Polymyxin Delivery Systems: Recent Advances and Challenges



{kind=link}
Abstract
1. Introduction
2. Chemical Structure of Polymyxins
3. Mechanism of Action and Side Effects
4. Polymyxin Delivery Systems
4.1. Polymeric Nano- and Microparticle Carriers
4.2. Lipid-Based Nanostructures (Liposomes and Niosomes)
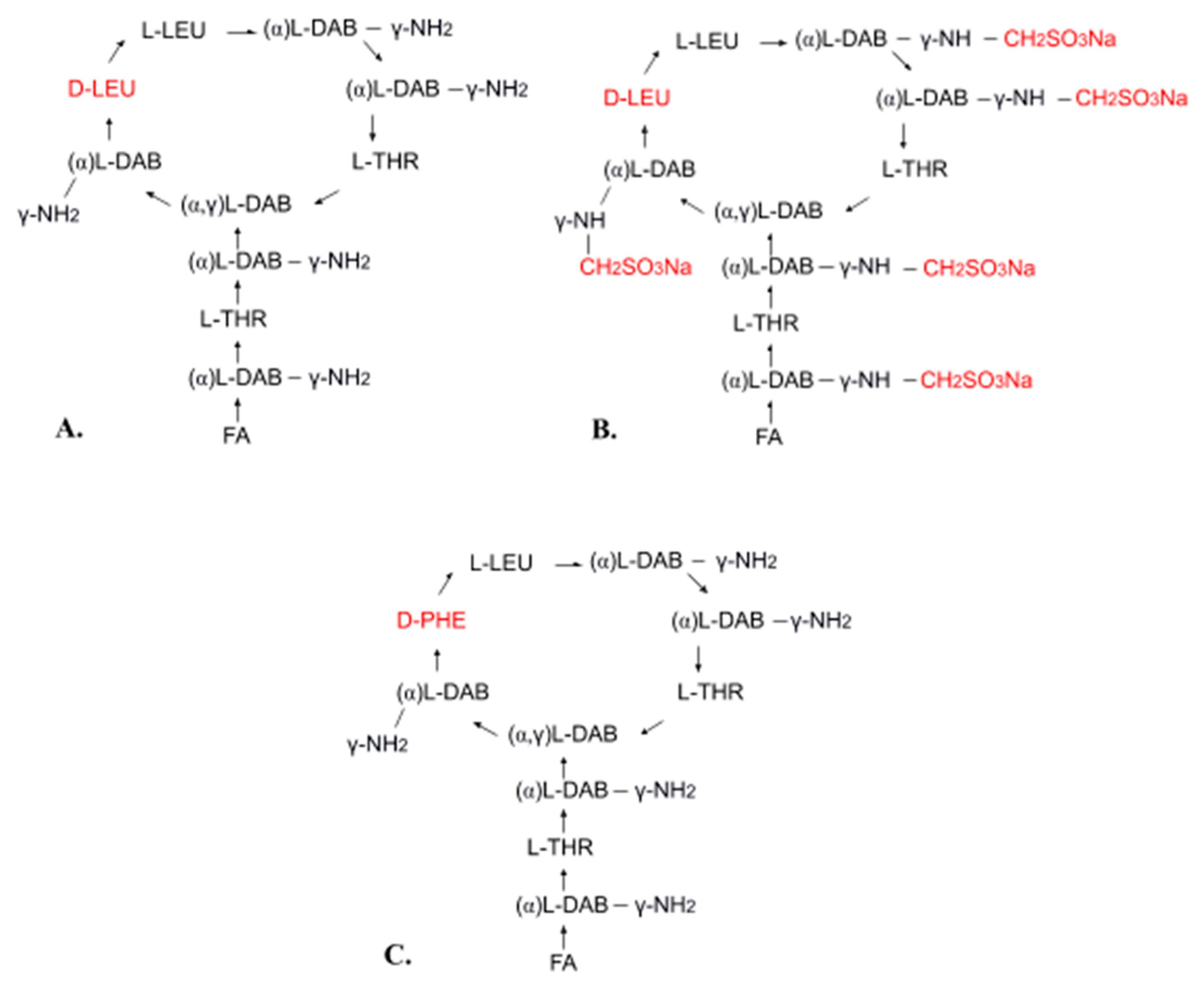
4.3. Conjugates
4.4. Structured Gels (Hydrogels and Microgels)
4.5. Polymeric Nanofibers and Membranes
4.6. Microneedles
5. Future Aspects
Author Contributions
Funding
Conflicts of Interest
References
- WHO Publishes List of Bacteria for Which New Antibiotics are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 12 April 2020).
