. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. Synergy and antagonism of promiscuous inhibition in multiple-compound mixtures. J. Med. Chem. 2006, 49, 2151–2154. [Google Scholar] [CrossRef]
- Hann, M.; Hudson, B.; Lewell, X.; Lifely, R.; Miller, L.; Ramsden, N. Strategic pooling of compounds for high-throughput screening. J. Chem. Inf. Comput. Sci. 1999, 39, 897–902. [Google Scholar] [CrossRef]
- Kainkaryam, R.M.; Woolf, P.J. Pooling in high-throughput drug screening. Curr. Opin. Drug Discov. Dev. 2009, 12, 339–350. [Google Scholar]
- Stroustrup, N.; Ulmschneider, B.E.; Nash, Z.M.; López-Moyado, I.F.; Apfeld, J.; Fontana, W. The Caenorhabditis elegans lifespan machine. Nat. Methods 2013, 10, 665–670. [Google Scholar] [CrossRef]
- Churgin, M.A.; Jung, S.K.; Yu, C.C.; Chen, X.; Raizen, D.M.; Fang-Yen, C. Longitudinal imaging of Caenorhabditis elegans in a microfabricated device reveals variation in behavioral decline during aging. Elife 2017, 6, e26652. [Google Scholar] [CrossRef] [PubMed]
- Xian, B.; Shen, J.; Chen, W.; Sun, N.; Qiao, N.; Jiang, D.; Yu, T.; Men, Y.; Han, Z.; Pang, Y.; et al. WormFarm: A quantitative control and measurement device toward automated Caenorhabditis elegans aging analysis. Aging Cell 2013, 12, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Knoefler, D.; Quarles, E.; Jakob, U.; Bazopoulou, D. Automated phenotyping and lifespan assessment of a C. elegans model of Parkinson’s disease. Transl. Med. Aging 2020, 4, 38–44. [Google Scholar] [CrossRef]
- Kamili, F.; Lu, H. Recent Advances and Trends in Microfluidic Platforms for C. elegans Biological Assays. Annu. Rev. Anal. Chem. 2018, 11, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Edwards, H.; Birze, N.; Gabrilska, R.; Rumbaugh, K.P.; Blawzdziewicz, J.; Szewczyk, N.J.; Driscoll, M.; Vanapalli, S.A. NemaLife: A structured microfluidic culture device optimized for aging studies in crawling C. elegans. bioRxiv 2019, 675827. [Google Scholar] [CrossRef]
- Banse, S.; Blue, B.; Robinson, K.; Jarrett, C.; Phillips, P. The Stress-Chip: A microfluidic platform for stress analysis in Caenorhabditis elegans. PLoS ONE 2019, 14, e0216283. [Google Scholar] [CrossRef]
- Midkiff, D.; San-Miguel, A. Microfluidic technologies for high throughput screening through sorting and on-chip culture of C. elegans. Molecules 2019, 24, 4292. [Google Scholar] [CrossRef]
- Letizia, M.C.; Cornaglia, M.; Trouillon, R.; Sorrentino, V.; Mouchiroud, L.; Bou Sleiman, M.S.; Auwerx, J.; Gijs, M.A.M. Microfluidics-enabled phenotyping of a whole population of C. elegans worms over their embryonic and post-embryonic development at single-organism resolution. Microsyst. Nanoeng. 2018, 4. [Google Scholar] [CrossRef]
- Lionaki, E.; Tavernarakis, N. High-Throughput and Longitudinal Analysis of Aging and Senescent Decline in Caenorhabditis elegans. In Cell Senescence: Methods and Protocols; Galluzzi, L., Vitale, I., Kepp, O., Kroemer, G., Eds.; Springer: New York, NY, USA, 2013; pp. 485–500. [Google Scholar]
- Doh, J.H.; Moore, A.B.; Celen, I.; Moore, M.T.; Sabanayagam, C.R. ChIP and Chips: Introducing the WormPharm for correlative studies employing pharmacology and genome-wide analyses in C. elegans. J. Biol. Methods 2016, 3, 44. [Google Scholar] [CrossRef]
- Wen, H.; Shi, W.; Qin, J. Multiparameter evaluation of the longevity in C. elegans under stress using an integrated microfluidic device. Biomed. Microdevices 2012, 14, 721–728. [Google Scholar] [CrossRef]
- Mathew, M.D.; Mathew, N.D.; Ebert, P.R. WormScan: A technique for high-throughput phenotypic analysis of Caenorhabditis elegans. PLoS ONE 2012, 7, 3–8. [Google Scholar] [CrossRef]
- Thompson, J.; Sathyamurthy, S.; Sukumar, S.; Shapira, T.; Ebert, P.; Szewczyk, N. Automated Wormscan [version 3; referees: 2 approved, 1 approved with reservations]. F1000Research 2019, 6, 1–19. [Google Scholar]
