Antibody Targeting of Eph Receptors in Cancer


Abstract
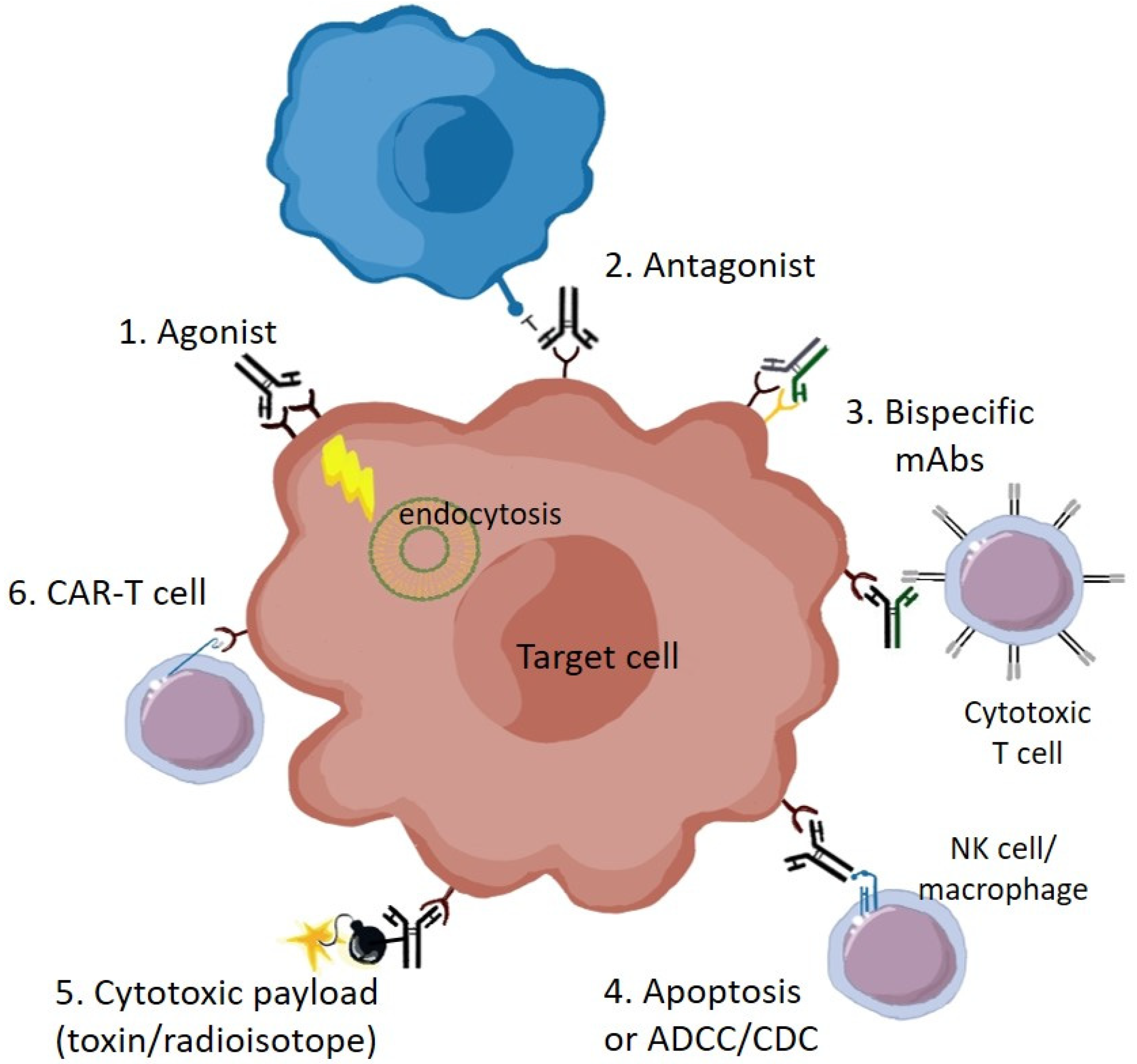
1. Introduction
2. EphA2
3. EphA3
4. EphBs
5. Ephrins
6. Co-Targeting and Bispecific Antibodies
7. Combination Therapies
8. Antibody Payloads
9. CAR-T Cells
10. In Vivo Imaging
11. Clinical Trials
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef]
- Wimmer-Kleikamp, S.H.; Nievergall, E.; Gegenbauer, K.; Adikari, S.; Mansour, M.; Yeadon, T.; Boyd, A.W.; Patani, N.R.; Lackmann, M. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood 2008, 112, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Nievergall, E.; Lackmann, M.; Janes, P.W. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell Mol. Life Sci. 2012, 69, 1813–1842. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, W.; Amato, K. Eph receptor tyrosine kinases in cancer stem cells. Cytokine Growth Factor Rev. 2015, 26, 1–6. [Google Scholar] [CrossRef]
- Cortina, C.; Palomo-Ponce, S.; Iglesias, M.; Fernandez-Masip, J.L.; Vivancos, A.; Whissell, G.; Huma, M.; Peiro, N.; Gallego, L.; Jonkheer, S.; et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat. Genet. 2007, 39, 1376–1383. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Hafeez, U.; Gan, H.K.; Scott, A.M. Monoclonal antibodies as immunomodulatory therapy against cancer and autoimmune diseases. Curr. Opin. Pharm. 2018, 41, 114–121. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Barquilla, A.; Pasquale, E.B. Eph receptors and ephrins: Therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Ireton, R.C.; Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Pasqualini, R.; Lindberg, R.A.; Kain, R.; Freeman, A.L.; Pasquale, E.B. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene 2000, 19, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.J.; Chen, J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J. Biol. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Fang, W.B.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Investig. 2008, 118, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I.S. Eph receptor signalling: From catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000, 2, 62–69. [Google Scholar] [CrossRef]
- Carles-Kinch, K.; Kilpatrick, K.E.; Stewart, J.C.; Kinch, M.S. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 2002, 62, 2840–2847. [Google Scholar]
- Coffman, K.T.; Hu, M.; Carles-Kinch, K.; Tice, D.; Donacki, N.; Munyon, K.; Kifle, G.; Woods, R.; Langermann, S.; Kiener, P.A.; et al. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003, 63, 7907–7912. [Google Scholar] [PubMed]