- Gajdacs, M. The concept of an ideal antibiotic: Implications for drug design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bello, C. Antibiotic adjuvants—A strategy to unlock bacterial resistance to antibiotics. Bioorg. Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pornpattananangku, D.; Hu, C.M.; Huang, C.M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef]
- Raza, A.; Sime, F.B.; Cabot, P.J.; Maqbool, F.; Roberts, J.A.; Falconer, J.R. Solid nanoparticles for oral antimicrobial drug delivery: A review. Drug Discov. Today 2019, 24, 858–866. [Google Scholar] [CrossRef]
- Dos Santos Ramos, M.A.; Da Silva, P.B.; Sposito, L.; De Toledo, L.G.; Bonifacio, B.V.; Rodero, C.F.; Dos Santos, K.C.; Chorilli, M.; Bauab, T.M. Nanotechnology-based drug delivery systems for control of microbial biofilms: A review. Int. J. Nanomed. 2018, 13, 1179–1213. [Google Scholar] [CrossRef]
- Martin-Serrano, A.; Gomez, R.; Ortega, P.; de la Mata, F.J. Nanosystems as vehicles for the delivery of antimicrobial peptides (AMPs). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef]
- Kelly, S.A.; Rodgers, A.M.; O’Brien, S.C.; Donnelly, R.F.; Gilmore, B.F. Gut check time: Antibiotic delivery strategies to reduce antimicrobial resistance. Trends Biotechnol. 2020, 38, 447–462. [Google Scholar] [CrossRef]
- Landman, D.; Georgescu, C.; Martin, D.A.; Quale, J. Polymyxins revisited. Clin. Microbiol. Rev. 2008, 21, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.X.; Wang, C.Y.; Li, Y.Y.; Li, J.; Wan, Q.Q.; Chen, J.H.; Tay, F.R.; Niu, L.N. Considerations and caveats in combating eskape pathogens against nosocomial infections. Adv. Sci. 2020, 7, 1901872. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Davin-Regli, A.; Lavigne, J.P.; Pages, J.M. Enterobacter spp.: Update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin. Microbiol. Rev. 2019, 32, e00002–e00019. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-how P. aeruginosa can escape antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef]
- Lupo, A.; Haenni, M.; Madec, J.Y. Antimicrobial resistance in Acinetobacter spp. and Pseudomonas spp. Microbiol. Spectr. 2018, 6, 377–393. [Google Scholar] [CrossRef]
- Felden, B.; Cattoir, V. Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrob. Agents Chemother 2018, 62, e02503–e02517. [Google Scholar] [CrossRef]
- Gaspar, M.C.; Couet, W.; Olivier, J.C.; Pais, A.A.; Sousa, J.J. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: A review. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1231–1252. [Google Scholar] [CrossRef]
- Friebel, C.; Steckel, H. Single-use disposable dry powder inhalers for pulmonary drug delivery. Expert Opin. Drug Deliv. 2010, 7, 1359–1372. [Google Scholar] [CrossRef]
- Safdar, A.; Shelburne, S.A.; Evans, S.E.; Dickey, B.F. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin. Drug Saf. 2009, 8, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.; Mills, N.; Whitaker, P. Nebuliser systems for drug delivery in cystic fibrosis. Cochrane Database Syst. Rev. 2013, 4. [Google Scholar] [CrossRef]
- Banerjee, D.; Stableforth, D. The treatment of respiratory pseudomonas infection in cystic fibrosis: What drug and which way? Drugs 2000, 60, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Novel formulations for antimicrobial peptides. Int. J. Mol. Sci. 2014, 15, 18040–18083. [Google Scholar] [CrossRef]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and their potential next generation as therapeutic antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure—Activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef]
- Martin, N.I.; Hu, H.; Moake, M.M.; Churey, J.J.; Whittal, R.; Worobo, R.W.; Vederas, J.C. Isolation, structural characterization, and properties of mattacin (polymyxin M), a cyclic peptide antibiotic produced by paenibacillus kobensis M. J. Biol. Chem. 2003, 278, 13124–13132. [Google Scholar] [CrossRef]
- Orwa, J.A.; Van Gerven, A.; Roets, E.; Hoogmartens, J. Development and validation of a liquid chromatography method for analysis of colistin sulphate. Chromatographia 2000, 51, 433–436. [Google Scholar] [CrossRef]
- Cao, G.; Ali, F.E.; Chiu, F.; Zavascki, A.P.; Nation, R.L.; Li, J. Development and validation of a reversed-phase high-performance liquid chromatography assay for polymyxin b in human plasma. J. Antimicrob. Chemother 2008, 62, 1009–1014. [Google Scholar] [CrossRef]
- Pfeifer, C.; Fassauer, G.; Gerecke, H.; Jira, T.; Remane, Y.; Frontini, R.; Byrne, J.; Reinhardt, R. Purity determination of amphotericin b, colistin sulfate and tobramycin sulfate in a hydrophilic suspension by HPLC. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 990, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Burkin, M.; Eremin, S.; Dias, A.C.P.; Zhang, X. Development of competitive ELISA and CLEIA for quantitative analysis of polymyxin B. Food Analyt. Methods 2019, 12, 1412–1419. [Google Scholar] [CrossRef]