- Kim, K.; Park, H.; Lim, K.M. Phototoxicity: Its mechanism and animal alternative test methods. Toxicol. Res. 2015, 31, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Jushaj, A.; Churgin, M.; Yao, B.; La Torre, M.D.; Fang-Yen, C.; Temmerman, L. Optimized criteria for locomotion-based healthspan evaluation in C. elegans using the WorMotel system. PLoS ONE 2020, 15, e0229583. [Google Scholar] [CrossRef] [PubMed]
- Churging, M.; Fang-Yen, C. An imaging system for C. elegans behavior. In C. elegans, Methods and Applications; Haspel, G., Biron, D., Eds.; Springer: New York, NY, USA, 2015; pp. 199–207. ISBN 978-1-4939-2842-2. [Google Scholar]
- Coleman-Hulbert, A.L.; Johnson, E.; Sedore, C.A.; Banse, S.A.; Guo, M.; Driscoll, M.; Lithgow, G.J.; Phillips, P.C. Caenorhabditis Intervention Testing Program: The tyrosine kinase inhibitor imatinib mesylate does not extend lifespan in nematodes. microPublication Biol. 2019, 2019. [Google Scholar] [CrossRef]
- Coleman-Hulbert, A.; Johnson, E.; Sedore, C.; Banse, S.; Guo, M.; Driscoll, M.; Lithgow, G.; Phillips, P. Caenorhabditis Intervention Testing Program: The creatine analog β-guanidinopropionic acid does not extend lifespan in nematodes. microPublication Biol. 2020, 2020. [Google Scholar] [CrossRef]
- Morshead, M.; Sedore, C.; Jones, E.; Hall, D.; Plummer, W.; Garrett, T.; Lucanic, M.; Guo, M.; Driscoll, M.; Phillips, P.; et al. Caenorhabditis Intervention Testing Program: The farnesoid X receptor agonist obeticholic acid does not robustly extend lifespan in nematodes. microPublication Biol. 2020, 2020. [Google Scholar] [CrossRef]
- Larsen, P.L. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1993, 90, 8905–8909. [Google Scholar] [CrossRef]
- Lithgow, G.J.; White, T.M.; Melov, S.; Johnson, T.E. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc. Natl. Acad. Sci. USA 1995, 92, 7540–7544. [Google Scholar] [CrossRef]
- Vanfleteren, J.R. Oxidative stress and ageing in Caenorhabditis elegans. Biochem. J. 1993, 292, 605–608. [Google Scholar] [CrossRef]
- Sampayo, J.N.; Jenkins, N.L.; Lithgow, G.J. Using stress resistance to isolate novel longevity mutations in Caenorhabditis elegans. Ann. N. Y. Acad. Sci. 2000, 908, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.J.; Riddle, D.L. Positive selection of Caenorhabditis elegans mutants with increased stress resistance and longevity. Genetics 2003, 163, 171–180. [Google Scholar] [PubMed]
- Miller, R.A. Cell stress and aging: New emphasis on multiplex resistance mechanisms. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64, 179–182. [Google Scholar] [CrossRef]
- Admasu, T.D.; Chaithanya Batchu, K.; Barardo, D.; Ng, L.F.; Lam, V.Y.M.; Xiao, L.; Cazenave-Gassiot, A.; Wenk, M.R.; Tolwinski, N.S.; Gruber, J. Drug Synergy Slows Aging and Improves Healthspan through IGF and SREBP Lipid Signaling. Dev. Cell 2018, 47, 67–79.e5. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Linton, J.M.; Schork, N.J.; Buck, L.B.; Petrascheck, M. A pharmacological network for lifespan extension in Caenorhabditis elegans. Aging Cell 2014, 13, 206–215. [Google Scholar] [CrossRef]
- Gill, M.S.; Olsen, A.; Sampayo, J.N.; Lithgow, G.J. An automated high-throughput assay for survival of the nematode Caenorhabditis elegans. Free. Radic. Biol. Med. 2003, 35, 558–565. [Google Scholar] [CrossRef]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar] [CrossRef]
- Moy, T.I.; Conery, A.L.; Larkins-Ford, J.; Wu, G.; Mazitschek, R.; Casadei, G.; Lewis, K.; Carpenter, A.E.; Ausubel, F.M. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem. Biol. 2009, 4, 527–533. [Google Scholar] [CrossRef]