- Landen, C.N., Jr.; Lu, C.; Han, L.Y.; Coffman, K.T.; Bruckheimer, E.; Halder, J.; Mangala, L.S.; Merritt, W.M.; Lin, Y.G.; Gao, C.; et al. Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J. Natl. Cancer Inst. 2006, 98, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Kiewlich, D.; Zhang, J.; Gross, C.; Xia, W.; Larsen, B.; Cobb, R.R.; Biroc, S.; Gu, J.M.; Sato, T.; Light, D.R.; et al. Anti-EphA2 antibodies decrease EphA2 protein levels in murine CT26 colorectal and human MDA-231 breast tumors but do not inhibit tumor growth. Neoplasia 2006, 8, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ansuini, H.; Meola, A.; Gunes, Z.; Paradisi, V.; Pezzanera, M.; Acali, S.; Santini, C.; Luzzago, A.; Mori, F.; Lazzaro, D.; et al. Anti-EphA2 Antibodies with Distinct In Vitro Properties Have Equal In Vivo Efficacy in Pancreatic Cancer. J. Oncol. 2009, 2009, 951917. [Google Scholar] [CrossRef]
- Wesa, A.K.; Herrem, C.J.; Mandic, M.; Taylor, J.L.; Vasquez, C.; Kawabe, M.; Tatsumi, T.; Leibowitz, M.S.; Finke, J.H.; Bukowski, R.M.; et al. Enhancement in specific CD8+ T cell recognition of EphA2+ tumors in vitro and in vivo after treatment with ligand agonists. J. Immunol. 2008, 181, 7721–7727. [Google Scholar] [CrossRef]
- Kawabe, M.; Mandic, M.; Taylor, J.L.; Vasquez, C.A.; Wesa, A.K.; Neckers, L.M.; Storkus, W.J. Heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin enhances EphA2+ tumor cell recognition by specific CD8+ T cells. Cancer Res. 2009, 69, 6995–7003. [Google Scholar] [CrossRef]
- Bruckheimer, E.M.; Fazenbaker, C.A.; Gallagher, S.; Mulgrew, K.; Fuhrmann, S.; Coffman, K.T.; Walsh, W.; Ready, S.; Cook, K.; Damschroder, M.; et al. Antibody-dependent cell-mediated cytotoxicity effector-enhanced EphA2 agonist monoclonal antibody demonstrates potent activity against human tumors. Neoplasia 2009, 11, 509–517, 2 p following 517. [Google Scholar] [CrossRef]
- Hasegawa, J.; Sue, M.; Yamato, M.; Ichikawa, J.; Ishida, S.; Shibutani, T.; Kitamura, M.; Wada, T.; Agatsuma, T. Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol. 2016, 17, 1158–1167. [Google Scholar] [CrossRef]
- Merritt, W.M.; Kamat, A.A.; Hwang, J.Y.; Bottsford-Miller, J.; Lu, C.; Lin, Y.G.; Coffey, D.; Spannuth, W.A.; Nugent, E.; Han, L.Y.; et al. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol. 2010, 10, 1306–1314. [Google Scholar] [CrossRef]
- Gokmen-Polar, Y.; Toroni, R.A.; Hocevar, B.A.; Badve, S.; Zhao, Q.; Shen, C.; Bruckheimer, E.; Kinch, M.S.; Miller, K.D. Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2-positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 375–384. [Google Scholar] [CrossRef]
- Shitara, K.; Satoh, T.; Iwasa, S.; Yamaguchi, K.; Muro, K.; Komatsu, Y.; Nishina, T.; Esaki, T.; Hasegawa, J.; Kakurai, Y.; et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the afucosylated, humanized anti-EPHA2 antibody DS-8895a: A first-in-human phase I dose escalation and dose expansion study in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Parakh, S.; Lee, F.T.; Tebbutt, N.C.; Ameratunga, M.; Lee, S.T.; O’Keefe, G.J.; Gong, S.J.; Vanrenen, C.; Caine, J.; et al. Manuscript in preparation.