- Decolin, D.; Leroy, P.; Nicolas, A.; Archimbault, P. Hyphenated liquid chromatographic method for the determination of colistin residues in bovine tissues. J. Chromatogr. Sci. 1997, 35, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Orwa, J.A.; Govaerts, C.; Busson, R.; Roets, E.; Van Schepdael, A.; Hoogmartens, J. Isolation and structural characterization of colistin components. J. Antibiot. 2001, 54, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.J.; Li, J.; Nation, R.L.; Prankerd, R.J.; Velkov, T.; Boyd, B.J. Self-assembly behavior of colistin and its prodrug colistin methanesulfonate: Implications for solution stability and solubilization. J. Phys. Chem. B 2010, 114, 4836–4840. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.D.; Stern, R.C.; O’Riordan, M.A.; Blumer, J.L. The pharmacokinetics of colistin in patients with cystic fibrosis. J. Clin. Pharmacol. 2001, 41, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Lopez, C.A.; Gnanakaran, S. Permeability barrier of gram-negative cell envelopes and approaches to bypass it. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef]
- Nishino, K.; Hsu, F.F.; Turk, J.; Cromie, M.J.; Wosten, M.M.; Groisman, E.A. Identification of the lipopolysaccharide modifications controlled by the Salmonella PmrA/PmrB system mediating resistance to Fe(III) and Al(III). Mol. Microbiol. 2006, 61, 645–654. [Google Scholar] [CrossRef]
- Hancock, R.E. Antibacterial peptides and the outer membranes of gram-negative bacilli. J. Med. Microbiol. 1997, 46, 1–3. [Google Scholar]
- Hancock, R.E.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.E.; Feola, D.J.; Rapp, R.P. Polymyxin B sulfate and colistin: Old antibiotics for emerging multiresistant gram-negative bacteria. Ann. Pharmacother. 1999, 33, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed. Res. Int. 2015, 2015, 679109. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.T.; Cajal, Y.; Skowronska, E.M.; Belkin, S.; Chen, J.; Van Dyk, T.K.; Sasser, M.; Jain, M.K. Cationic peptide antimicrobials induce selective transcription of micF and osmY in Escherichia coli. Biochim. Biophys. Acta 2000, 1463, 43–54. [Google Scholar] [CrossRef][Green Version]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Khondker, A.; Alsop, R.J.; Dhaliwal, A.; Saem, S.; Moran-Mirabal, J.M.; Rheinstadter, M.C. Membrane cholesterol reduces polymyxin b nephrotoxicity in renal membrane analogs. Biophys. J. 2017, 113, 2016–2028. [Google Scholar] [CrossRef]
- Khondker, A.; Dhaliwal, A.K.; Saem, S.; Mahmood, A.; Fradin, C.; Moran-Mirabal, J.; Rheinstadter, M.C. Membrane charge and lipid packing determine polymyxin-induced membrane damage. Commun. Biol. 2019, 2, 67. [Google Scholar] [CrossRef]
- Khondker, A.; Rheinstadter, M.C. How do bacterial membranes resist polymyxin antibiotics? Commun. Biol. 2020, 3, 77. [Google Scholar] [CrossRef]
- Fu, L.; Wan, M.; Zhang, S.; Gao, L.; Fang, W. Polymyxin B loosens lipopolysaccharide bilayer but stiffens phospholipid bilayer. Biophys. J. 2020, 118, 138–150. [Google Scholar] [CrossRef]
- Dupuy, F.G.; Pagano, I.; Andenoro, K.; Peralta, M.F.; Elhady, Y.; Heinrich, F.; Tristram-Nagle, S. Selective interaction of colistin with lipid model membranes. Biophys. J. 2018, 114, 919–928. [Google Scholar] [CrossRef]
- Zhang, H.; Srinivas, S.; Xu, Y.; Wei, W.; Feng, Y. Genetic and biochemical mechanisms for bacterial lipid a modifiers associated with polymyxin resistance. Trends Biochem. Sci. 2019, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wei, W.; Lei, S.; Lin, J.; Srinivas, S.; Feng, Y. An evolutionarily conserved mechanism for intrinsic and transferable polymyxin resistance. mBio 2018, 9, e02317. [Google Scholar] [CrossRef] [PubMed]
- Nang, S.C.; Li, J.; Velkov, T. The rise and spread of mcr plasmid-mediated polymyxin resistance. Crit. Rev. Microbiol. 2019, 45, 131–161. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, Z.; Gholizadeh, P.; Ganbarov, K.; Bialvaei, A.Z.; Mahmood, S.S.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Molecular mechanisms related to colistin resistance in enterobacteriaceae. Infect. Drug Resist. 2019, 12, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Kiani, M.H.; Imran, M.; Raza, A.; Shahnaz, G. Multi-functionalized nanocarriers targeting bacterial reservoirs to overcome challenges of multi drug-resistance. Daru 2020. [Google Scholar] [CrossRef] [PubMed]
- Yeom, J.H.; Lee, B.; Kim, D.; Lee, J.K.; Kim, S.; Bae, J.; Park, Y.; Lee, K. Gold nanoparticle-DNA aptamer conjugate-assisted delivery of antimicrobial peptide effectively eliminates intracellular Salmonella enterica serovar Typhimurium. Biomaterials 2016, 104, 43–51. [Google Scholar] [CrossRef]