- Coburn, C.; Allman, E.; Mahanti, P.; Benedetto, A.; Cabreiro, F.; Pincus, Z.; Matthijssens, F.; Araiz, C.; Mandel, A.; Vlachos, M.; et al. Anthranilate Fluorescence Marks a Calcium-Propagated Necrotic Wave That Promotes Organismal Death in C. elegans. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef]
- Galimov, E.R.; Pryor, R.E.; Poole, S.E.; Benedetto, A.; Pincus, Z.; Gems, D. Coupling of Rigor Mortis and Intestinal Necrosis during C. elegans Organismal Death. Cell Rep. 2018, 22, 2730–2741. [Google Scholar] [CrossRef]
- Janssens, G.E.; Houtkooper, R.H. Identification of longevity compounds with minimized probabilities of side effects. Biogerontology 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Barardo, D.; Thornton, D.; Thoppil, H.; Walsh, M.; Sharifi, S.; Ferreira, S.; Anžič, A.; Fernandes, M.; Monteiro, P.; Grum, T.; et al. The DrugAge database of aging-related drugs. Aging Cell 2017, 16, 594–597. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P. Why genes extending lifespan in model organisms have not been consistently associated with human longevity and what it means to translation research. Cell Cycle 2014, 13, 2671–2673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Magalhães, J.P.; Stevens, M.; Thornton, D. The Business of Anti-Aging Science. Trends Biotechnol. 2017, 35, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Wählby, C.; Riklin-Raviv, T.; Ljosa, V.; Conery, A.L.; Golland, P.; Ausubel, F.M.; Carpenter, A.E. Resolving clustered worms via probabilistic shape models. In Proceedings of the 2010 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Rotterdam, The Netherlands, 14–17 April 2010; pp. 552–555. [Google Scholar] [CrossRef]
- Martin, G.M. Geroscience: Addressing the mismatch between its exciting research opportunities, its economic imperative and its current funding crisis. Exp. Gerontol. 2017, 94, 46–51. [Google Scholar] [CrossRef]
- Hahm, J.H.; Kim, S.; Diloreto, R.; Shi, C.; Lee, S.J.V.; Murphy, C.T.; Nam, H.G. C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Le, K.N.; Zhan, M.; Cho, Y.; Wan, J.; Patel, D.S.; Lu, H. An automated platform to monitor long-term behavior and healthspan in Caenorhabditis elegans under precise environmental control. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Olshansky, S.J. Articulating the Case for the Longevity Dividend. Cold Spring Harb. Perspect. Med. 2016, 6, a025940. [Google Scholar] [CrossRef]
- Goldman, D.P.; Cutler, D.; Rowe, J.W.; Michaud, P.C.; Sullivan, J.; Peneva, D.; Olshansky, S.J. Substantial health and economic returns from delayed aging may warrant a new focus for medical research. Health Aff. 2013, 32, 1698–1705. [Google Scholar] [CrossRef]


{kind=link}
| Target-Based Screening | Phenotypic Screening in Cells | Phenotypic Screening in Small Organisms (e.g., C. elegans) | ||||
|---|---|---|---|---|---|---|
| Strengths | Weaknesses | Strengths | Weaknesses | Strengths | Weaknesses | |
| Target | Known target selected for screen. | Cannot find new targets. | Target agnostic. | Target identification can be cumbersome. | Target agnostic. | Target identification can be cumbersome. |
| Human relevance | In vitro study on isolated targets. | In vitro but on whole cells. Cells used can be of human origin. Even patient-derived primary cells or in vitro reprogrammed cells from patient-derived fibroblasts. | Access to disease relevant cell types can be difficult. Diseases cannot always be easily recapitulated in isolated cells because they depend on interactions of various cells and/or systemic factors. | In vivo, small organisms contain multiple cell types and even organ systems thus better capturing disease processes that depend on cell interactions and/or systemic factors. | Small model organisms may not fully capture human biology. | |