- Charmsaz, S.; Beckett, K.; Smith, F.M.; Bruedigam, C.; Moore, A.S.; Al-Ejeh, F.; Lane, S.W.; Boyd, A.W. EphA2 Is a Therapy Target in EphA2-Positive Leukemias but Is Not Essential for Normal Hematopoiesis or Leukemia. PLoS ONE 2015, 10, e0130692. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Gooya, J.; Mao, S.; Kinneer, K.; Xu, L.; Camara, M.; Fazenbaker, C.; Fleming, R.; Swamynathan, S.; Meyer, D.; et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008, 68, 9367–9374. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Han, H.D.; Shahzad, M.M.; Kim, S.W.; Mangala, L.S.; Nick, A.M.; Lu, C.; Langley, R.R.; Schmandt, R.; Kim, H.S.; et al. EphA2 immunoconjugate as molecularly targeted chemotherapy for ovarian carcinoma. J. Natl. Cancer Inst. 2009, 101, 1193–1205. [Google Scholar] [CrossRef]
- Lee, J.W.; Stone, R.L.; Lee, S.J.; Nam, E.J.; Roh, J.W.; Nick, A.M.; Han, H.D.; Shahzad, M.M.; Kim, H.S.; Mangala, L.S.; et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin. Cancer Res. 2010, 16, 2562–2570. [Google Scholar] [CrossRef]
- Cai, W.; Ebrahimnejad, A.; Chen, K.; Cao, Q.; Li, Z.B.; Tice, D.A.; Chen, X. Quantitative radioimmunoPET imaging of EphA2 in tumor-bearing mice. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 2024–2036. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Kohn, E.C.; LoRusso, P.; Houston, N.D.; Coleman, R.L.; Buzoianu, M.; Robbie, G.; Lechleider, R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Investig. New Drugs 2013, 31, 77–84. [Google Scholar] [CrossRef]
- Puttick, S.; Stringer, B.W.; Day, B.W.; Bruce, Z.C.; Ensbey, K.S.; Mardon, K.; Cowin, G.J.; Thurecht, K.J.; Whittaker, A.K.; Fay, M.; et al. EphA2 as a Diagnostic Imaging Target in Glioblastoma: A Positron Emission Tomography/Magnetic Resonance Imaging Study. Mol. Imaging 2015, 14, 385–399. [Google Scholar] [CrossRef]
- Kamoun, W.S.; Kirpotin, D.B.; Huang, Z.R.; Tipparaju, S.K.; Noble, C.O.; Hayes, M.E.; Luus, L.; Koshkaryev, A.; Kim, J.; Olivier, K.; et al. Antitumour activity and tolerability of an EphA2-targeted nanotherapeutic in multiple mouse models. Nat. Biomed. Eng. 2019, 3, 264–280. [Google Scholar] [CrossRef]
- Vail, M.E.; Murone, C.; Tan, A.; Hii, L.; Abebe, D.; Janes, P.W.; Lee, F.-T.; Baer, M.; Palath, V.; Bebbington, C.; et al. Targeting EphA3 Inhibits Cancer Growth by Disrupting the Tumor Stromal Microenvironment. Cancer Res. 2014, 74, 4470–4481. [Google Scholar] [CrossRef]
- Vearing, C.; Lee, F.T.; Wimmer-Kleikamp, S.; Spirkoska, V.; To, C.; Stylianou, C.; Spanevello, M.; Brechbiel, M.; Boyd, A.W.; Scott, A.M.; et al. Concurrent binding of anti-EphA3 antibody and ephrin-A5 amplifies EphA3 signaling and downstream responses: Potential as EphA3-specific tumor-targeting reagents. Cancer Res. 2005, 65, 6745–6754. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Miller, K.J.; Day, B.W.; El-Ajeh, F.; Yarranton, G.T.; Bebbington, C.R.; Scott, A.M.; Lackmann, M.; Boyd, A.W. EphA3 as a target for monoclonal antibody therapy for acute leukemia. Blood (Ash Annu. Meet. Abstr.) 2013, 122, 5013. [Google Scholar] [CrossRef]