- Domalaon, R.; De Silva, P.M.; Kumar, A.; Zhanel, G.G.; Schweizer, F. The anthelmintic drug niclosamide synergizes with colistin and reverses colistin resistance in gram-negative bacilli. Antimicrob. Agents Chemother 2019, 63. [Google Scholar] [CrossRef]
- Domalaon, R.; Okunnu, O.; Zhanel, G.G.; Schweizer, F. Synergistic combinations of anthelmintic salicylanilides oxyclozanide, rafoxanide, and closantel with colistin eradicates multidrug-resistant colistin-resistant gram-negative bacilli. J. Antibiot. 2019, 72, 605–616. [Google Scholar] [CrossRef]
- Ayerbe-Algaba, R.; Gil-Marques, M.L.; Miro-Canturri, A.; Parra-Millan, R.; Pachon-Ibanez, M.E.; Jimenez-Mejias, M.E.; Pachon, J.; Smani, Y. The anthelmintic oxyclozanide restores the activity of colistin against colistin-resistant gram-negative bacilli. Int. J. Antimicrob. Agents 2019, 54, 507–512. [Google Scholar] [CrossRef]
- Zimmerman, S.M.; Lafontaine, A.J.; Herrera, C.M.; McLean, A.B.; Trent, M.S. A whole-cell screen identifies small bioactives that synergize with polymyxin and exhibit antimicrobial activities against multidrug-resistant bacteria. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Niu, H.; Wang, R.; Cai, Y. Safety and efficacy of colistin alone or in combination in adults with Acinetobacter baumannii infection: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2019, 53, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Ioannidou, E.; Falagas, M.E. Colistin monotherapy vs. combination therapy: Evidence from microbiological, animal and clinical studies. Clin. Microbiol. Infect. 2008, 14, 816–827. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I.; Kasiakou, S.K.; Hatzopoulou, P.; Michalopoulos, A. Effectiveness and nephrotoxicity of colistin monotherapy vs. colistin-meropenem combination therapy for multidrug-resistant gram-negative bacterial infections. Clin. Microbiol. Infect. 2006, 12, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Ribera, A.; Benavent, E.; Lora-Tamayo, J.; Tubau, F.; Pedrero, S.; Cabo, X.; Ariza, J.; Murillo, O. Osteoarticular infection caused by MDR Pseudomonas aeruginosa: The benefits of combination therapy with colistin plus β-lactams. J. Antimicrob. Chemother 2015, 70, 3357–3365. [Google Scholar] [PubMed]
- Zhang, X.; Guo, F.; Shao, H.; Zheng, X. Clinical translation of polymyxin-based combination therapy: Facts, challenges and future opportunities. J. Infect. 2017, 74, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Falagas, M.E. Colistin versus polymyxin b for the treatment of patients with multidrug-resistant gram-negative infections: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2017, 49, 233–238. [Google Scholar] [CrossRef]
- Liu, Q.; Li, W.; Feng, Y.; Tao, C. Efficacy and safety of polymyxins for the treatment of Acinectobacter baumannii infection: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98091. [Google Scholar] [CrossRef]
- Zusman, O.; Altunin, S.; Koppel, F.; Dishon Benattar, Y.; Gedik, H.; Paul, M. Polymyxin monotherapy or in combination against carbapenem-resistant bacteria: Systematic review and meta-analysis. J. Antimicrob. Chemother 2017, 72, 29–39. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, J.; Liu, H.X.; Zhu, Y.G.; Qu, J.M. Intravenous combined with aerosolised polymyxin versus intravenous polymyxin alone in the treatment of pneumonia caused by multidrug-resistant pathogens: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2015, 46, 603–609. [Google Scholar] [CrossRef]
- Terayama, T.; Yamakawa, K.; Umemura, Y.; Aihara, M.; Fujimi, S. Polymyxin B hemoperfusion for sepsis and septic shock: A systematic review and meta-analysis. Surg. Infect. 2017, 18, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, S.C.; Wong, H.; Hop, C.E.C.A. Oral absorption. In Drug Metabolism and Pharmacokinetics Quick Guide; Springer: New York, NY, USA, 2011; pp. 47–56. [Google Scholar]
- Sorli, L.; Luque, S.; Li, J.; Campillo, N.; Danes, M.; Montero, M.; Segura, C.; Grau, S.; Horcajada, J.P. Colistin for the treatment of urinary tract infections caused by extremely drug-resistant Pseudomonas aeruginosa: Dose is critical. J. Infect. 2019, 79, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.K.; Nichols, B.L.B.; Edgar, K.J.; Murgia, X.; Loretz, B.; Lehr, C.M. Challenges and strategies in drug delivery systems for treatment of pulmonary infections. Eur. J. Pharm. Biopharm. 2019, 144, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef] [PubMed]
- Oliota, A.F.; Penteado, S.T.; Tonin, F.S.; Fernandez-Llimos, F.; Sanches, A.C. Nephrotoxicity prevalence in patients treated with polymyxins: A systematic review with meta-analysis of observational studies. Diagn. Microbiol. Infect. Dis. 2019, 94, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, F.; Dashti-Khavidaki, S. Colistin: Efficacy and safety in different populations. Expert Rev. Clin. Pharmacol. 2015, 8, 423–448. [Google Scholar] [CrossRef]