| False positives | False positives due to nonspecific mechanisms (fluorescence quenching, aggregation). | False positives due to compounds that target generic mechanisms such as protein synthesis which affect the assayed phenotype but are not specific enough to be used as drug leads. | False positives due to compounds that target generic mechanisms such as protein synthesis which affect the assayed phenotype but are not specific enough to be used as drug leads. | |||
| Hit identification | Will identify all hits that modify the target of interest. | Hits will include molecules that cannot be used as drug leads (such as cytotoxic compounds). | Initial screen may already inform about toxicity of compounds (cell viability). Hits already have “drug-like” properties. | If the library is screened at high concentrations, low-potency effects could cloud the interpretation of the results. | Initial screen already informs about toxicity of compounds (organism viability). Hits already have “drug-like” properties. | If the library is screened at high concentrations, low-potency effects could cloud the interpretation of the results. However, if too low concentrations are used, then no effect may be seen because drug concentrations in the organism tend to be much smaller than those in the medium. Toxic compounds are eliminated even though they might have pharmacological properties and less toxic variants could possibly be made. |
| Lead optimization | Very amendable for lead optimization (SAR). | Exclusion of hits that have poor pharmacokinetic and pharmacodynamic properties but that could still be amendable to medicinal chemistry optimization. In addition, lead optimization (SAR) can be more difficult. | Exclusion of hits that have poor pharmacokinetic and pharmacodynamic properties but that could still be amendable to medicinal chemistry optimization. In addition, lead optimization (SAR) can be more difficult. | |||
| Amount of compound required | Low amounts of compound required. | Low amounts of compound required. | Large amounts of compound required. | |||
| Throughput | Very high throughput | High throughput | Low throughput | |||
| Manual | Wormbot | Automated Lifespan Machines | WorMotel | Microfluidics | LFASS | |
|---|---|---|---|---|---|---|
| Culture medium | Liquid or NGM | NGM | Modified version of NGM | NGM | Liquid | See manual, assay in liquid |
| FUdR | Optional | Needed | Needed | Needed | Not needed | Generally needed |
| Throughput | Very low | High (144 wells) | Moderate (16 Petri plates) | High (240 wells) | Low (depends on used chip) | Very high (96- or 384-well plates) |
| Temporal resolution | Very low | High | High | High | High | Very high |
| Individual/population | Both are possible | Population | Population | Individual | Both are possible | Population |
| Equipment cost (excluding labor cost) | Very low | High | Moderate | Very high | Moderate | Moderate |
| Automated data collection and analysis | No | Yes | Yes | Yes | Depends | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulterijs, S.; Braeckman, B.P. Phenotypic Screening in C. elegans as a Tool for the Discovery of New Geroprotective Drugs. Pharmaceuticals 2020, 13, 164. https://doi.org/10.3390/ph13080164
Bulterijs S, Braeckman BP. Phenotypic Screening in C. elegans as a Tool for the Discovery of New Geroprotective Drugs. Pharmaceuticals. 2020; 13(8):164. https://doi.org/10.3390/ph13080164
Chicago/Turabian StyleBulterijs, Sven, and Bart P. Braeckman. 2020. "Phenotypic Screening in C. elegans as a Tool for the Discovery of New Geroprotective Drugs" Pharmaceuticals 13, no. 8: 164. https://doi.org/10.3390/ph13080164
APA StyleBulterijs, S., & Braeckman, B. P. (2020). Phenotypic Screening in C. elegans as a Tool for the Discovery of New Geroprotective Drugs. Pharmaceuticals, 13(8), 164. https://doi.org/10.3390/ph13080164