- La Rocca, F.; Airoldi, I.; Di Carlo, E.; Marotta, P.; Falco, G.; Simeon, V.; Laurenzana, I.; Trino, S.; De Luca, L.; Todoerti, K.; et al. EphA3 targeting reduces in vitro adhesion and invasion and in vivo growth and angiogenesis of multiple myeloma cells. Cell Oncol. (Dordr) 2017, 40, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Cher, L.; Inglis, P.; Lwin, Z.; Lau, E.; Ackermann, U.; Coombs, N.; Remen, K.; Guo, N.; Ting Lee, S.; et al. ATIM-23. Preliminary Findings of A Phase I Safety and Bioimaging Trial of KB004 (Ifabotuzumab) In Patients with Glioblastoma. Neuro-Oncol. 2019, 21, vi6. [Google Scholar] [CrossRef]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; Fan, A.C.; et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: Results from a phase 1 study. Leuk Res. 2016, 50, 123–131. [Google Scholar] [CrossRef]
- Charmsaz, S.; Al-Ejeh, F.; Yeadon, T.M.; Miller, K.J.; Smith, F.M.; Stringer, B.W.; Moore, A.S.; Lee, F.T.; Cooper, L.T.; Stylianou, C.; et al. EphA3 as a target for antibody immunotherapy in acute lymphoblastic leukemia. Leukemia 2017, 31, 1779–1787. [Google Scholar] [CrossRef]
- Offenhauser, C.; Al-Ejeh, F.; Puttick, S.; Ensbey, K.S.; Bruce, Z.C.; Jamieson, P.R.; Smith, F.M.; Stringer, B.W.; Carrington, B.; Fuchs, A.V.; et al. EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers (Basel) 2018, 10, 519. [Google Scholar] [CrossRef]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhauser, C.; et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef]
- Chu, L.; Wang, A.; Ni, L.; Yan, X.; Song, Y.; Zhao, M.; Sun, K.; Mu, H.; Liu, S.; Wu, Z.; et al. Nose-to-brain delivery of temozolomide-loaded PLGA nanoparticles functionalized with anti-EPHA3 for glioblastoma targeting. Drug Deliv. 2018, 25, 1634–1641. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M.K.; Savage, N.; Yang, J.; et al. Cotargeting Ephrin Receptor Tyrosine Kinases A2 and A3 in Cancer Stem Cells Reduces Growth of Recurrent Glioblastoma. Cancer Res. 2018, 78, 5023–5037. [Google Scholar] [CrossRef]
- Taki, S.; Kamada, H.; Inoue, M.; Nagano, K.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S. A Novel Bispecific Antibody against Human CD3 and Ephrin Receptor A10 for Breast Cancer Therapy. PLoS ONE 2015, 10, e0144712. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Luis, E.; Ross, S.; Silva, J.; Tan, C.; Crowley, C.; Chui, C.; Franz, G.; Senter, P.; Koeppen, H.; et al. EphB2 as a therapeutic antibody drug target for the treatment of colorectal cancer. Cancer Res. 2004, 64, 781–788. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krasnoperov, V.; Kumar, S.R.; Ley, E.; Li, X.; Scehnet, J.; Liu, R.; Zozulya, S.; Gill, P.S. Novel EphB4 monoclonal antibodies modulate angiogenesis and inhibit tumor growth. Am. J. Pathol. 2010, 176, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, S.-A.; Douglas, E.L.; Mertens-Walker, I.; Lisle, J.E.; Maharaj, M.S.N.; Herington, A.C. Anti-tumour effects of antibodies targeting the extracellular cysteine-rich region of the receptor tyrosine kinase EphB4. Oncotarget 2015, 6, 7554–7569. [Google Scholar] [CrossRef]