- Dai, C.; Xiao, X.; Li, J.; Ciccotosto, G.D.; Cappai, R.; Tang, S.; Schneider-Futschik, E.K.; Hoyer, D.; Velkov, T.; Shen, J. Molecular mechanisms of neurotoxicity induced by polymyxins and chemoprevention. ACS Chem. Neurosci. 2019, 10, 120–131. [Google Scholar] [CrossRef]
- Pinto-Alphandary, H.; Andremont, A.; Couvreur, P. Targeted delivery of antibiotics using liposomes and nanoparticles: Research and applications. Int. J. Antimicrob. Agents 2000, 13, 155–168. [Google Scholar] [CrossRef]
- Falciani, C.; Zevolini, F.; Brunetti, J.; Riolo, G.; Gracia, R.; Marradi, M.; Loinaz, I.; Ziemann, C.; Cossio, U.; Llop, J.; et al. Antimicrobial peptide-loaded nanoparticles as inhalation therapy for Pseudomonas aeruginosa infections. Int. J. Nanomed. 2020, 15, 1117–1128. [Google Scholar] [CrossRef]
- Weers, J.G.; Miller, D.P.; Tarara, T.E. Spray-dried pulmosphere formulations for inhalation comprising crystalline drug particles. AAPS Pharm. Sci. Tech. 2019, 20, 103. [Google Scholar] [CrossRef]
- Woods, A.; Rahman, K.M. Antimicrobial molecules in the lung: Formulation challenges and future directions for innovation. Future Med. Chem. 2018, 10, 575–604. [Google Scholar] [CrossRef] [PubMed]
- Faustino, C.; Pinheiro, L. Lipid systems for the delivery of amphotericin b in antifungal therapy. Pharmaceutics 2020, 12, 29. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Diep, D.B.; Tonnesen, H.H. Topical antimicrobial peptide formulations for wound healing: Current developments and future prospects. Acta Biomater. 2020, 103, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Ohnstedt, E.; Lofton Tomenius, H.; Vagesjo, E.; Phillipson, M. The discovery and development of topical medicines for wound healing. Expert Opin. Drug Discov. 2019, 14, 485–497. [Google Scholar] [CrossRef]
- Ambekar, R.S.; Kandasubramanian, B. Advancements in nanofibers for wound dressing: A review. Eur. Polym. J. 2019, 117, 304–336. [Google Scholar] [CrossRef]
- Mofazzal Jahromi, M.A.; Sahandi Zangabad, P.; Moosavi Basri, S.M.; Sahandi Zangabad, K.; Ghamarypour, A.; Aref, A.R.; Karimi, M.; Hamblin, M.R. Nanomedicine and advanced technologies for burns: Preventing infection and facilitating wound healing. Adv. Drug Deliv. Rev. 2018, 123, 33–64. [Google Scholar] [CrossRef]
- Kurmi, B.D.; Tekchandani, P.; Paliwal, R.; Paliwal, S.R. Transdermal drug delivery: Opportunities and challenges for controlled delivery of therapeutic agents using nanocarriers. Curr. Drug Metab. 2017, 18, 481–495. [Google Scholar] [CrossRef]
- Nematpour, N.; Farhadian, N.; Ebrahimi, K.S.; Arkan, E.; Seyedi, F.; Khaledian, S.; Shahlaei, M.; Moradi, S. Sustained release nanofibrous composite patch for transdermal antibiotic delivery. Colloids Surfaces A Physicochem. Eng. Aspects 2020, 586, 124267. [Google Scholar] [CrossRef]
- Kodoth, A.K.; Ghate, V.M.; Lewis, S.A.; Prakash, B.; Badalamoole, V. Pectin-based silver nanocomposite film for transdermal delivery of donepezil. Int. J. Biol. Macromol. 2019, 134, 269–279. [Google Scholar] [CrossRef]
- Gonzalez-Vazquez, P.; Larraneta, E.; McCrudden, M.T.C.; Jarrahian, C.; Rein-Weston, A.; Quintanar-Solares, M.; Zehrung, D.; McCarthy, H.; Courtenay, A.J.; Donnelly, R.F. Transdermal delivery of gentamicin using dissolving microneedle arrays for potential treatment of neonatal sepsis. J. Control Release 2017, 265, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Tsioris, K.; Raja, W.K.; Pritchard, E.M.; Panilaitis, B.; Kaplan, D.L.; Omenetto, F.G. Fabrication of silk microneedles for controlled-release drug delivery. Adv. Funct. Mater. 2012, 22, 330–335. [Google Scholar] [CrossRef]
- Lee, N.Y.; Ko, W.C.; Hsueh, P.R. Nanoparticles in the treatment of infections caused by multidrug-resistant organisms. Front. Pharmacol. 2019, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Giri, T.K. (Eds.) Recent advances of chitosan nanoparticles as a carrier for delivery of antimicrobial drugs. In Polysaccharide Based Nano-Biocarrier in Drug Delivery; CRC Press: Boca Raton, FL, USA, 2018; pp. 637–679. [Google Scholar]
- Parisi, O.I.; Scrivano, L.; Sinicropi, M.S.; Puoci, F. Polymeric nanoparticle constructs as devices for antibacterial therapy. Curr. Opin. Pharmacol. 2017, 36, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Opatz, T.; Landfester, K.; Wurm, F.R. Carbohydrate nanocarriers in biomedical applications: Functionalization and construction. Chem. Soc. Rev. 2015, 44, 8301–8325. [Google Scholar] [CrossRef]
- D’ANGELO, I.; Conte, C.; Miro, A.; Quaglia, F.; Ungaro, F. Pulmonary drug delivery: A role for polymeric nanoparticles? Curr. Top. Med. Chem. 2015, 15, 386–400. [Google Scholar] [CrossRef][Green Version]
- Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-based local antimicrobial drug delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57. [Google Scholar] [CrossRef]
- Yasar, H.; Ho, D.K.; De Rossi, C.; Herrmann, J.; Gordon, S.; Loretz, B.; Lehr, C.M. Starch-chitosan polyplexes: A versatile carrier system for anti-infectives and gene delivery. Polymers 2018, 10, 252. [Google Scholar] [CrossRef]
- Raik, S.V.; Gasilova, E.R.; Dubashynskaya, N.V.; Dobrodumov, A.V.; Skorik, Y.A. Diethylaminoethyl chitosan-hyaluronic acid polyelectrolyte complexes. Int. J. Biol. Macromol. 2020, 146, 1161–1168. [Google Scholar] [CrossRef]
- Deacon, J.; Abdelghany, S.M.; Quinn, D.J.; Schmid, D.; Megaw, J.; Donnelly, R.F.; Jones, D.S.; Kissenpfennig, A.; Elborn, J.S.; Gilmore, B.F.; et al. Antimicrobial efficacy of tobramycin polymeric nanoparticles for pseudomonas aeruginosa infections in cystic fibrosis: Formulation, characterisation and functionalisation with dornase alfa (dnase). J. Control Release 2015, 198, 55–61. [Google Scholar] [CrossRef]
- Balmayor, E.R.; Baran, E.T.; Azevedo, H.S.; Reis, R.L. Injectable biodegradable starch/chitosan delivery system for the sustained release of gentamicin to treat bone infections. Carbohydr. Polym. 2012, 87, 32–39. [Google Scholar] [CrossRef]
- Coppi, G.; Iannuccelli, V.; Sala, N.; Bondi, M. Alginate microparticles for polymyxin B peyer’s patches uptake: Microparticles for antibiotic oral administration. J. Microencapsul. 2004, 21, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Coppi, G.; Sala, N.; Bondi, M.; Sergi, S.; Iannuccelli, V. Ex-vivo evaluation of alginate microparticles for polymyxin B oral administration. J. Drug Target. 2006, 14, 599–606. [Google Scholar] [CrossRef]
- Coppi, G.; Montanari, M.; Rossi, T.; Bondi, M.; Iannuccelli, V. Cellular uptake and toxicity of microparticles in a perspective of polymyxin b oral administration. Int. J. Pharm. 2010, 385, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Kuo, S.C.; Yao, B.Y.; Fang, Z.S.; Lee, Y.T.; Chang, Y.C.; Chen, T.L.; Hu, C.J. Colistin nanoparticle assembly by coacervate complexation with polyanionic peptides for treating drug-resistant gram-negative bacteria. Acta Biomater. 2018, 82, 133–142. [Google Scholar] [CrossRef]
- Zashikhina, N.N.; Yudin, D.V.; Tarasenko, I.I.; Osipova, O.M.; Korzhikova-Vlakh, E.G. Multilayered particles based on biopolyelectrolytes as potential peptide delivery systems. Polym. Sci. Ser. A 2020, 62, 43–53. [Google Scholar] [CrossRef]
- Souza Ribeiro Costa, J.; Medeiros, M.; Yamashiro-Kanashiro, E.H.; Rocha, M.C.; Cotrim, P.C.; Stephano, M.A.; Lancellotti, M.; Tavares, G.D.; Oliveira-Nascimento, L. Biodegradable nanocarriers coated with polymyxin B: Evaluation of leishmanicidal and antibacterial potential. PLoS Negl. Trop. Dis. 2019, 13, e0007388. [Google Scholar] [CrossRef] [PubMed]
- Abouelmagd, S.A.; Abd Ellah, N.H.; Amen, O.; Abdelmoez, A.; Mohamed, N.G. Self-assembled tannic acid complexes for ph-responsive delivery of antibiotics: Role of drug-carrier interactions. Int. J. Pharm. 2019, 562, 76–85. [Google Scholar] [CrossRef]
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I. Mesoporous silica nanoparticles in biomedical applications. Chem. Soc. Rev. 2012, 41, 2590–2605. [Google Scholar] [CrossRef]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.Y. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Gounani, Z.; Asadollahi, M.A.; Pedersen, J.N.; Lyngso, J.; Skov Pedersen, J.; Arpanaei, A.; Meyer, R.L. Mesoporous silica nanoparticles carrying multiple antibiotics provide enhanced synergistic effect and improved biocompatibility. Colloids Surf. B Biointerfaces 2019, 175, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Gounani, Z.; Asadollahi, M.A.; Meyer, R.L.; Arpanaei, A. Loading of polymyxin B onto anionic Mesoporous silica nanoparticles retains antibacterial activity and enhances biocompatibility. Int. J. Pharm. 2018, 537, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Naeem, S.; Viswanathan, G.; Misran, M.B. Liposomes as colloidal nanovehicles: On the road to success in intravenous drug delivery. Rev. Chem. Eng. 2018, 34, 365–383. [Google Scholar] [CrossRef]
- Wang, D.Y.; van der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-based antimicrobial delivery-systems for the treatment of bacterial infections. Front. Chem. 2019, 7, 872. [Google Scholar] [CrossRef] [PubMed]
- Menina, S.; Eisenbeis, J.; Kamal, M.A.M.; Koch, M.; Bischoff, M.; Gordon, S.; Loretz, B.; Lehr, C.M. Bioinspired liposomes for oral delivery of colistin to combat intracellular infections by Salmonella enterica. Adv. Healthc. Mater. 2019, 8, e1900564. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, L.; Tang, C.; Zhang, E.; Ding, L.; Yang, L. Preparation and characterisation of the colistin-entrapped liposome driven by electrostatic interaction for intravenous administration. J. Microencapsul. 2016, 33, 427–437. [Google Scholar] [CrossRef]