- Liu, S.; Li, D.; Park, R.; Liu, R.; Xia, Z.; Guo, J.; Krasnoperov, V.; Gill, P.S.; Li, Z.; Shan, H.; et al. PET imaging of colorectal and breast cancer by targeting EphB4 receptor with 64Cu-labeled hAb47 and hAb131 antibodies. J. Nucl. Med. 2013, 54, 1094–1100. [Google Scholar] [CrossRef]
- Damelin, M.; Bankovich, A.; Park, A.; Aguilar, J.; Anderson, W.; Santaguida, M.; Aujay, M.; Fong, S.; Khandke, K.; Pulito, V.; et al. Anti-EFNA4 Calicheamicin Conjugates Effectively Target Triple-Negative Breast and Ovarian Tumor-Initiating Cells to Result in Sustained Tumor Regressions. Clin. Cancer Res. 2015, 21, 4165–4173. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Krop, I.; Burris, H.A., 3rd; Hamilton, E.; Braiteh, F.; Weise, A.M.; Abu-Khalaf, M.; Werner, T.L.; Pirie-Shepherd, S.; Zopf, C.J.; et al. First-in-human, phase I study of PF-06647263, an anti-EFNA4 calicheamicin antibody-drug conjugate, in patients with advanced solid tumors. Int. J. Cancer 2019, 145, 1798–1808. [Google Scholar] [CrossRef]
- Abengozar, M.A.; de Frutos, S.; Ferreiro, S.; Soriano, J.; Perez-Martinez, M.; Olmeda, D.; Marenchino, M.; Canamero, M.; Ortega, S.; Megias, D.; et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 2012, 119, 4565–4576. [Google Scholar] [CrossRef]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef]
- Boyd, A.W.; Ward, L.D.; Wicks, I.P.; Simpson, R.J.; Salvaris, E.; Wilks, A.; Welch, K.; Loudovaris, M.; Rockman, S.; Busmanis, I. Isolation and characterization of a novel receptor-type protein tyrosine kinase (hek) from a human pre-B cell line. J. Biol. Chem. 1992, 267, 3262–3267. [Google Scholar]
- Chiari, R.; Hames, G.; Stroobant, V.; Texier, C.; Maillere, B.; Boon, T.; Coulie, P.G. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res. 2000, 60, 4855–4863. [Google Scholar] [PubMed]
- Arruga, F.; Messa, F.; Carturan, S.; Pradatto, M.; Maff, C.; Pautasso, M.; Panuzzo, C.; Iacobucci, I.; Bracco, E.; Messa, E.; et al. EphA3 is abnormally expressed in chronic myeloproliferative disorders and could represent a new molecular target. Proc. Am. Assoc. Cancer Res. 2009, AACR 2009, Abstract nr 2866. [Google Scholar]
- Keane, N.; Freeman, C.; Swords, R.; Giles, F.J. EPHA3 as a novel therapeutic target in the hematological malignancies. Exp. Rev. Hematol. 2012, 5, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ferluga, S.; Tome, C.M.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Farnsworth, R.; Vail, M.; Chheang, C.; Gargett, C.; Murone, C.; Llerena, C.; Major, A.; Scott, A.; Janes, P.; et al. Hypoxia-controlled EphA3 marks a human endometrium derived multipotent mesenchymal stromal cell that supports vascular growth. PLoS ONE 2014, 9, e112106. [Google Scholar] [CrossRef]
- Salvucci, O.; Tosato, G. Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv. Cancer Res. 2012, 114, 21–57. [Google Scholar]
- Solanas, G.; Batlle, E. Control of cell adhesion and compartmentalization in the intestinal epithelium. Exp. Cell Res. 2011, 317, 2695–2701. [Google Scholar] [CrossRef]
- Stephenson, S.A.; Slomka, S.; Douglas, E.L.; Hewett, P.J.; Hardingham, J.E. Receptor protein tyrosine kinase EphB4 is up-regulated in colon cancer. BMC Mol. Biol. 2001, 2, 15. [Google Scholar] [CrossRef]
- Merchant, A.A.; Jorapur, A.; McManus, A.; Liu, R.; Krasnoperov, V.; Chaudhry, P.; Singh, M.; Harton, L.; Agajanian, M.; Kim, M.; et al. EPHB4 is a therapeutic target in AML and promotes leukemia cell survival via AKT. Blood Adv. 2017, 1, 1635–1644. [Google Scholar] [CrossRef]
- Hammond, S.A.; Lutterbuese, R.; Roff, S.; Lutterbuese, P.; Schlereth, B.; Bruckheimer, E.; Kinch, M.S.; Coats, S.; Baeuerle, P.A.; Kufer, P.; et al. Selective targeting and potent control of tumor growth using an EphA2/CD3-Bispecific single-chain antibody construct. Cancer Res. 2007, 67, 3927–3935. [Google Scholar] [CrossRef]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem. Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Brantley-Sieders, D.M.; Vaught, D.; Yu, J.; Xie, L.; Wells, S.; Jackson, D.; Muraoka-Cook, R.; Arteaga, C.; Chen, J. Elevation of Receptor Tyrosine Kinase EphA2 Mediates Resistance to Trastuzumab Therapy. Cancer Res. 2010, 70, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef]
- Ruff, M.; Sakemura, R.; Cox, M.; Hefazi Torghabeh, M.; Roman Moreno, P.; Schick, K.; Sarkaria, J.; Kenderian, S. Exth-32. Development of EphA3 directed chimeric antigen receptor T cell therapy for the treatment of Glioblastoma Multiforme. Neuro-Oncol. 2019, 21, vi88–vi89. [Google Scholar] [CrossRef]
- Burvenich, I.J.; Parakh, S.; Gan, H.K.; Lee, F.T.; Guo, N.; Rigopoulos, A.; Lee, S.T.; Gong, S.; O’Keefe, G.J.; Tochon-Danguy, H.; et al. Molecular Imaging and Quantitation of EphA2 Expression in Xenograft Models with 89Zr-DS-8895a. J. Nucl. Med. 2016, 57, 974–980. [Google Scholar] [CrossRef]
- Jacobson, O.; Li, Q.; Chen, H.; Niu, G.; Kiesewetter, D.O.; Xu, L.; Cook, K.; Yang, G.; Dall’Acqua, W.; Tsui, P.; et al. PET-Guided Evaluation and Optimization of Internalized Antibody-Drug Conjugates Targeting Erythropoietin-Producing Hepatoma A2 Receptor. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1838–1844. [Google Scholar] [CrossRef]
- Li, D.; Liu, S.; Liu, R.; Zhou, Y.; Park, R.; Naga, K.; Krasnoperov, V.; Gill, P.S.; Li, Z.; Shan, H.; et al. EphB4-targeted imaging with antibody h131, h131-F(ab’)2 and h131-Fab. Mol. Pharm. 2013, 10, 4527–4533. [Google Scholar] [CrossRef]


{kind=link}
| Target | Antibody | Effects/Application | Clinic | Ref. |
|---|---|---|---|---|
| EphA2 | EA1.2 | Agonist, receptor phosphorylation and degradation. Inhibition of tumor cell growth/tube formation | No | [20] |
| EphA2 | EA2, B233 | Agonist, tumor cell selective, inhibition of MDA-MB-231 breast tumor growth in vivo | No | [21] |