- Wang, S.; Yu, S.; Lin, Y.; Zou, P.; Chai, G.; Yu, H.H.; Wickremasinghe, H.; Shetty, N.; Ling, J.; Li, J.; et al. Co-delivery of ciprofloxacin and colistin in liposomal formulations with enhanced in vitro antimicrobial activities against multidrug resistant Pseudomonas aeruginosa. Pharm. Res. 2018, 35, 187. [Google Scholar] [CrossRef]
- Wallace, S.J.; Li, J.; Nation, R.L.; Prankerd, R.J.; Boyd, B.J. Interaction of colistin and colistin methanesulfonate with liposomes: Colloidal aspects and implications for formulation. J. Pharm. Sci. 2012, 101, 3347–3359. [Google Scholar] [CrossRef]
- Yu, S.; Wang, S.; Zou, P.; Chai, G.; Lin, Y.W.; Velkov, T.; Li, J.; Pan, W.; Zhou, Q.T. Inhalable liposomal powder formulations for co-delivery of synergistic ciprofloxacin and colistin against multi-drug resistant gram-negative lung infections. Int. J. Pharm. 2020, 575, 118915. [Google Scholar] [CrossRef]
- Jadon, P.S.; Gajbhiye, V.; Jadon, R.S.; Gajbhiye, K.R.; Ganesh, N. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS Pharm. Sci. Tech. 2009, 10, 1186–1192. [Google Scholar] [CrossRef]
- Chauhan, M.K.; Bhatt, N. Bioavailability enhancement of polymyxin b with novel drug delivery: Development and optimization using quality-by-design approach. J. Pharm. Sci. 2019, 108, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Laing, B. Bioconjugates for targeted delivery of therapeutic oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Turos, E.; Shim, J.Y.; Wang, Y.; Greenhalgh, K.; Reddy, G.S.; Dickey, S.; Lim, D.V. Antibiotic-conjugated polyacrylate nanoparticles: New opportunities for development of anti-MRSA agents. Bioorg. Med. Chem. Lett. 2007, 17, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Page, M.G. Siderophore conjugates. Ann N. Y. Acad. Sci. 2013, 1277, 115–126. [Google Scholar] [CrossRef]
- Berezin, A.S.; Lomkova, E.A.; Skorik, Y.A. Chitosan conjugates with biologically active compounds: Design strategies, properties, and targeted drug delivery. Rus. Chem. Bull. 2012, 61, 781–795. [Google Scholar] [CrossRef]
- Berezin, A.S.; Skorik, Y.A. Chitosan-isoniazid conjugates: Synthesis, evaluation of tuberculostatic activity, biodegradability and toxicity. Carbohydr. Polym. 2015, 127, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Dizdarevic, A.; Efiana, N.A.; Phan, T.N.Q.; Matuszczak, B.; Bernkop-Schnurch, A. Imine bond formation: A novel concept to incorporate peptide drugs in self-emulsifying drug delivery systems (SEDDS). Eur. J. Pharm. Biopharm. 2019, 142, 92–100. [Google Scholar] [CrossRef]
- Varache, M.; Powell, L.C.; Aarstad, O.A.; Williams, T.L.; Wenzel, M.N.; Thomas, D.W.; Ferguson, E.L. Polymer masked-unmasked protein therapy: Identification of the active species after amylase activation of dextrin-colistin conjugates. Mol. Pharm. 2019, 16, 3199–3207. [Google Scholar] [CrossRef]
- Roberts, J.L.; Cattoz, B.; Schweins, R.; Beck, K.; Thomas, D.W.; Griffiths, P.C.; Ferguson, E.L. In vitro evaluation of the interaction of dextrin-colistin conjugates with bacterial lipopolysaccharide. J. Med. Chem. 2016, 59, 647–654. [Google Scholar] [CrossRef]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. Development and validation of an in vitro pharmacokinetic/pharmacodynamic model to test the antibacterial efficacy of antibiotic polymer conjugates. Antimicrob. Agents Chemother 2015, 59, 1837–1843. [Google Scholar] [CrossRef]
- Ferguson, E.L.; Azzopardi, E.; Roberts, J.L.; Walsh, T.R.; Thomas, D.W. Dextrin-colistin conjugates as a model bioresponsive treatment for multidrug resistant bacterial infections. Mol. Pharm. 2014, 11, 4437–4447. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Schneider, E.K.; Nikolaou, V.; Klein, T.; Li, J.; Davis, T.P.; Whittaker, M.R.; Wilson, P.; Kempe, K.; Velkov, T.; et al. Hydrolyzable poly[poly(ethylene glycol) methyl ether acrylate]-colistin prodrugs through copper-mediated photoinduced living radical polymerization. Bioconjug. Chem. 2017, 28, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.E.; Bell, C.S.; Mejias, R.; McClain, M.S.; Cover, T.L.; Giorgio, T.D. Colistin-functionalized nanoparticles for the rapid capture of Acinetobacter baumannii. J. Biomed. Nanotechnol. 2016, 12, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control Release 2007, 119, 5–24. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Petrova, V.A.; Elokhovskiy, V.Y.; Raik, S.V.; Poshina, D.N.; Romanov, D.P.; Skorik, Y.A. Alginate gel reinforcement with chitin nanowhiskers modulates rheological properties and drug release profile. Biomolecules 2019, 9, 291. [Google Scholar] [CrossRef]