| EphA2 | EA5 | EphA2 degradation in ovarian tumors in vivo, inhibition of tumor growth | No | [22,29,30] |
| EphA2 | IgG25 | Agonist, inhibitory in a pancreatic xenograft | No | [24] |
| EphA2 | IgG28 | Ligand blocking, inhibitory in a pancreatic xenograft | No | [24] |
| EphA2 | mAb208 | Agonist, leading to receptor degradation, promoting CD8 T cell-mediated lysis | No | [25] |
| EphA2 | 3F2-3M (B233) | Agonist, induction of ADCC | No | [27] |
| EphA2 | DS-8895a | Antagonist, induction of ADCC; radiolabeled for biodistribution | Yes | [28,31,32] |
| EphA2 | IF7-Lu-177 | Radiolabeled mAb inhibited murine leukemia model | No | [33] |
| EphA2 | 1C1/MEDI-547 | Agonist, receptor phosphorylation and internalization; Auristatin-1C1 ADC (not 1C1 alone) inhibited tumor growth and metastasis in vivo; radiolabeled for biodistribution | Yes | [34,35,36,37,38] |
| EphA2 | 4B3 | Radio-labeled for PET/MRI biodistribution imaging | No | [39] |
| EphA2 | MM-310 | Pro-docetaxol loaded immunoliposomes, tumor targeting and growth inhibition, low organ toxicity | Begun | [40] |
| EphA3 | IIIA4/KB004/ Ifabotuzumab | Agonist, receptor phosphorylation and internalization; Targeted stem-like tumor and TME cells, inhibited growth and vascularity of solid and hematopoietic tumors | Yes | [41,42,43,44,45,46] |
| EphA3 | IIIA4 conjugates | Radioactive or drug payloads inhibited tumor growth in leukemic and GBM models; also in vivo imaging | No | [47,48,49,50] |
| EphA2/A3 | A2/A3 BsAb | Reduced GBM stem-cell qualities and tumor growth in vivo | No | [51] |
| EphA10 | EphA10/CD3 BsAb | Promoted T cell-mediated tumor cell lysis and inhibited breast cancer xenografts | No | [52] |
| EphB2 | 2H9 | Antagonist. Auristatin-ADC inhibited colon xenograft growth | No | [53,54] |
| EphB4 | C2 | Inhibits tumor angiogenesis and growth | No | [55] |
| EphB4 | Mab131, Mab47 | Inhibit tumor angiogenesis and growth; radio- labeled to image; 131 induces receptor degradation | No | [54,56] |
| EphrinA4 | PF-06647263 | ADC with calicheamicin-γ1, inhibited breast and ovarian xenografts | Yes | [57,58] |
| EphrinB2 | scFv B11, 2B1 | Inhibited angiogenesis and pancreatic xenografts | No | [59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janes, P.W.; Vail, M.E.; Gan, H.K.; Scott, A.M. Antibody Targeting of Eph Receptors in Cancer. Pharmaceuticals 2020, 13, 88. https://doi.org/10.3390/ph13050088
Janes PW, Vail ME, Gan HK, Scott AM. Antibody Targeting of Eph Receptors in Cancer. Pharmaceuticals. 2020; 13(5):88. https://doi.org/10.3390/ph13050088
Chicago/Turabian StyleJanes, Peter W., Mary E. Vail, Hui K. Gan, and Andrew M. Scott. 2020. "Antibody Targeting of Eph Receptors in Cancer" Pharmaceuticals 13, no. 5: 88. https://doi.org/10.3390/ph13050088
APA StyleJanes, P. W., Vail, M. E., Gan, H. K., & Scott, A. M. (2020). Antibody Targeting of Eph Receptors in Cancer. Pharmaceuticals, 13(5), 88. https://doi.org/10.3390/ph13050088