- Malmsten, M.; Bysell, H.; Hansson, P. Biomacromolecules in microgels—Opportunities and challenges for drug delivery. Curr. Opin. Colloid Interface Sci. 2010, 15, 435–444. [Google Scholar] [CrossRef]
- Obuobi, S.; Voo, Z.X.; Low, M.W.; Czarny, B.; Selvarajan, V.; Ibrahim, N.L.; Yang, Y.Y.; Ee, P.L.R. Phenylboronic acid functionalized polycarbonate hydrogels for controlled release of polymyxin B in pseudomonas aeruginosa infected burn wounds. Adv. Healthc. Mater. 2018, 7, e1701388. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Sun, T.; Tsou, Y.H.; Chen, H.; Xu, X. Dual-functional dextran-peg hydrogel as an antimicrobial biomedical material. Macromol. Biosci. 2018, 18, 1700325. [Google Scholar] [CrossRef]
- Bayat, F.; Karimi, A.R. Design of photodynamic chitosan hydrogels bearing phthalocyanine-colistin conjugate as an antibacterial agent. Int. J. Biol. Macromol. 2019, 129, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Borro, B.C.; Bohr, A.; Bucciarelli, S.; Boetker, J.P.; Foged, C.; Rantanen, J.; Malmsten, M. Microfluidics-based self-assembly of peptide-loaded microgels: Effect of three dimensional (3D) printed micromixer design. J. Colloid Interface Sci. 2019, 538, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Wendorff, J.H.; Greiner, A. Use of electrospinning technique for biomedical applications. Polymer 2008, 49, 5603–5621. [Google Scholar] [CrossRef]
- Kim, K.; Luu, Y.K.; Chang, C.; Fang, D.; Hsiao, B.S.; Chu, B.; Hadjiargyrou, M. Incorporation and controlled release of a hydrophilic antibiotic using poly(lactide-co-glycolide)-based electrospun nanofibrous scaffolds. J. Control Release 2004, 98, 47–56. [Google Scholar] [CrossRef]
- Liang, D.; Hsiao, B.S.; Chu, B. Functional electrospun nanofibrous scaffolds for biomedical applications. Adv. Drug Deliv. Rev. 2007, 59, 1392–1412. [Google Scholar] [CrossRef]
- Garg, T.; Singh, O.; Arora, S.; Murthy, R. Scaffold: A novel carrier for cell and drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 1–63. [Google Scholar] [CrossRef]
- Zienkiewicz-Strzalka, M.; Derylo-Marczewska, A.; Skorik, Y.A.; Petrova, V.A.; Choma, A.; Komaniecka, I. Silver nanoparticles on chitosan/silica nanofibers: Characterization and antibacterial activity. Int. J. Mol. Sci. 2020, 21, 166. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, R.; Xu, J.; Lan, Y.; Jiao, Y.; Zhou, C.; Zhao, Y. Poly(l-lactide)/halloysite nanotube electrospun mats as dual-drug delivery systems and their therapeutic efficacy in infected full-thickness burns. J. Biomater. Appl. 2015, 30, 512–525. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, Z.; Zhao, D.; Wang, F.; Zhang, K.; Li, Y.; Wen, Y.; Wang, C. Polymyxin b immobilized nanofiber sponge for endotoxin adsorption. Eur. Polym. J. 2019, 110, 69–75. [Google Scholar] [CrossRef]
- Shi, R.; Niu, Y.; Gong, M.; Ye, J.; Tian, W.; Zhang, L. Antimicrobial gelatin-based elastomer nanocomposite membrane loaded with ciprofloxacin and polymyxin b sulfate in halloysite nanotubes for wound dressing. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 87, 128–138. [Google Scholar] [CrossRef]
- Ita, K. Dissolving microneedles for transdermal drug delivery: Advances and challenges. Biomed. Pharmacother. 2017, 93, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Hughes, H.; O’Reilly, N.J.; Allender, C.J.; Barrow, D.A.; McLoughlin, P. Dissolving microneedle based transdermal delivery of therapeutic peptide analogues. Int. J. Pharm. 2019, 565, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Hughes, H.; O’Reilly, N.J.; McLoughlin, P. Formulation and characterisation of dissolving microneedles for the transdermal delivery of therapeutic peptides. Int. J. Pharm. 2017, 526, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Pamornpathomkul, B.; Ngawhirunpat, T.; Tekko, I.A.; Vora, L.; McCarthy, H.O.; Donnelly, R.F. Dissolving polymeric microneedle arrays for enhanced site-specific acyclovir delivery. Eur. J. Pharm. Sci. 2018, 121, 200–209. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubashynskaya, N.V.; Skorik, Y.A. Polymyxin Delivery Systems: Recent Advances and Challenges. Pharmaceuticals 2020, 13, 83. https://doi.org/10.3390/ph13050083
Dubashynskaya NV, Skorik YA. Polymyxin Delivery Systems: Recent Advances and Challenges. Pharmaceuticals. 2020; 13(5):83. https://doi.org/10.3390/ph13050083
Chicago/Turabian StyleDubashynskaya, Natallia V., and Yury A. Skorik. 2020. "Polymyxin Delivery Systems: Recent Advances and Challenges" Pharmaceuticals 13, no. 5: 83. https://doi.org/10.3390/ph13050083
APA StyleDubashynskaya, N. V., & Skorik, Y. A. (2020). Polymyxin Delivery Systems: Recent Advances and Challenges. Pharmaceuticals, 13(5), 83. https://doi.org/10.3390/ph13050083